Molecular Neuropathology of TDP-43 Proteinopathies

Abstract
:1. Introduction
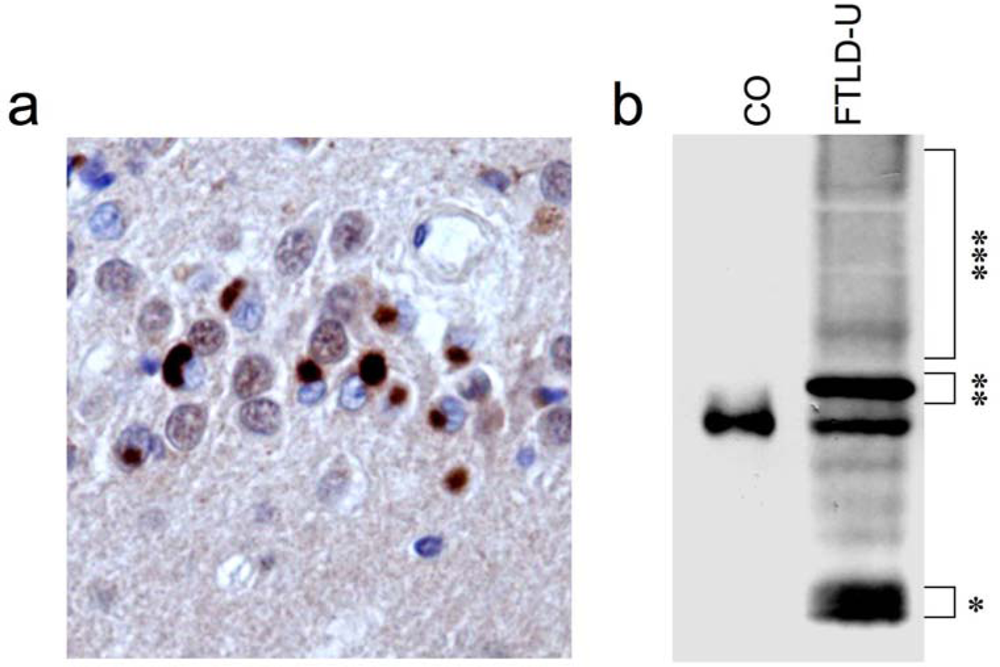
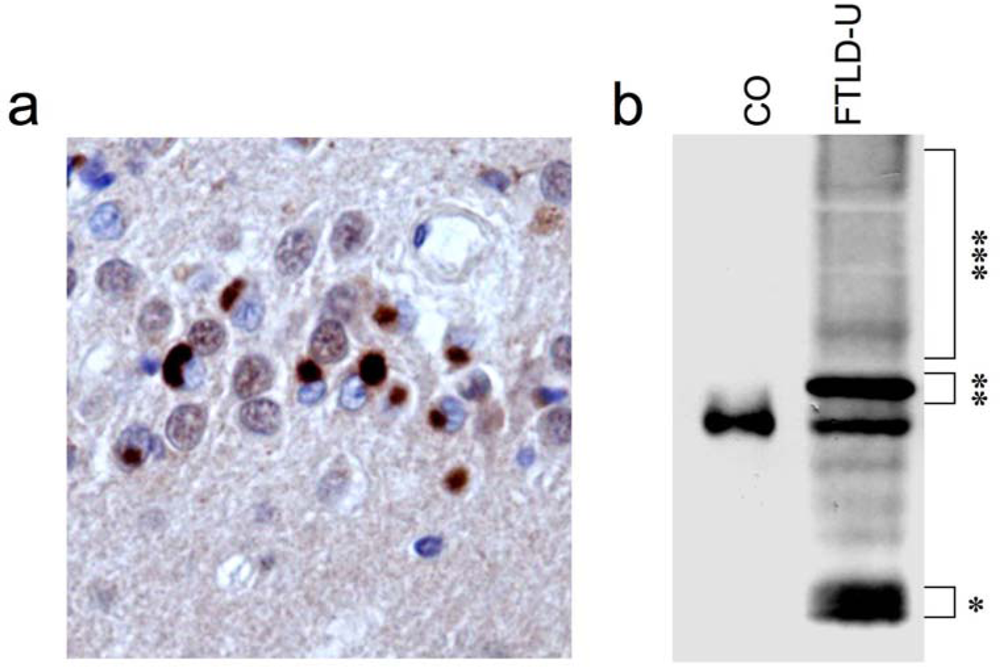
2. Identification of TDP-43 as disease protein in FTLD-U and ALS
3. TDP-43 pathology in sporadic and familial forms of FTLD-U
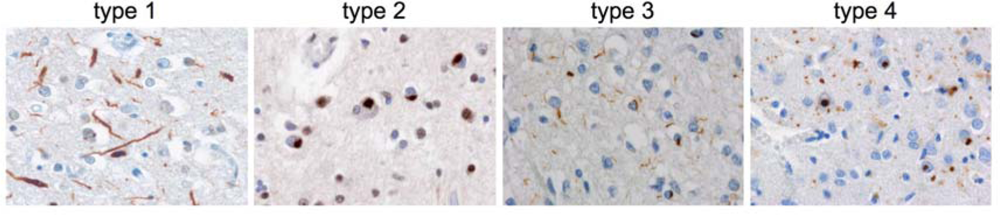
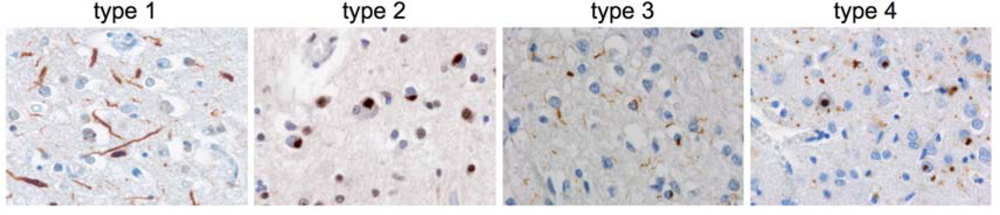
3.1. Heterogeneity among TDP-43-positive FTLD-U cases
3.2. Not all FTLD-U cases show TDP-43 pathology
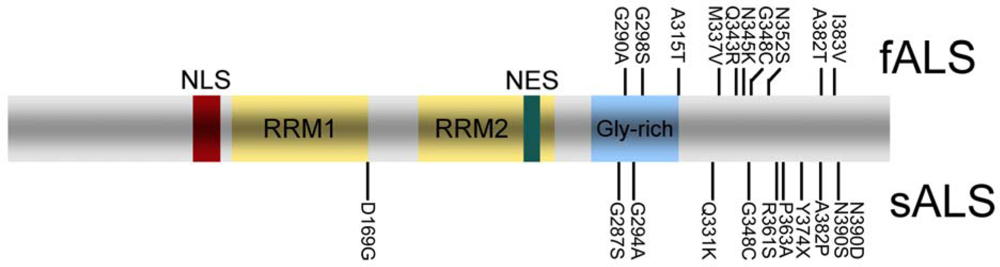
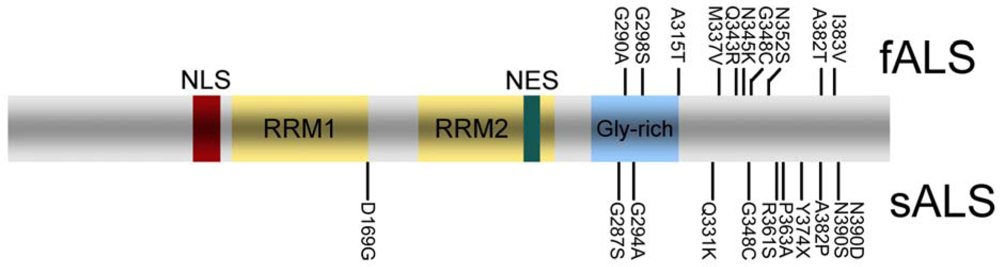
4. TDP-43 pathology in sporadic and familial ALS
5. TDP-43 pathology in other neurodegenerative diseases
6. Biology and Pathobiology of TDP-43
7. Conclusions
Acknowledgments
References
- Neary, D; Snowden, JS; Gustafson, L; Passant, U; Stuss, D; Black, S; Freedman, M; Kertesz, A; Robert, PH; Albert, M; Boone, K; Miller, BL; Cummings, J; Benson, DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar]
- Lomen-Hoerth, C; Anderson, T; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar]
- Forman, MS; Farmer, J; Johnson, JK; Clark, CM; Arnold, SE; Coslett, HB; Chatterjee, A; Hurtig, HI; Karlawish, JH; Rosen, HJ; Van Deerlin, V; Lee, VM; Miller, BL; Trojanowski, JQ; Grossman, M. Frontotemporal dementia: Clinicopathological correlations. Ann. Neurol 2006, 59, 952–962. [Google Scholar]
- Lee, VM; Goedert, M; Trojanowski, JQ. Neurodegenerative tauopathies. Ann. Rev. Neurosci 2001, 24, 1121–1159. [Google Scholar]
- Neumann, M; Sampathu, DM; Kwong, LK; Truax, AC; Micsenyi, MC; Chou, TT; Bruce, J; Schuck, T; Grossman, M; Clark, CM; McCluskey, LF; Miller, BL; Masliah, E; Mackenzie, IR; Feldman, H; Feiden, W; Kretzschmar, HA; Trojanowski, JQ; Lee, VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar]
- Arai, T; Hasegawa, M; Akiyama, H; Ikeda, K; Nonaka, T; Mori, H; Mann, D; Tsuchiya, K; Yoshida, M; Hashizume, Y; Oda, M. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun 2006, 351, 602–611. [Google Scholar]
- Mackenzie, IR; Bigio, EH; Ince, PG; Geser, F; Neumann, M; Cairns, NJ; Kwong, LK; Forman, MS; Ravits, J; Stewart, H; Eisen, A; McClusky, L; Kretzschmar, HA; Monoranu, CM; Highley, JR; Kirby, J; Siddique, T; Shaw, PJ; Lee, VM; Trojanowski, JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol 2007, 61, 427–434. [Google Scholar]
- Kwong, LK; Neumann, M; Sampathu, DM; Lee, VM; Trojanowski, JQ. TDP-43 proteinopathy: The neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. (Berl) 2007, 114, 63–70. [Google Scholar]
- Neumann, M; Kwong, LK; Sampathu, DM; Trojanowski, JQ; Lee, VM. TDP-43 Proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: Protein misfolding diseases without amyloidosis. Arch. Neurol 2007, 64, 1388–1394. [Google Scholar]
- Hasegawa, M; Arai, T; Nonaka, T; Kametani, F; Yoshida, M; Hashizume, Y; Beach, TG; Buratti, E; Baralle, F; Morita, M; Nakano, I; Oda, T; Tsuchiya, K; Akiyama, H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol 2008, 64, 60–70. [Google Scholar]
- Inukai, Y; Nonaka, T; Arai, T; Yoshida, M; Hashizume, Y; Beach, TG; Buratti, E; Baralle, FE; Akiyama, H; Hisanaga, S; Hasegawa, M. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett 2008, 582, 2899–2904. [Google Scholar]
- Sampathu, DM; Neumann, M; Kwong, LK; Chou, TT; Micsenyi, M; Truax, A; Bruce, J; Grossman, M; Trojanowski, JQ; Lee, VM. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol 2006, 169, 1343–1352. [Google Scholar]
- Davidson, Y; Kelley, T; Mackenzie, IR; Pickering-Brown, S; Du Plessis, D; Neary, D; Snowden, JS; Mann, DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. (Berl) 2007, 113, 521–533. [Google Scholar]
- Neumann, M; Mackenzie, IR; Cairns, NJ; Boyer, PJ; Markesbery, WR; Smith, CD; Taylor, JP; Kretzschmar, HA; Kimonis, VE; Forman, MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol 2007, 66, 152–157. [Google Scholar]
- Cairns, NJ; Neumann, M; Bigio, EH; Holm, IE; Troost, D; Hatanpaa, KJ; Foong, C; White, CL, 3rd; Schneider, JA; Kretzschmar, HA; Carter, D; Taylor-Reinwald, L; Paulsmeyer, K; Strider, J; Gitcho, M; Goate, AM; Morris, JC; Mishra, M; Kwong, LK; Stieber, A; Xu, Y; Forman, MS; Trojanowski, JQ; Lee, VM; Mackenzie, IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol 2007, 171, 227–240. [Google Scholar]
- Hatanpaa, KJ; Bigio, EH; Cairns, NJ; Womack, KB; Weintraub, S; Morris, JC; Foong, C; Xiao, G; Hladik, C; Mantanona, TY; White, CL, 3rd. TAR DNA-Binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: A midwest-southwest consortium for FTLD study. J. Neuropathol. Exp. Neurol 2008, 67, 271–279. [Google Scholar]
- Neumann, M; Igaz, LM; Kwong, LK; Nakashima-Yasuda, H; Kolb, SJ; Dreyfuss, G; Kretzschmar, HA; Trojanowski, JQ; Lee, VM. Absence of heterogeneous nuclear ribonucleoproteins and survival motor neuron protein in TDP-43 positive inclusions in frontotemporal lobar degeneration. Acta Neuropathol. (Berl) 2007, 113, 543–548. [Google Scholar]
- Neumann, M; Kwong, LK; Truax, AC; Vanmassenhove, B; Kretzschmar, HA; Van Deerlin, VM; Clark, CM; Grossman, M; Miller, BL; Trojanowski, JQ; Lee, VM. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol 2007, 66, 177–183. [Google Scholar]
- Brandmeir, NJ; Geser, F; Kwong, LK; Zimmerman, E; Qian, J; Lee, VM; Trojanowski, JQ. Severe subcortical TDP-43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta Neuropathol 2008, 115, 123–131. [Google Scholar]
- Igaz, LM; Kwong, LK; Xu, Y; Truax, AC; Uryu, K; Neumann, M; Clark, CM; Elman, LB; Miller, BL; Grossman, M; McCluskey, LF; Trojanowski, JQ; Lee, VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol 2008, 173, 182–194. [Google Scholar]
- Mackenzie, IR; Baborie, A; Pickering-Brown, S; Plessis, DD; Jaros, E; Perry, RH; Neary, D; Snowden, JS; Mann, DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: Classification and relation to clinical phenotype. Acta Neuropathol. (Berl) 2006, 112, 539–549. [Google Scholar]
- Mackenzie, IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol 2007, 114, 49–54. [Google Scholar]
- Forman, MS; Mackenzie, IR; Cairns, NJ; Swanson, E; Boyer, PJ; Drachman, DA; Jhaveri, BS; Karlawish, JH; Pestronk, A; Smith, TW; Tu, PH; Watts, GD; Markesbery, WR; Smith, CD; Kimonis, VE. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J. Neuropathol. Exp. Neurol 2006, 65, 571–581. [Google Scholar]
- Holm, IE; Englund, E; Mackenzie, IR; Johannsen, P; Isaacs, AM. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J. Neuropathol. Exp. Neurol 2007, 66, 884–891. [Google Scholar]
- Roeber, S; Mackenzie, IR; Kretzschmar, HA; Neumann, M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 2008, 116, 147–157. [Google Scholar]
- Mackenzie, IR; Foti, D; Woulfe, J; Hurwitz, TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 2008, 131, 1282–1293. [Google Scholar]
- Pikkarainen, M; Hartikainen, P; Alafuzoff, I. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J. Neuropathol. Exp. Neurol 2008, 67, 280–298. [Google Scholar]
- Mackenzie, IR; Neumann, M; Bigio, EH; Cairns, NJ; Alafuzoff, I; Kril, J; Kovacs, GG; Ghetti, B; Halliday, G; Holm, IE; Ince, PG; Kamphorst, W; Revesz, T; Rozemuller, AJ; Kumar-Singh, S; Akiyama, H; Baborie, A; Spina, S; Dickson, DW; Trojanowski, JQ; Mann, DM. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: Consensus recommendations. Acta Neuropathol 2009, 117, 15–18. [Google Scholar]
- Rosen, DR; Siddique, T; Patterson, D; Figlewicz, DA; Sapp, P; Hentati, A; Donaldson, D; Goto, J; O’Regan, JP; Deng, HX; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar]
- Tan, CF; Eguchi, H; Tagawa, A; Onodera, O; Iwasaki, T; Tsujino, A; Nishizawa, M; Kakita, A; Takahashi, H. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. (Berl) 2007, 113, 535–542. [Google Scholar]
- Robertson, J; Sanelli, T; Xiao, S; Yang, W; Horne, P; Hammond, R; Pioro, EP; Strong, MJ. Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci. Lett 2007, 420, 128–132. [Google Scholar]
- Turner, BJ; Baumer, D; Parkinson, NJ; Scaber, J; Ansorge, O; Talbot, K. TDP-43 expression in mouse models of amyotrophic lateral sclerosis and spinal muscular atrophy. BMC Neurosci 2008, 9, 104. [Google Scholar]
- Geser, F; Brandmeir, NJ; Kwong, LK; Martinez-Lage, M; Elman, L; McCluskey, L; Xie, SX; Lee, VM; Trojanowski, JQ. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch. Neurol 2008, 65, 636–641. [Google Scholar]
- Rollinson, S; Snowden, JS; Neary, D; Morrison, KE; Mann, DM; Pickering-Brown, SM. TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci. Lett 2007, 419, 1–4. [Google Scholar]
- Gijselinck, I; Sleegers, K; Engelborghs, S; Robberecht, W; Martin, JJ; Vandenberghe, R; Sciot, R; Dermaut, B; Goossens, D; van der Zee, J; De Pooter, T; Del-Favero, J; Santens, P; De Jonghe, P; De Deyn, PP; Van Broeckhoven, C; Cruts, M. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol. Aging 2007. [Google Scholar]
- Schumacher, A; Friedrich, P; Diehl-Schmid, J; Ibach, B; Perneczky, R; Eisele, T; Vukovich, R; Foerstl, H; Riemenschneider, M. No association of TDP-43 with sporadic frontotemporal dementia. Neurobiol. Aging 2009, 30, 157–159. [Google Scholar]
- Pamphlett, R; Luquin, N; McLean, C; Jew, SK; Adams, L. TDP-43 neuropathology is similar in sporadic amyotrophic lateral sclerosis with or without TDP-43 mutations. Neuropathol. Appl. Neurobiol. 2008. [Google Scholar]
- Rutherford, NJ; Zhang, YJ; Baker, M; Gass, JM; Finch, NA; Xu, YF; Stewart, H; Kelley, BJ; Kuntz, K; Crook, RJ; Sreedharan, J; Vance, C; Sorenson, E; Lippa, C; Bigio, EH; Geschwind, DH; Knopman, DS; Mitsumoto, H; Petersen, RC; Cashman, NR; Hutton, M; Shaw, CE; Boylan, KB; Boeve, B; Graff-Radford, NR; Wszolek, ZK; Caselli, RJ; Dickson, DW; Mackenzie, IR; Petrucelli, L; Rademakers, R. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 2008, 4, e1000193. [Google Scholar]
- Gitcho, MA; Baloh, RH; Chakraverty, S; Mayo, K; Norton, JB; Levitch, D; Hatanpaa, KJ; White, CL, 3rd; Bigio, EH; Caselli, R; Baker, M; Al-Lozi, MT; Morris, JC; Pestronk, A; Rademakers, R; Goate, AM; Cairns, NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol 2008, 63, 535–538. [Google Scholar]
- Kuhnlein, P; Sperfeld, AD; Vanmassenhove, B; Van Deerlin, V; Lee, VM; Trojanowski, JQ; Kretzschmar, HA; Ludolph, AC; Neumann, M. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch. Neurol 2008, 65, 1185–1189. [Google Scholar]
- Sreedharan, J; Blair, IP; Tripathi, VB; Hu, X; Vance, C; Rogelj, B; Ackerley, S; Durnall, JC; Williams, KL; Buratti, E; Baralle, F; de Belleroche, J; Mitchell, JD; Leigh, PN; Al-Chalabi, A; Miller, CC; Nicholson, G; Shaw, CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar]
- Van Deerlin, VM; Leverenz, JB; Bekris, LM; Bird, TD; Yuan, W; Elman, LB; Clay, D; Wood, EM; Chen-Plotkin, AS; Martinez-Lage, M; Steinbart, E; McCluskey, L; Grossman, M; Neumann, M; Wu, IL; Yang, WS; Kalb, R; Galasko, DR; Montine, TJ; Trojanowski, JQ; Lee, VM; Schellenberg, GD; Yu, CE. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 2008, 7, 409–416. [Google Scholar]
- Kabashi, E; Valdmanis, PN; Dion, P; Spiegelman, D; McConkey, BJ; Vande Velde, C; Bouchard, JP; Lacomblez, L; Pochigaeva, K; Salachas, F; Pradat, PF; Camu, W; Meininger, V; Dupre, N; Rouleau, GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet 2008, 40, 572–574. [Google Scholar]
- Daoud, H; Valdmanis, PN; Kabashi, E; Dion, P; Dupre, N; Camu, W; Meininger, V; Rouleau, GA. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2008. [Google Scholar]
- Yokoseki, A; Shiga, A; Tan, CF; Tagawa, A; Kaneko, H; Koyama, A; Eguchi, H; Tsujino, A; Ikeuchi, T; Kakita, A; Okamoto, K; Nishizawa, M; Takahashi, H; Onodera, O. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol 2008, 63, 538–542. [Google Scholar]
- Hasegawa, M; Arai, T; Akiyama, H; Nonaka, T; Mori, H; Hashimoto, T; Yamazaki, M; Oyanagi, K. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain 2007, 130, 1386–1394. [Google Scholar]
- Geser, F; Winton, MJ; Kwong, LK; Xu, Y; Xie, SX; Igaz, LM; Garruto, RM; Perl, DP; Galasko, D; Lee, VM; Trojanowski, JQ. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 2008, 115, 133–145. [Google Scholar]
- Freeman, SH; Spires-Jones, T; Hyman, BT; Growdon, JH; Frosch, MP. TAR-DNA binding protein 43 in Pick disease. J. Neuropathol. Exp. Neurol 2008, 67, 62–67. [Google Scholar]
- Fujishiro, H; Uchikado, H; Arai, T; Hasegawa, M; Akiyama, H; Yokota, O; Tsuchiya, K; Togo, T; Iseki, E; Hirayasu, Y. Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol 2008. [Google Scholar]
- Schwab, C; Arai, T; Hasegawa, M; Yu, S; McGeer, PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol 2008, 67, 1159–1165. [Google Scholar]
- Uryu, K; Nakashima-Yasuda, H; Forman, MS; Kwong, LK; Clark, CM; Grossman, M; Miller, BL; Kretzschmar, HA; Lee, VM; Trojanowski, JQ; Neumann, M. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol 2008, 67, 555–564. [Google Scholar]
- Nakashima-Yasuda, H; Uryu, K; Robinson, J; Xie, SX; Hurtig, H; Duda, JE; Arnold, SE; Siderowf, A; Grossman, M; Leverenz, JB; Woltjer, R; Lopez, OL; Hamilton, R; Tsuang, DW; Galasko, D; Masliah, E; Kaye, J; Clark, CM; Montine, TJ; Lee, VM; Trojanowski, JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. (Berl) 2007, 114, 221–229. [Google Scholar]
- Amador-Ortiz, C; Lin, WL; Ahmed, Z; Personett, D; Davies, P; Duara, R; Graff-Radford, NR; Hutton, ML; Dickson, DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol 2007, 61, 435–445. [Google Scholar]
- Higashi, S; Iseki, E; Yamamoto, R; Minegishi, M; Hino, H; Fujisawa, K; Togo, T; Katsuse, O; Uchikado, H; Furukawa, Y; Kosaka, K; Arai, H. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 2007, 1184, 284–294. [Google Scholar]
- Ou, SH; Wu, F; Harrich, D; Garcia-Martinez, LF; Gaynor, RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol 1995, 69, 3584–3596. [Google Scholar]
- Buratti, E; Dork, T; Zuccato, E; Pagani, F; Romano, M; Baralle, FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 2001, 20, 1774–1784. [Google Scholar]
- Mercado, PA; Ayala, YM; Romano, M; Buratti, E; Baralle, FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res 2005, 33, 6000–6010. [Google Scholar]
- Buratti, E; Brindisi, A; Giombi, M; Tisminetzky, S; Ayala, YM; Baralle, FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem 2005, 280, 37572–37584. [Google Scholar]
- Strong, MJ; Volkening, K; Hammond, R; Yang, W; Strong, W; Leystra-Lantz, C; Shoesmith, C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci 2007, 35, 320–327. [Google Scholar]
- Wang, IF; Wu, LS; Chang, HY; Shen, CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem 2008, 105, 797–806. [Google Scholar]
- Buratti, E; Baralle, FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci 2008, 13, 867–878. [Google Scholar]
- Wang, IF; Reddy, NM; Shen, CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc. Natl. Acad. Sci. USA 2002, 99, 13583–13588. [Google Scholar]
- Winton, MJ; Igaz, LM; Wong, MM; Kwong, LK; Trojanowski, JQ; Lee, VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem 2008, 283, 13302–13309. [Google Scholar]
- Ayala, YM; Zago, P; D’Ambrogio, A; Xu, YF; Petrucelli, L; Buratti, E; Baralle, FE. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell. Sci 2008, 121, 3778–3785. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| type 1 | type 2 | type 3 | type 4 | |
|---|---|---|---|---|
| Pathology | predominance of long neurites, NIIs absent - rare | predominance of cytoplasmic inclusions, often preinclusions, NIIs absent - few | small neurites and cytoplasmic inclusions, NIIs absent - abundant | numerous NIIs and small neurites |
| laminar distribution | upper layer > lower | upper = lower layers | upper layer >> lower | upper layer > lower |
| Glial inclusions | absent - rare
| moderate - frequent
| moderate - frequent
| absent
|
| Clinical symptoms | SD
| FTD often with MND
| FTD or PNFA
| IBMPFD
|
| Genetic defect in familial forms | / | Chrom 9p | GRN | VCP |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Neumann, M. Molecular Neuropathology of TDP-43 Proteinopathies. Int. J. Mol. Sci. 2009, 10, 232-246. https://doi.org/10.3390/ijms10010232
Neumann M. Molecular Neuropathology of TDP-43 Proteinopathies. International Journal of Molecular Sciences. 2009; 10(1):232-246. https://doi.org/10.3390/ijms10010232
Chicago/Turabian StyleNeumann, Manuela. 2009. "Molecular Neuropathology of TDP-43 Proteinopathies" International Journal of Molecular Sciences 10, no. 1: 232-246. https://doi.org/10.3390/ijms10010232
APA StyleNeumann, M. (2009). Molecular Neuropathology of TDP-43 Proteinopathies. International Journal of Molecular Sciences, 10(1), 232-246. https://doi.org/10.3390/ijms10010232