Results and Discussion
Remote intramolecular free radical functionalization [
3] provides an elegant method for the construction of bis-spiroacetal ring systems from spiroacetal precursors. Our first example of this approach [
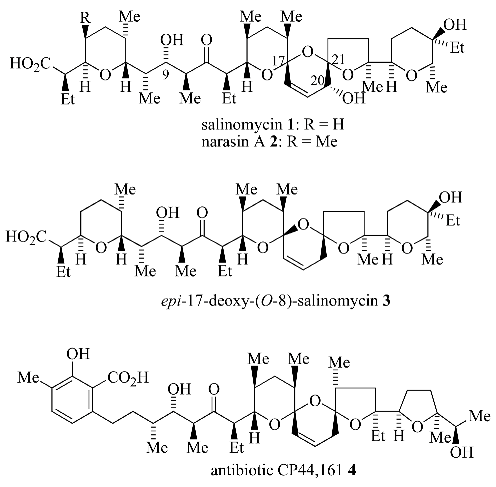
4] was the assembly of the unsaturated 6,6,5-bis-spiroacetal ring system present in the polyether antibiotics salinomycin (
1, [
5]), narasin A (
2, [
6]),
epi-deoxysalinomycin (
3, [
7]) and antibiotic CP44,161 (
4, [
8]).
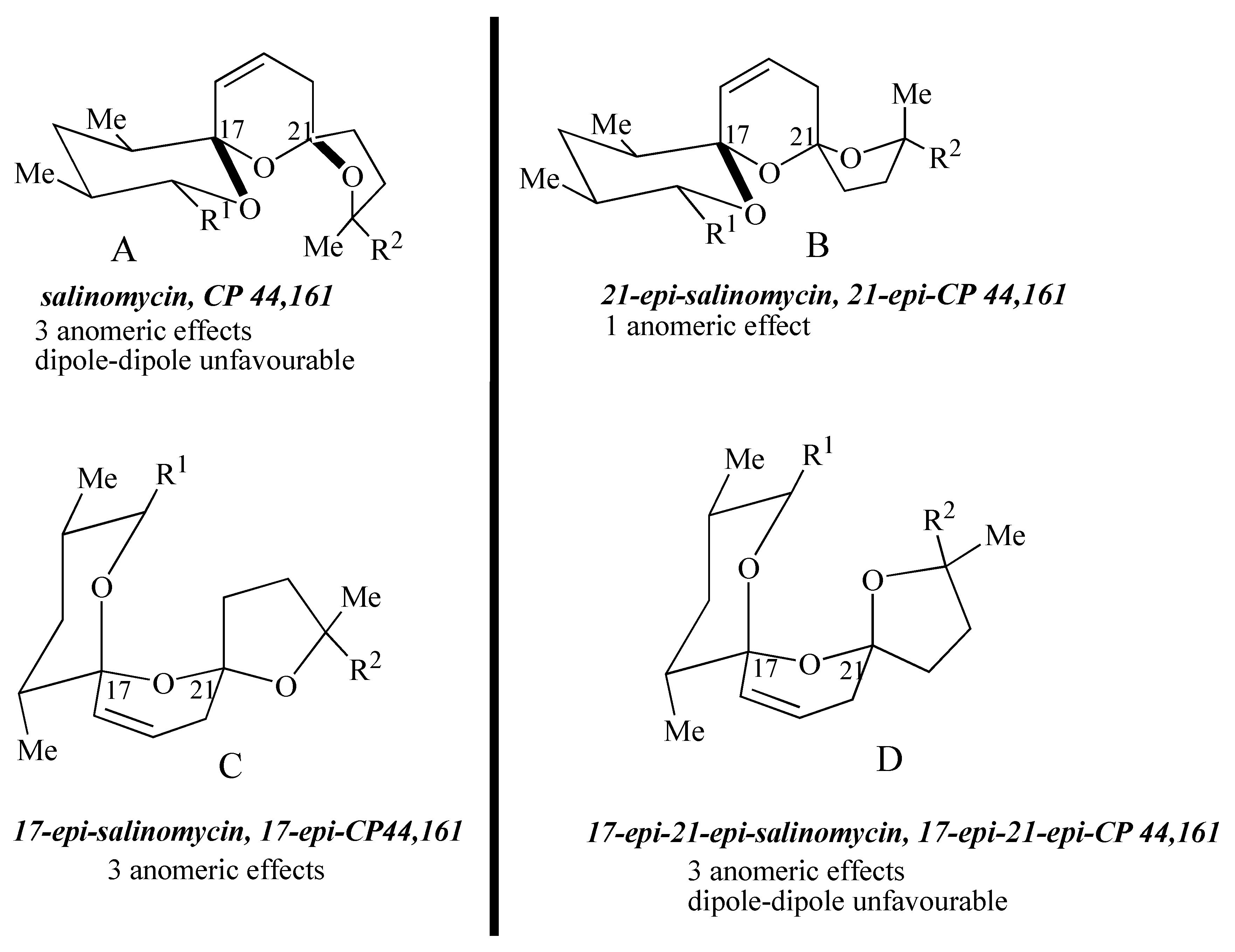
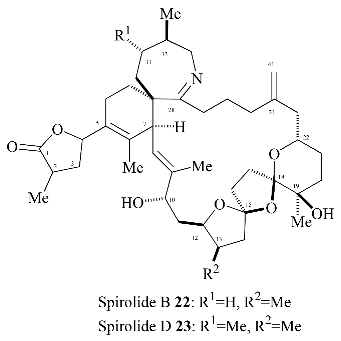
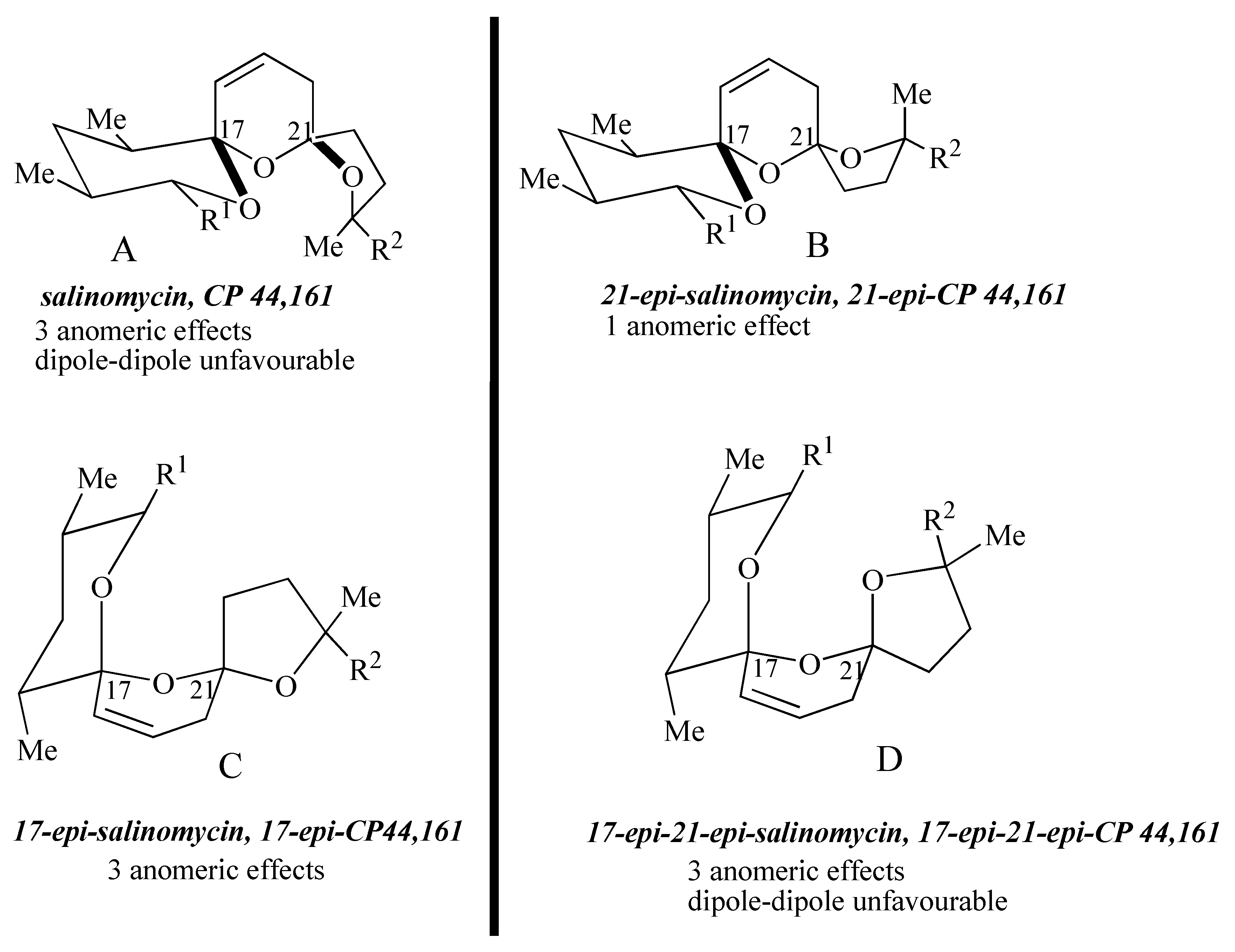
The four possible stereoisomers of the 1,6,8-trioxadispiro[4.1.5.3]pentadec-13-ene ring system present in the salinomycin family of polyether antibiotics are depicted in
Figure 1. Diastereomer A represents the stereochemistry adopted by salinomycin (
1) and CP44,161 (
4) and has three stabilizing anomeric effects but exhibits unfavourable 1,3-dipole-dipole interactions. 21-
epi-salinomycin B has only one anomeric effect and is the thermodynamically least stable diastereomer. The 17-
epi-diastereomer C exhibits three anomeric effects and although it exhibits unfavourable 1,3-diaxial interactions between the C17 oxygen atom and the C21 methylene it lacks the unfavourable 1,3-dipole-dipole interactions exhibited by diastereomer A and 17-
epi-21-
epi-diastereomer D. This qualitative analysis leads to the assumption that 17-
epi-diastereomer C exhibits the most thermodynamically stable configuration. It transpires that in the cyclic structure that salinomycin (
1) adopts, the repulsive 1,3-dipolar interactions in diastereomer A are compensated for by a hydrogen bond between the C9 and C20 hydroxy groups. Bis-spiroacetals whose structures preclude this remote hydrogen bond do not adopt the salinomycin configuration A.
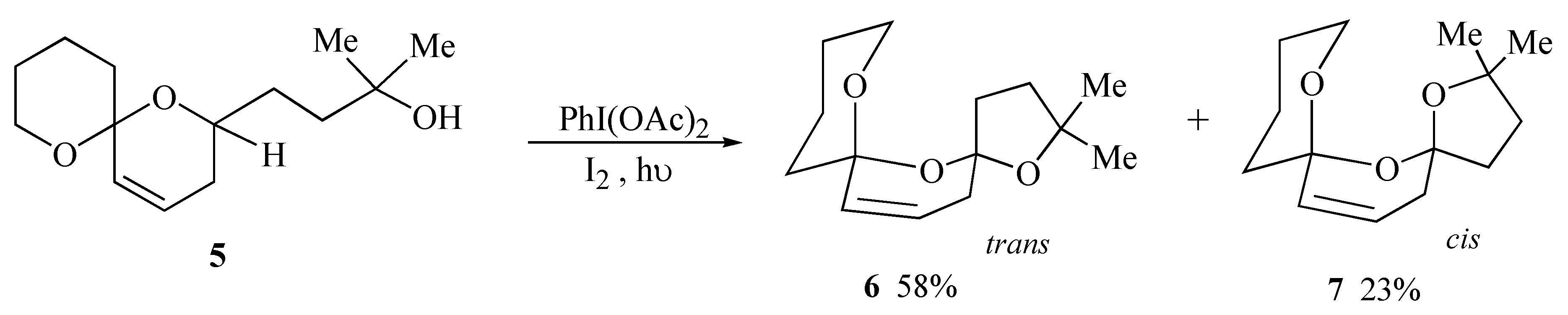
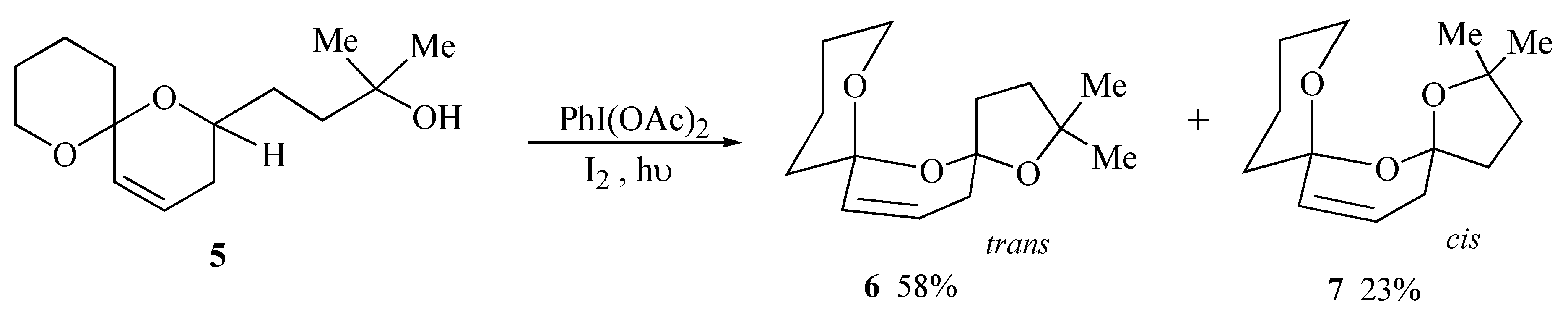
During initial model studies directed towards the synthesis of salinomycin (
1) it was observed that irradiation of spiroacetal alcohol
5 in the presence of iodobenzene diacetate and iodine yielded a 2.5:1 separable mixture of the major
trans bis-spiroacetal
6 together with the minor
cis isomer
7 in 81% yield (
Scheme 1). The
trans isomer is stabilized by only three out of the four possible anomeric effects but is the thermodynamically isomer due to the absence of unfavourable 1,3-dipole interactions between carbon-oxygen bonds on the central ring [
9].
In light of the fact that the major isomer obtained in this model oxidative cyclization was the
trans isomer
6 which has the same stereochemistry as that present in
epi-deoxysalinomycin (
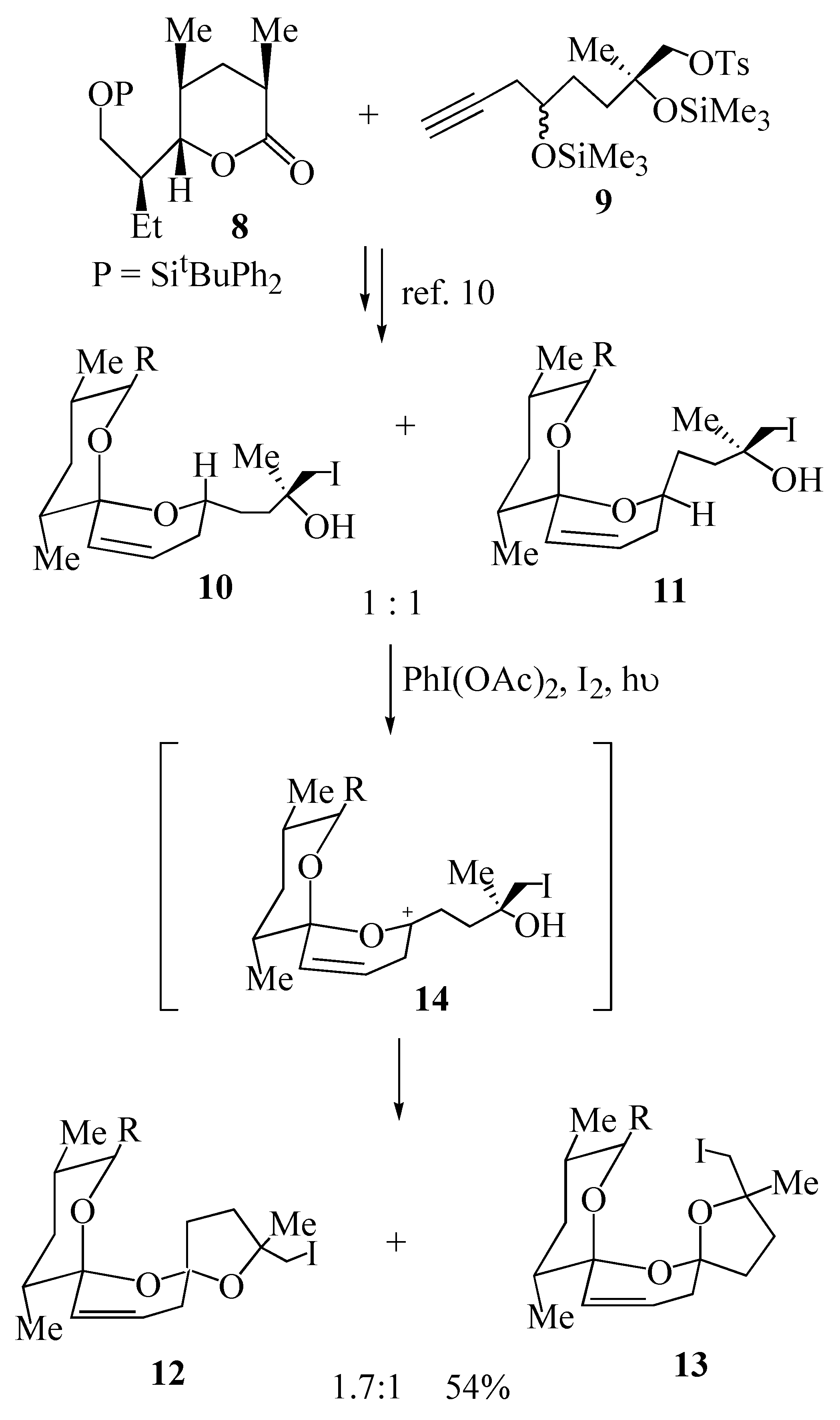
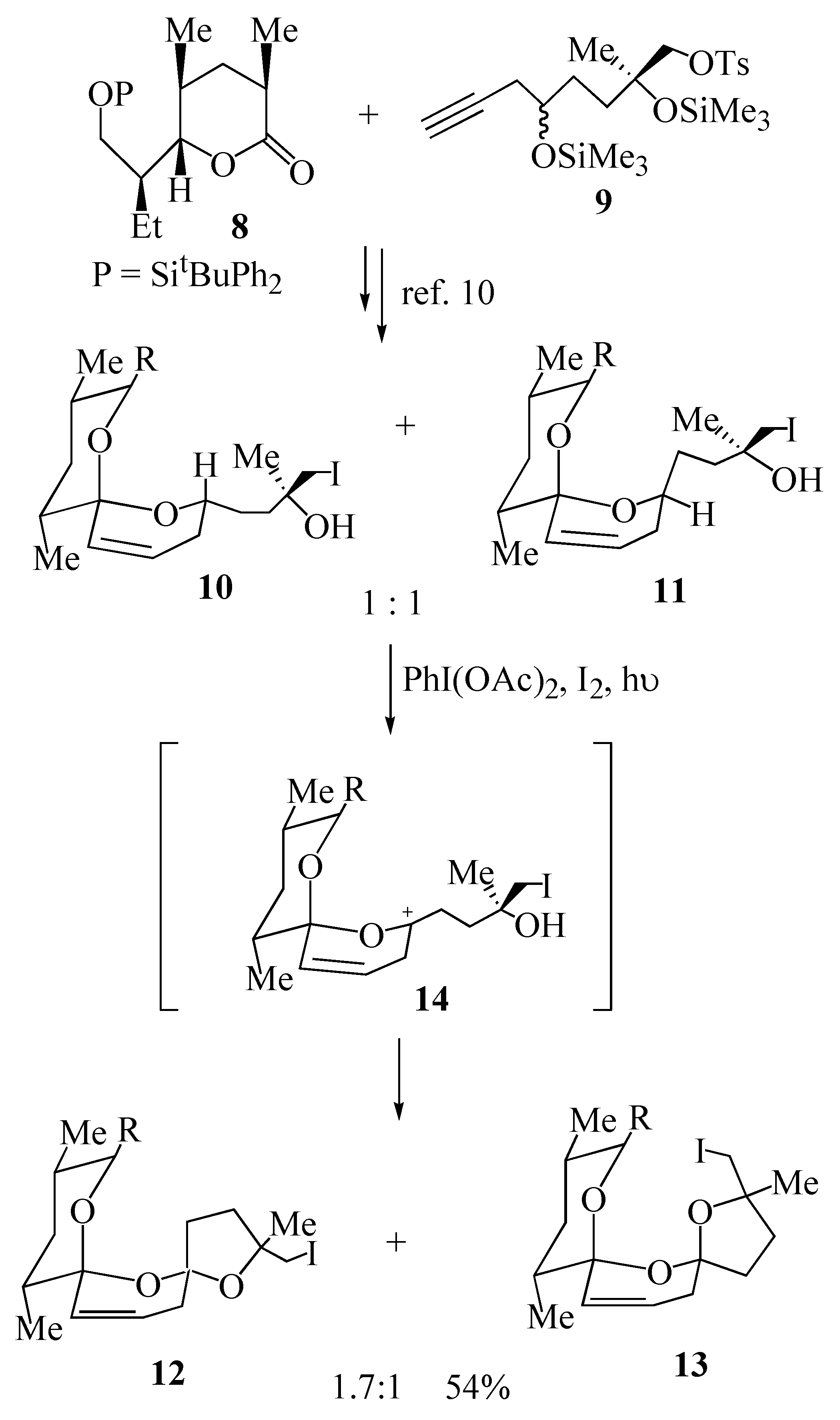
3) we embarked on the synthesis of this polyether antibiotic using this methodology. Union of lactone
8 with acetylene
9 provided the key cyclization precursors
10 and
11 bearing an iodomethyl group for further elaboration of the resultant bis-spiroacetals
12 and
13 (
Scheme 2). The key oxidative cyclization was then achieved upon irradiation of either iodohydrin
10 or iodohydrin
11 in a solution of cyclohexane, containing iodine and iodobenzene diacetate affording the
trans bis-spiroacetal
12 and the
cis bis-spiroacetal
13 in a ratio of 1.7:1 [
10]. This outcome may be explained by the proposed mechanism wherein both diastereomeric precursors
10 and
11 form the same carbocation intermediate
14 (derived from the corresponding free radical), that is subsequently trapped predominantly from the least hindered α-face, to give the
trans bis-spiroacetal
12 as the major product.
Pleasingly the major isomer obtained from the above radical induced oxidative cyclization had the same
trans stereochemistry as that present in
epi-17-deoxysalinomycin (
3). We therefore set about appending the terminal tetrahydropyran E ring to this key bis-spiroacetal intermediate
12. After considerable effort we were forced to abandon this work due to the incompatibility of the sensitive bis-spiroacetal moiety with a key ring expansion reaction planned for introduction of the tetrahydropyran E ring [
11]. We therefore turned our attention to the synthesis of the related antibiotic CP44,161
4 that contained a terminal tetrahydrofuran ring whose synthesis did not require recourse to this ring expansion step.
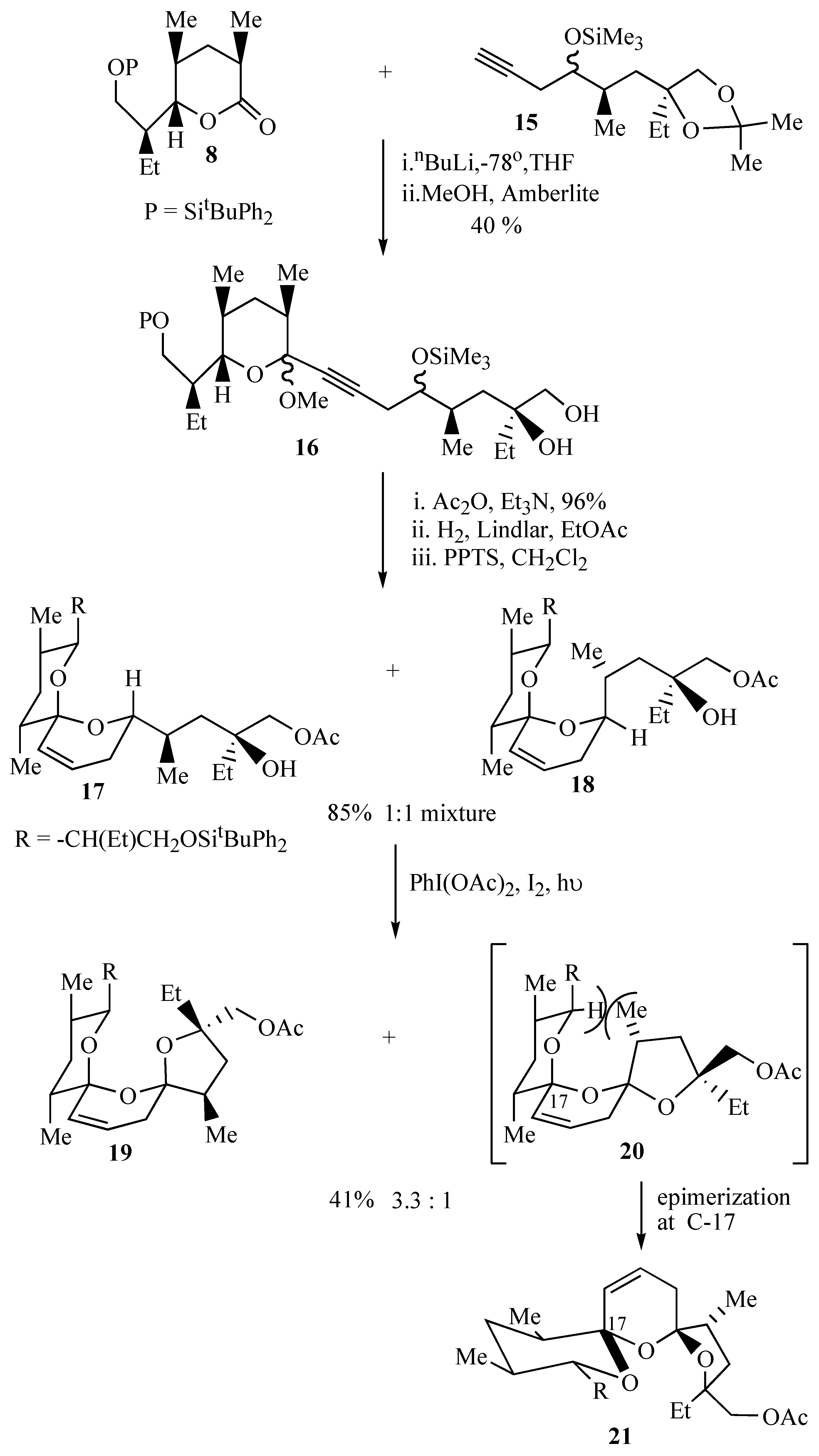
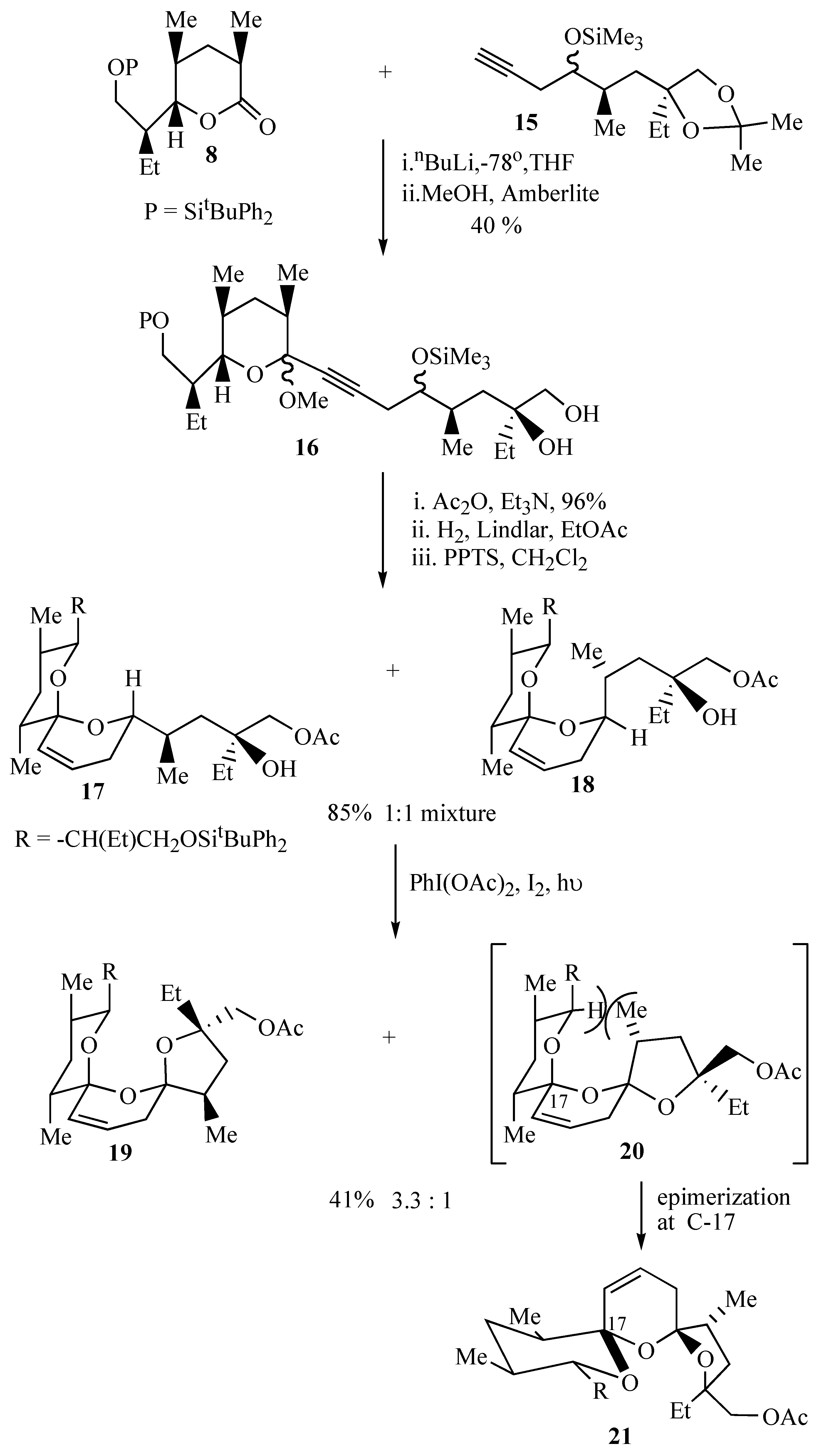
Assembly of the bis-spiroacetal core of antibiotic CP44,161 (
4) was effected using methodology previously applied to the synthesis of the bis-spiroacetal moiety of
epi-17-deoxy-(
O-8)-salinomycin (
3), starting from acetylene
15 and lactone
8 as used previously (
Scheme 3). Addition of the lithium acetylide derived from acetylene
15 to lactone
8 followed by treatment of the intermediate lactol with methanol and Amberlite IR 118 resin effected hydrolysis of the acetonide group and formation of more stable methyl acetals as a mixture of diastereomers that were not separated.
Protection of the primary hydroxyl group as an acetate allowed ready purification of the acetates by flash chromatography. Partial hydrogenation of the alkyne 16 to a cis-olefin followed by acid catalyzed cyclization resulted in a 1:1 mixture of spiroacetals 17 and 18 that were inseparable by flash chromatography. This initial spirocyclisation is carried out under thermodynamically controlled conditions such that the most stable configuration is afforded at the newly formed spiro centre owing to maximum stabilization by the anomeric effect. The two isomeric spiroacetals 17 and 18 therefore only differ in the position that the side chain adopts.
Spiroacetals
17 and
18 were treated with iodobenzene diacetate and iodine to afford a 3.3:1 mixture of tricyclic bis-spiroacetals
19 and
20. The preference for
cis bis-spiroacetal
19 in this cyclisation reaction can be attributed to the presence of the additional methyl group in the tetrahydrofuran ring, which causes unfavourable steric interactions upon formation of the minor
trans bis-spiroacetal
20.
Trans bis-spiroacetal
20 therefore undergoes rapid epimerisation at the allylic spirocentre to
cis bis-spiroacetal
21. The presence of the additional methyl group exhibited a marked effect on the stereochemical outcome of the oxidative cyclisation in that the oxidative cyclisation of the related spiroacetals
10 and
11 which lack this additional methyl group provided the
trans isomer as the major product (
Scheme 2).
The major bis-spiroacetal
19 isolated from this oxidative cyclisation has the 17-
epi-21-
epi-salinomycin stereochemistry (
Figure 1) whereas the minor
cis isomer
21 has the correct bis-spiroacetal stereochemistry for salinomycin (
1) and CP44,161 (
4). Given that it is well established that long range hydrogen bonding in the final molecule can dramatically alter the position of the bis-spiroacetal equilibrium, it was envisaged that thermodynamically controlled cyclization after the whole carbon skeleton of the natural product has been assembled, will allow convergence of the bis-spiroacetal stereochemistry to that depicted in
cis isomer
21.
The successful use of an intramolecular radical cyclization strategy to construct the five membered ring of the 6,6,5-bis-spiroacetal moiety of the polyether antibiotics epi-17-deoxy-O-8-salinomycin (3) and CP44,161 (4) prompted us to extend this methodology to the synthesis of the novel 6,5,5-bis-spiroacetal present in the shellfish toxins, spirolide B (22) and D (23).
Spirolides B (
22) and D (
23) are members of a novel family of marine macrolides isolated from mussels (
Mytilus edulis) and scallops (
Placopecten magellanicus) during routine monitoring for diarrhetic shellfish toxins in Nova Scotia, Canada in 1995 [
12]. They induce characteristic symptoms in the mouse bioassay possibly through muscarinic acetylcholine receptor antagonism and are weak activators of L-type transmembrane Ca
2+ channels. The relative and absolute stereochemistry has not been established to date, but a tentative assignment based on NMR studies has been reported [
13]. In common with the pinnatoxins [
14], the spirolides possess a seven-membered spiro-linked cyclic imine together with the novel 1,7,9-trioxadispiro[4.1.5.2]tetradecane bis-spiroacetal ring system.
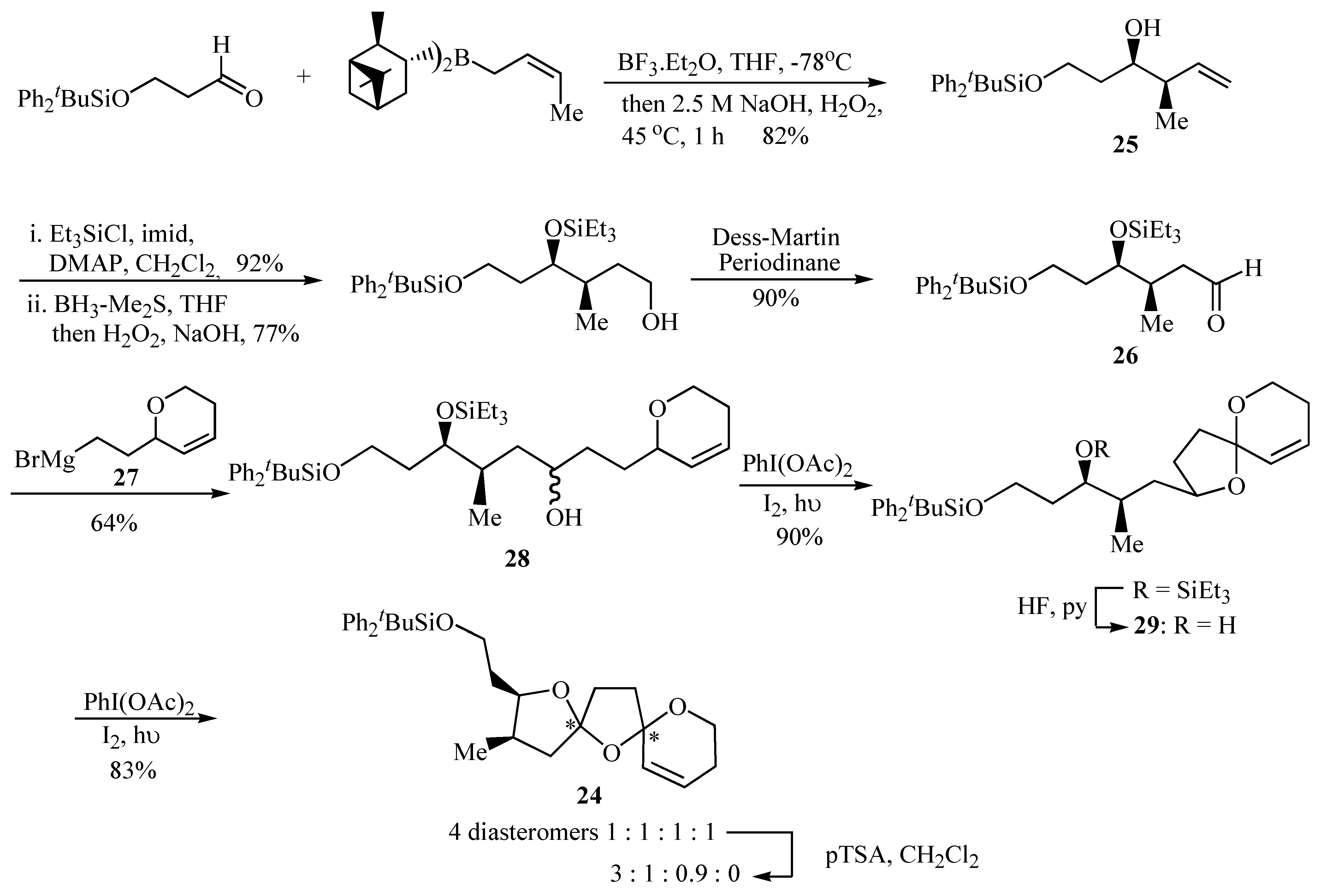
The 6,5,5-bis-spiroacetal unit of the spirolides B (
22) and D (
23) is challenging due to the presence of two five-membered rings, for which the anomeric effect is considerably reduced as compared with six-membered rings. It was envisaged that the two spiroacetal centres in the key bis-spiroacetal fragment
24 provide an excellent synthetic target for using an iterative oxidative radical cyclisation strategy to construct the two five-membered rings in a stepwise fashion (
Scheme 4).
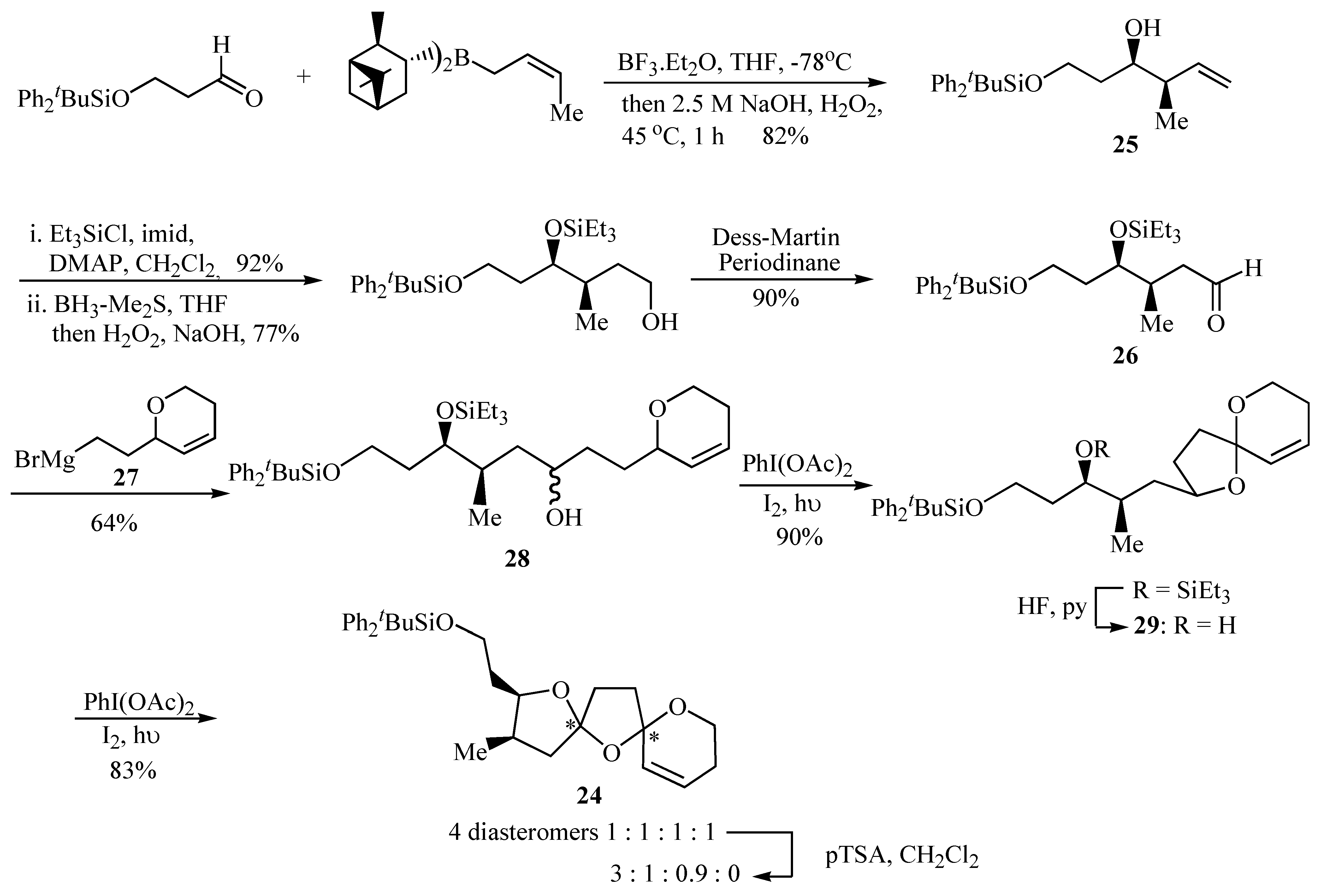
Formation of oxaspiro rings by hydrogen abstraction promoted by hypervalent iodine led to the spirocyclization precursors
28 and
29 as an effective strategy for the synthesis of the key 6,5,5-bis-spiroacetal
24 present in compounds
22 and
23. Aldehyde
26 provided the substituents required for the substituted tetrahydrofuran ring of the 6,5,5-bis-spiroacetal ring system and these were introduced in a stereocontrolled manner by crotyl metallation of a protected 3-hydroxypropanal that afforded the desired homoallylic alcohol
25 in 82% yield and >97% d.e. The
R,
R configuration was selected both by analogy with pinnatoxin A [
14] and the relative stereochemistry of the spirolides proposed by Falk
et al [
13]. The resultant homoallylic alcohol
25 was then converted into aldehyde
26 in good yield using standard transformations. The synthesis of aldehyde
26 is also flexible enabling access to bis-spiroacetals of varying relative and absolute stereochemistry by the appropriate choice of chiral crotyl borane used to prepare the homoallylic alcohol intermediate
25. Coupling of aldehyde
26 with the Grignard reagent
27 proved problematic. Standard Grignard reaction conditions failed to yield the desired product and only mediocre yields were obtained using an organolithium species derived from the analogous iodide. Alcohol
28 was finally prepared reproducibly in 64% yield using Barbier conditions in diethyl ether, together with the undesired Wurtz coupling product of
27. Cyclisation of
28 using iodobenzene diacetate and iodine with irradiation afforded spiroacetal
29 in 90% yield after removal of the triethylsilyl ether by portionwise addition of HF·pyridine over several hours. Finally, bicyclic spiroacetal
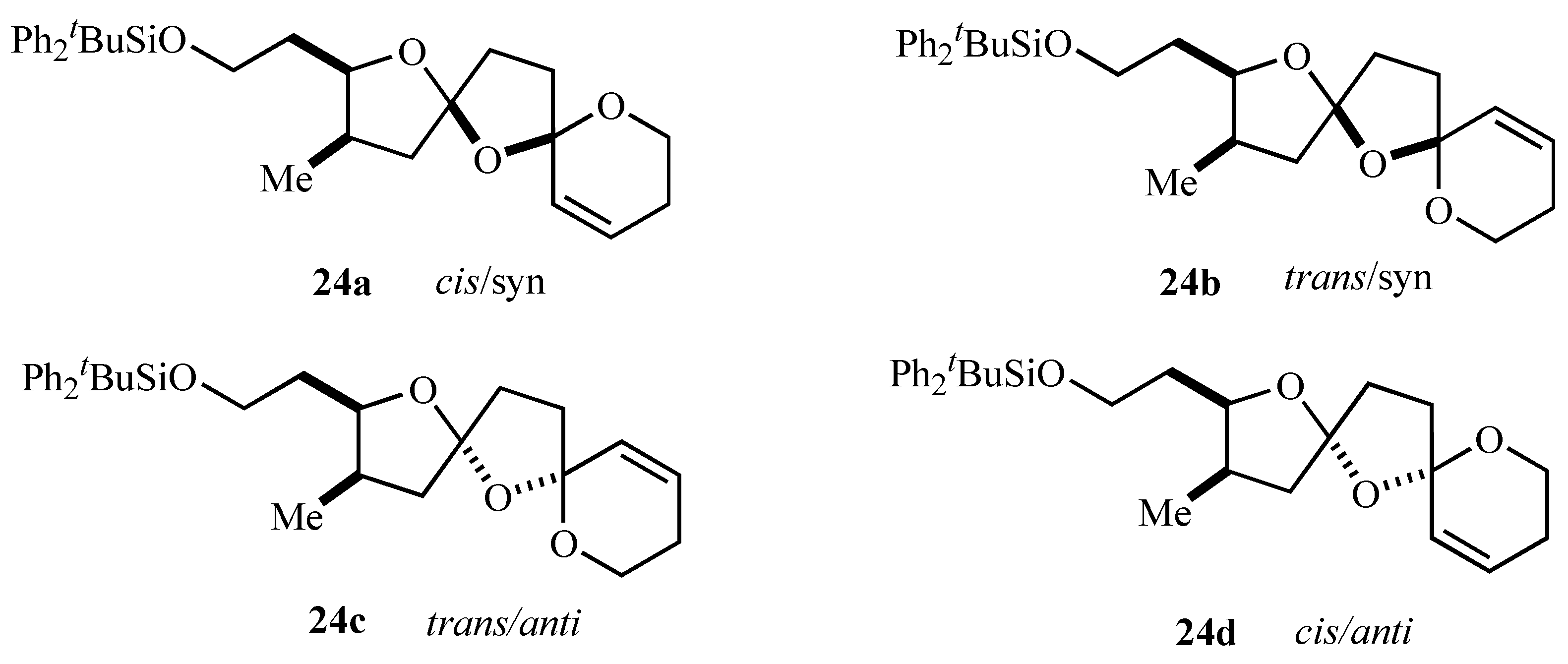
29 underwent smooth radical oxidative cyclisation to a 1:1:1:1 mixture of the four possible diastereomers of bis-spiroacetal
24 upon treatment with iodobenzene diacetate and iodine with irradiation. Prolonged stirring of the initial 1:1:1:1 product mixture with a catalytic quantity of
p-toluenesulfonic acid in dichloromethane gave a 3:1:0.9 mixture (
24a:
24b:
24c,
Figure 2) of three of the four possible isomers and these were then separable by careful chromatography [
15].
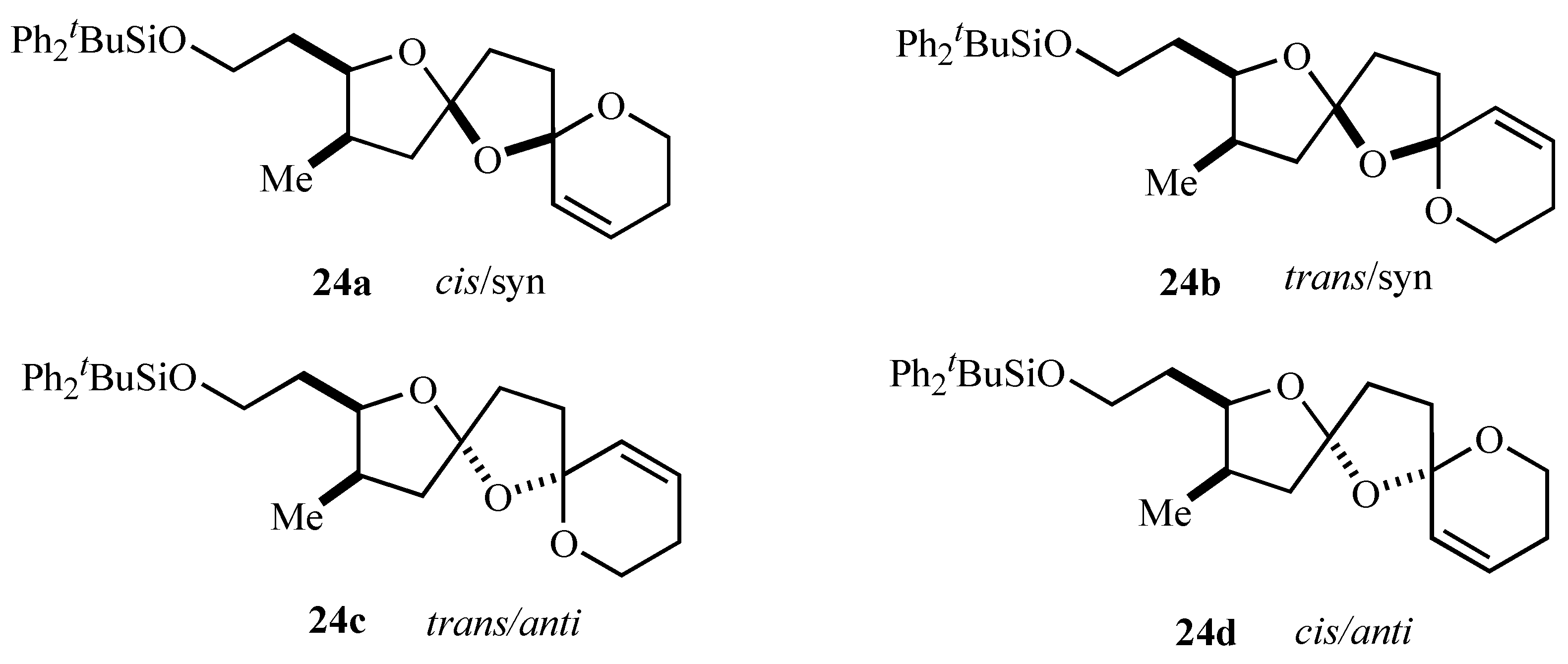
Figure 2.
| Note: | The cis/trans notation refers to the arrangement of oxygens across the central ring and syn/anti to the arrangement of the central oxygen and the alkyl substituent, respectively. |
The stereochemistry assigned to the bis-spiroacetal isomers
24a-c was assigned on the basis of 2D NMR and
ab initio molecular modeling [
15] and it was satisfying that the major bis-spiroacetal isomer
24a had the same stereochemistry as that assigned by NMR spectroscopy to the bis-spiroacetal ring system of spirolides B (
22) and D (
23) [
13]. Notably the initial oxidative radical cyclizations did not provide any stereocontrol in the initially formed isomeric mixture of 1,7,9-trioxadispiro[4.1.5.2]tetradecanes supporting the concept that control of stereochemistry in systems containing two five-membered rings is more problematic than similar bis-spiroacetals containing six-membered rings.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}