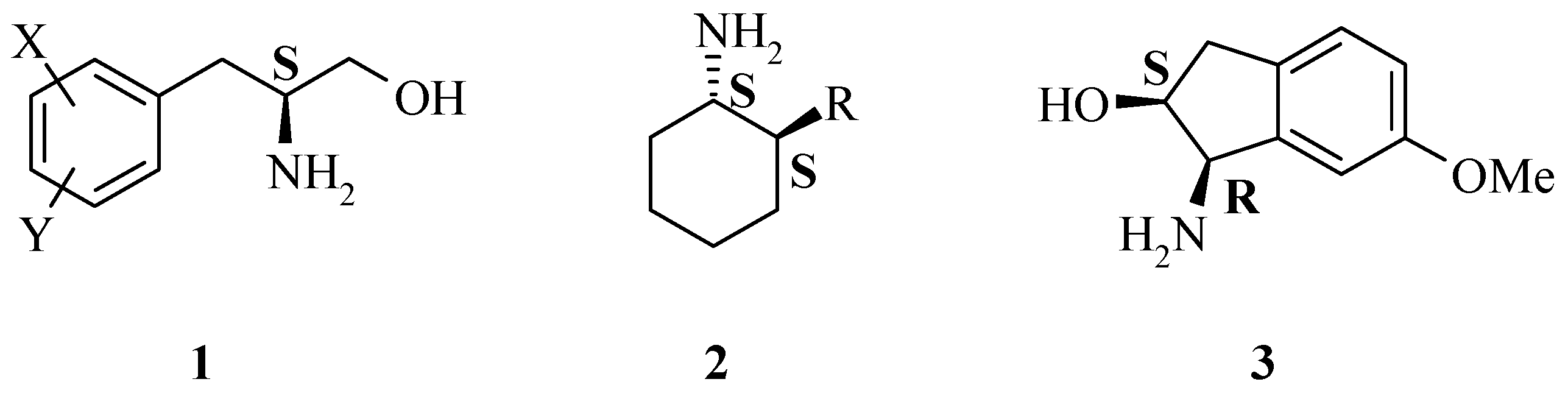
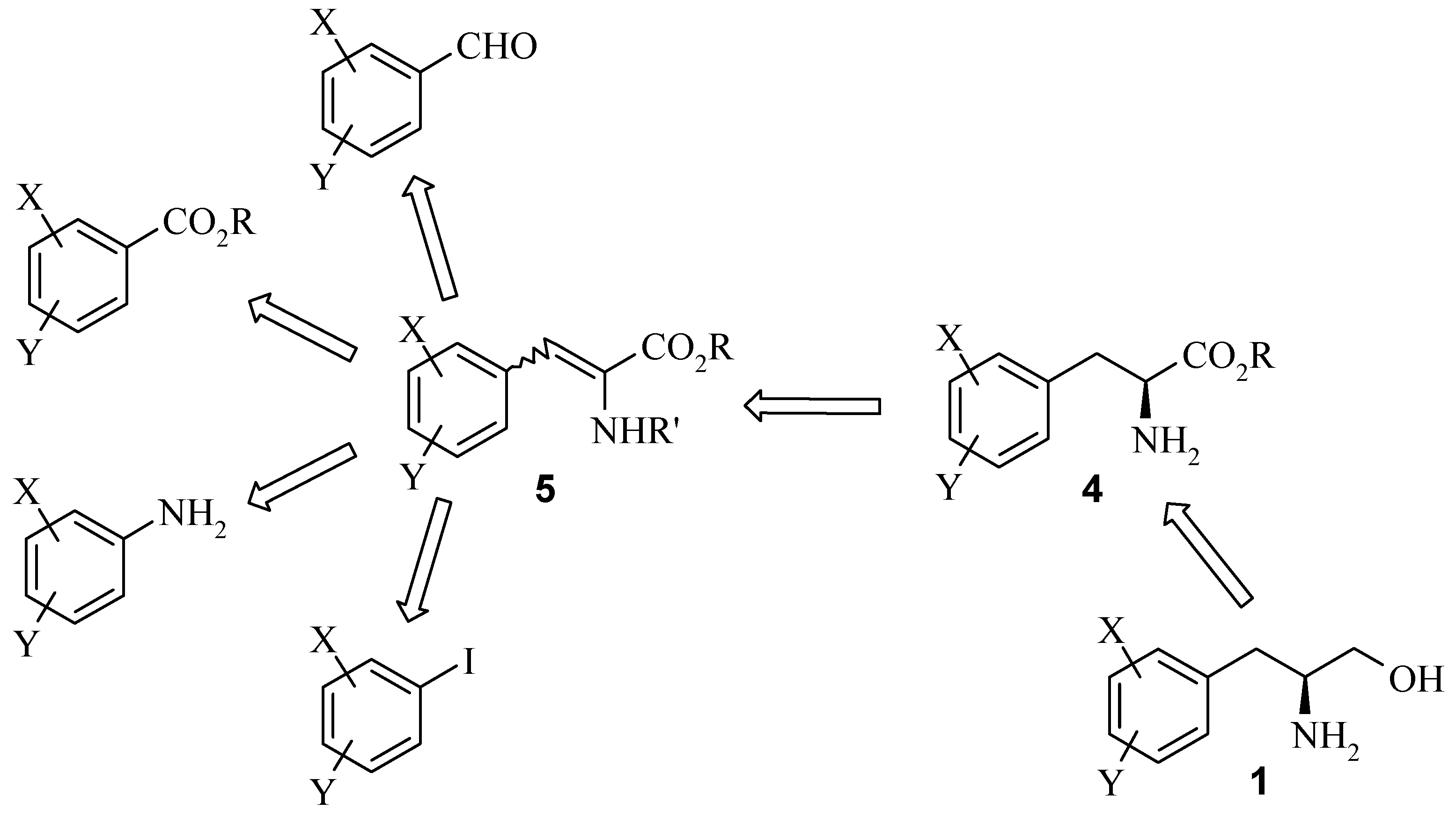
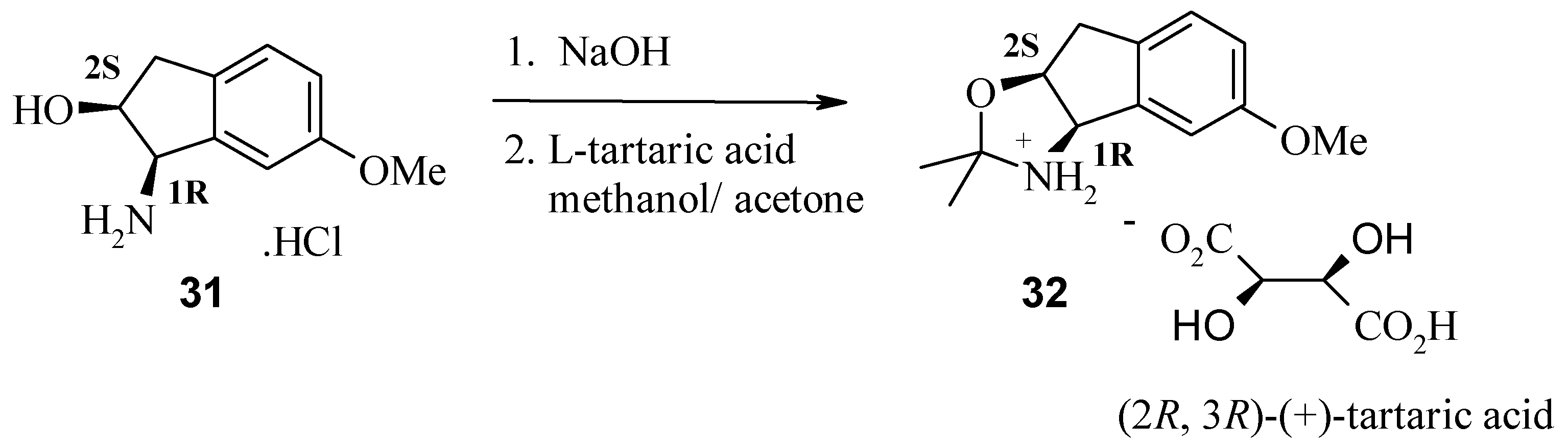
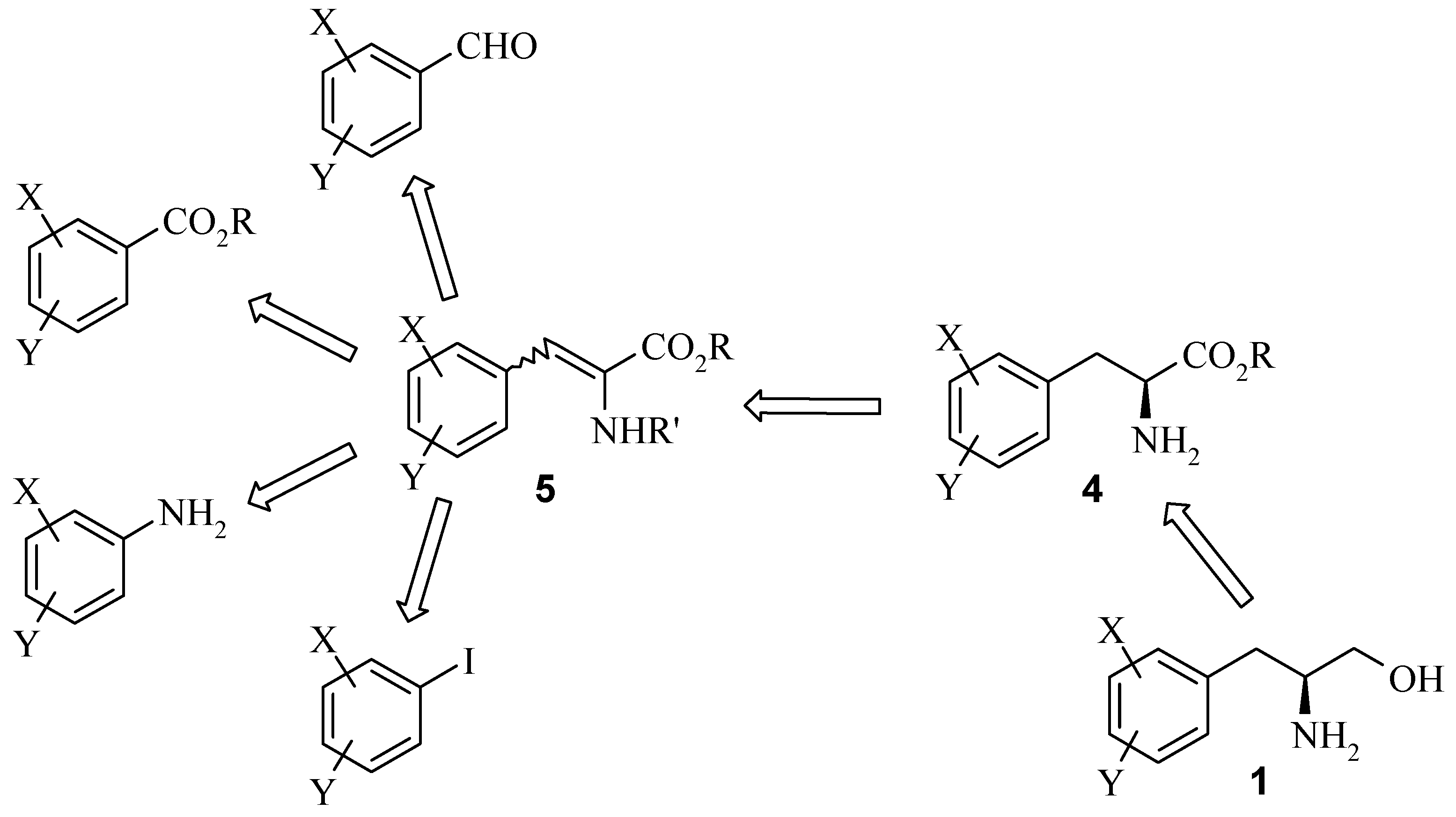
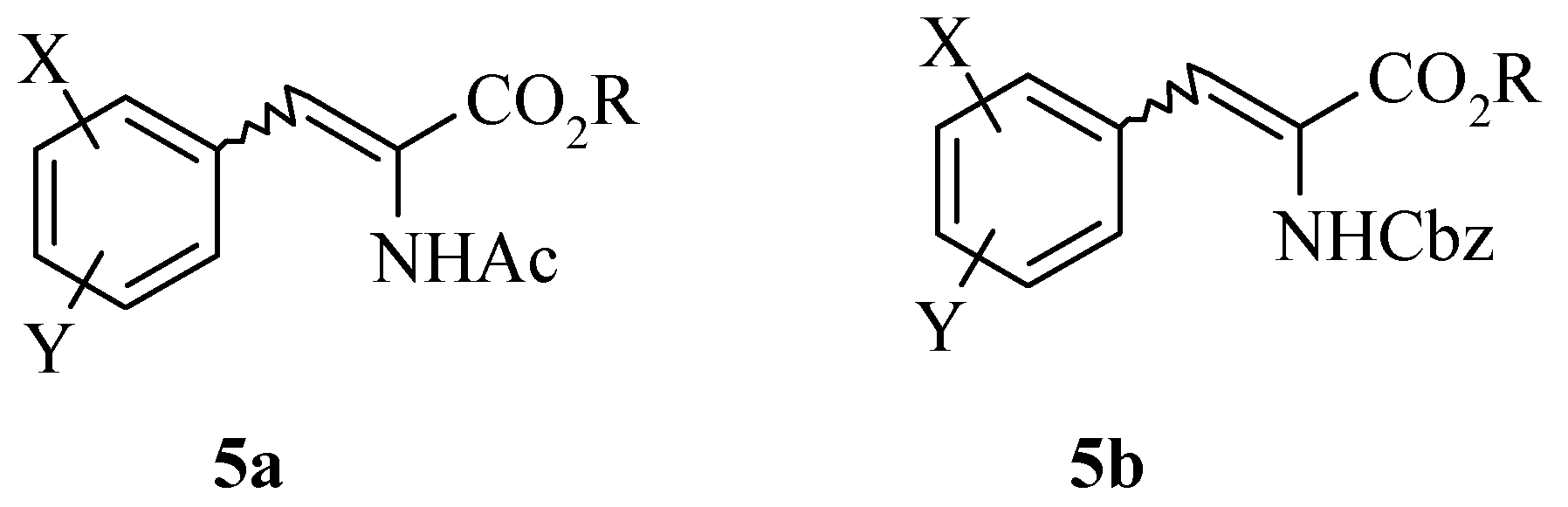Results and Discussion
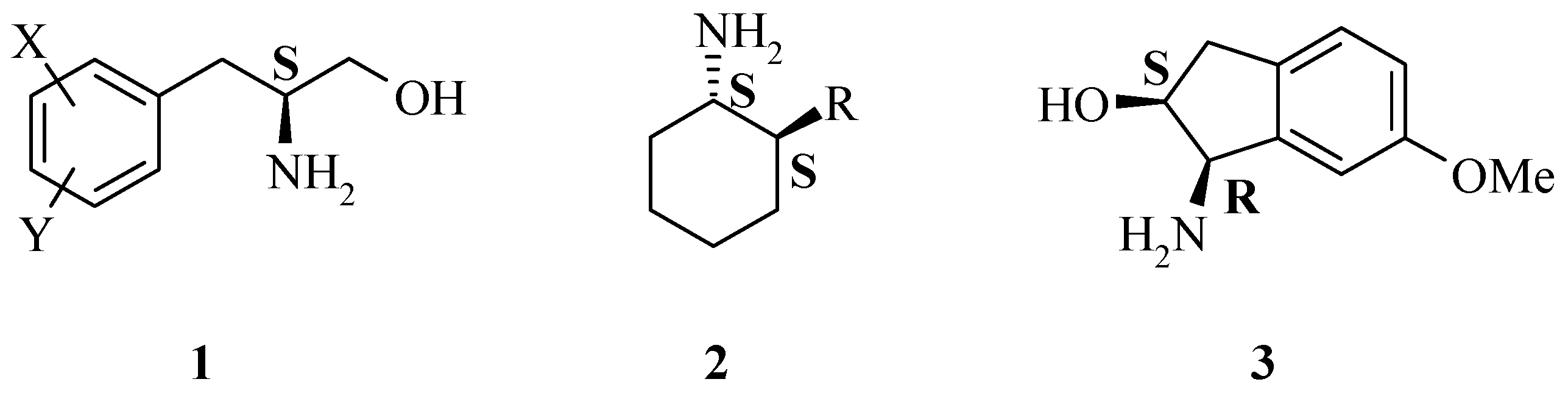
A generic synthesis to access large numbers of diverse chiral α-amino acids
4 and β-amino alcohols
1 has been developed. There are many known methods of preparing the dehydrophenylalanine intermediate
5 from commercial starting materials (
Scheme 1).
This, coupled with selective reduction of the dehydrophenylalanine intermediate 5 using an asymmetric hydrogenation catalyst, such as DuPHOS, provides a very powerful route. This reaction synthesis is high yielding overall and provides the final chiral 2-amino-3-phenylpropanol building blocks 1 in high enantiomeric excess.
The preparation of the dehydro amino acid derivatives was accessed by several routes depending on the analogue and available starting materials. Dehydrophenylalanine intermediates
5a and
5b (
Figure 2) were prepared from either an Erlenmeyer reaction with benzaldehydes and
N-acetyl glycine or a Horner-Emmons reaction with Cbz-glycine phosphonate respectively. Alternatively, a Heck reaction between an aryl iodide (where the aldehyde is unavailable) and an acrylate also provides amidoacrylate
5a.
Figure 2.
Dehydrophenylalanine intermediates
Figure 2.
Dehydrophenylalanine intermediates
The dehydrophenylalanine intermediate
5 was subjected to selective reduction with a chiral DuPHOS Rh-catalyst commercially available from Strem. The advantage of this catalyst is that it is predictable. The relative stereochemistry of the product is determined by the chiral catalyst ie [(COD)Rh((
S,S)-Et-DuPHOS)]
+ will always give the (
S)-enantiomer and [(COD)Rh((
R,R)-Et-DuPHOS)]
+ gives the corresponding (
R)-enantiomer [
1,
2]. In addition, the double bond geometry of the starting alkene, whether it be
E or
Z gives the same absolute configuration in high enantiomeric excess.
Chiral 2-amino-3-phenylpropanol building blocks
1 were then prepared following several functional group manipulations. Confirmation of optical purity (stereochemistry obligated by the catalyst employed) was determined by preparation of either the Mosher amide or Mosher ester derivative. The diastereomeric excess (de) of the Mosher amide is representative of the enantiomeric excess (ee) of the precusor. The epimeric compounds were prepared in a similar manner in most cases. In general,
19F-NMR integrals indicated >98% ee. In some cases the minor isomer was not observed at all. The absolute configuration of 2-substituted-1-propanols can be assigned by the
1H-NMR splitting patterns of the Mosher’s ester [
3]. Reverse phase HPLC was also used to distinguish between Mosher diastereoisomers.
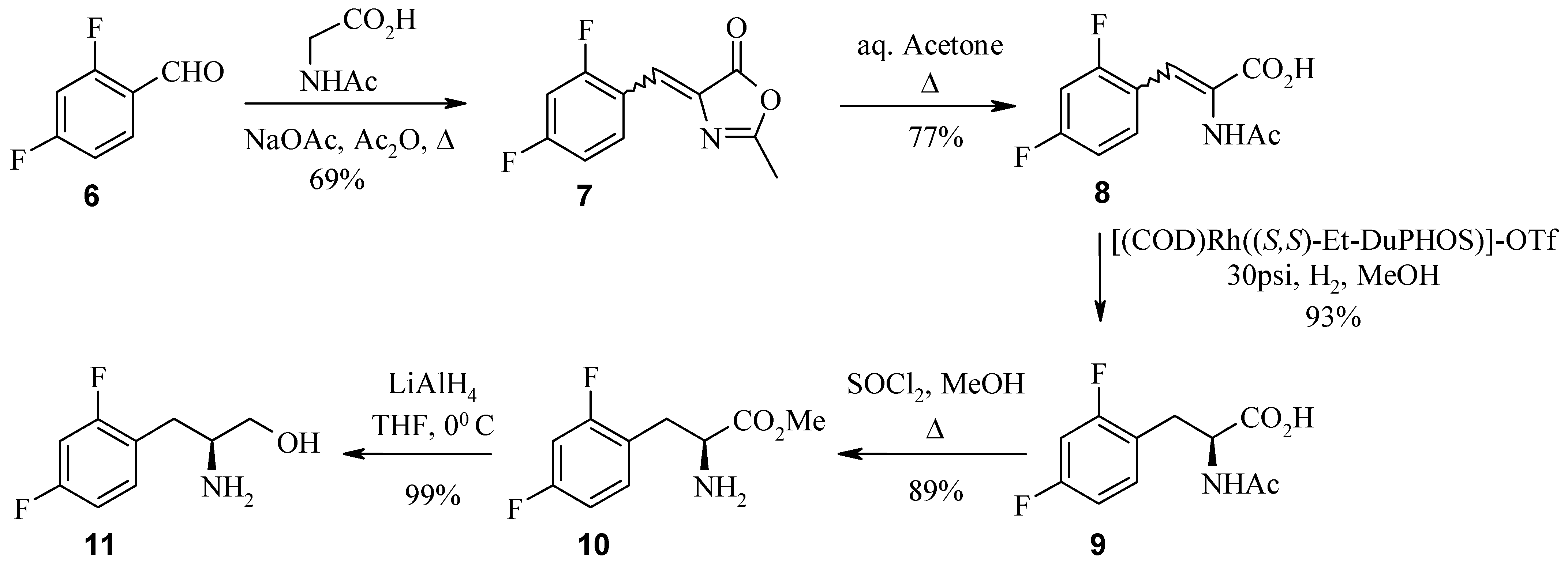
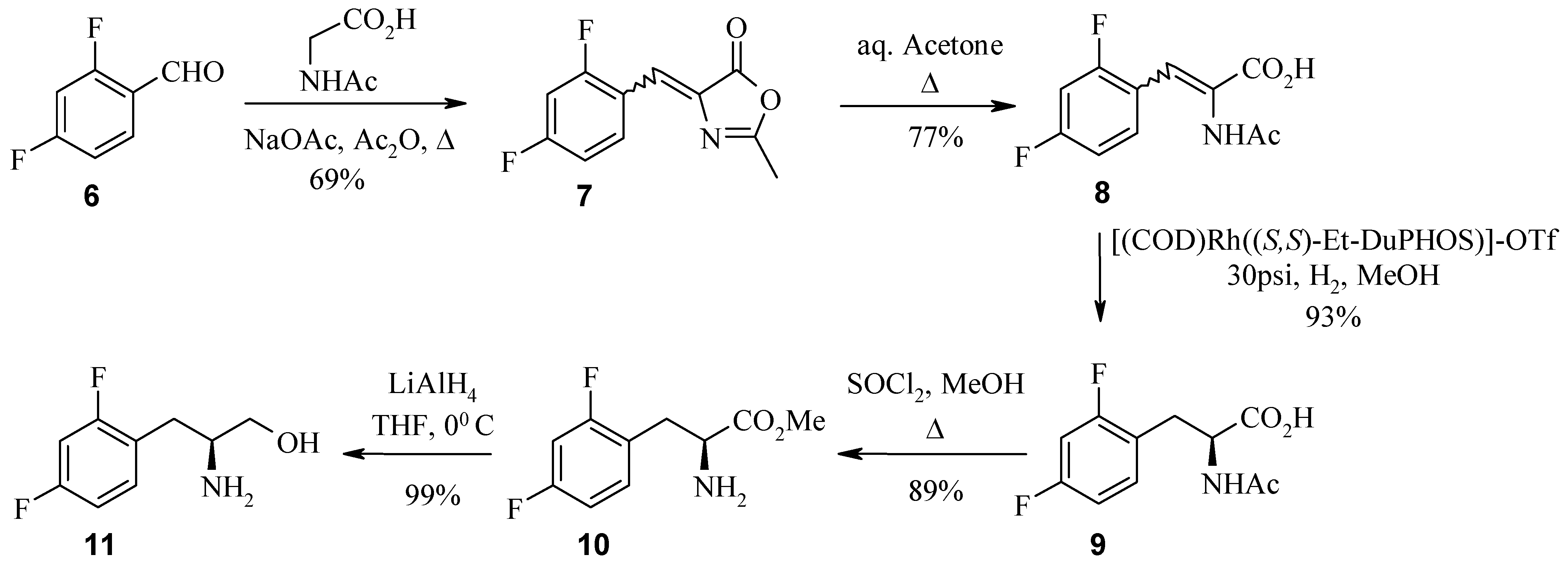
In the first example, the 2,4-difluoro analogue was best accessed via the Erlenmeyer reaction of benzaldehyde 6 with N-acetyl glycine. The intermediate azlactone 7 was subsequently ring opened by reflux in acetone. Asymmetric hydrogenation of acrylic acid 8 with (S,S)-DuPHOS catalyst at 30 psi then installed the desired S stereochemistry in acetylamino acid 9. The next step in the synthesis was esterification of acid 9 with methanolic hydrogen chloride. It was observed here, that with longer reaction times the acetyl group was also cleaved. Thus, the installation and removal of protecting groups was achieved in a one pot synthesis in high yield. Reduction of (S)-amino ester 10 to (S)-amino alcohol 11 was achieved with lithium aluminium hydride. Preparation of the Mosher amide of 11 and analysis by 19F-NMR (ratio 97:0.065) and HPLC indicated >98% ee.
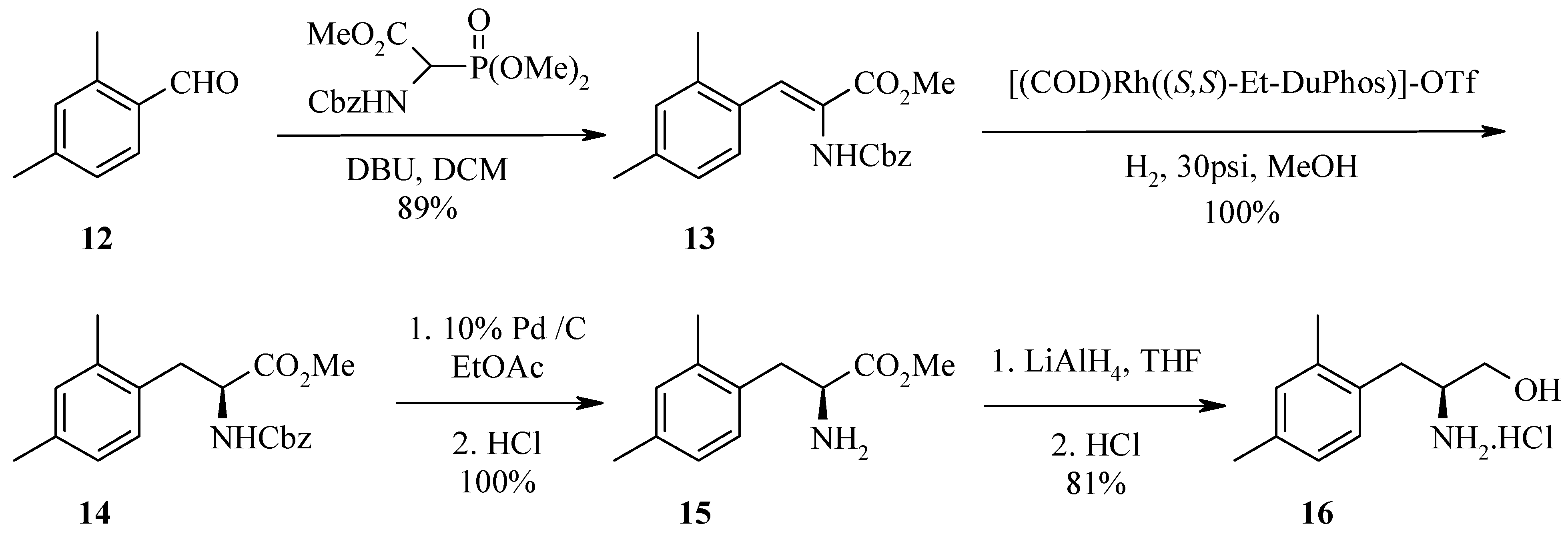
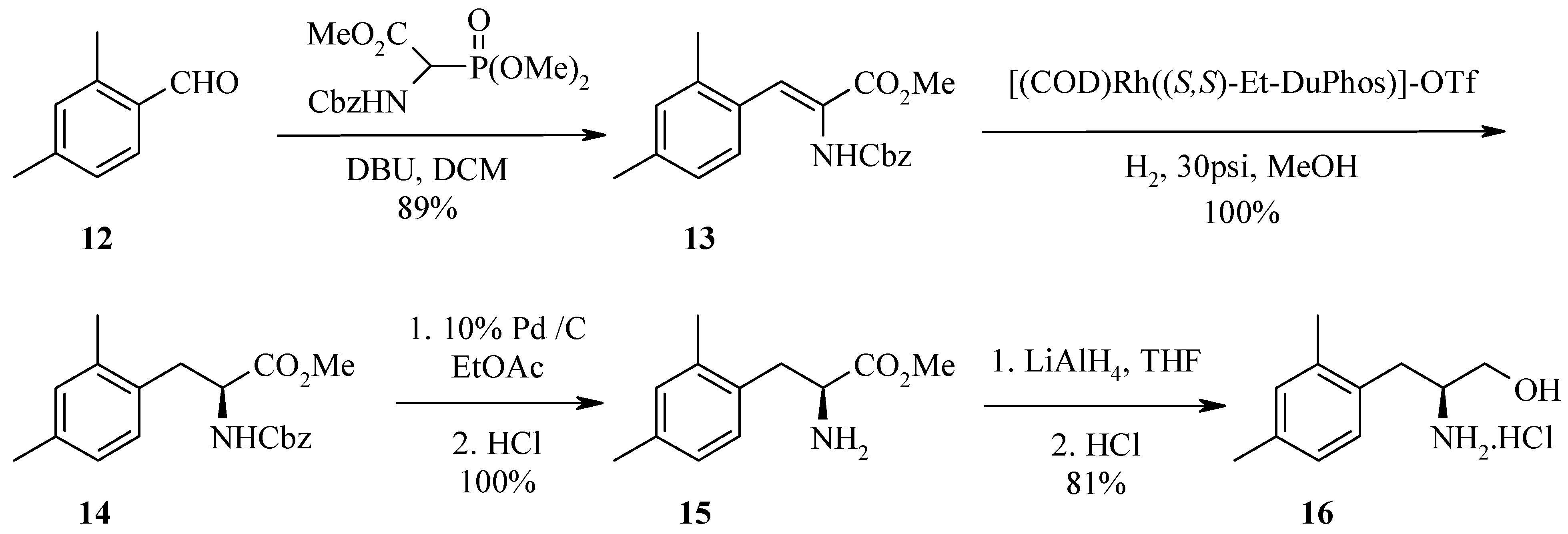
In a second example, Horner-Emmons reaction of 2,4-dimethylbenzaldehyde 12 with N-Cbz-glycine phosphonate gave amidoacrylate 13. Catalytic hydrogenation with (S,S)-DuPHOS provided the (S)-amido ester 14. Hydrogenation of Cbz-amide 14 with Pd/C followed by reduction of ester 15 with lithium aluminium hydride afforded the desired (S)-amino alcohol 16. LCMS and reverse phase HPLC of the (R)-Mosher amide derivative estimate the enantiomeric excess to be >98%, as the alternate (R,R)-diastereoisomer was undetectable.
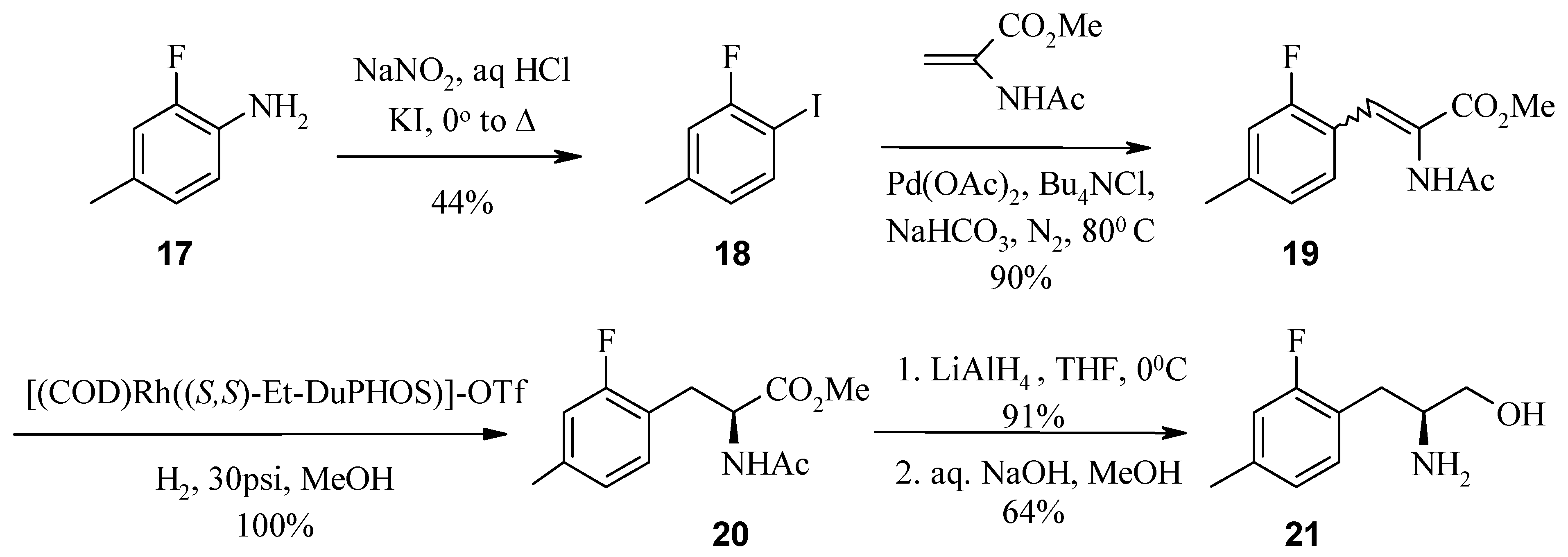
In a third example, 2-fluoro-4-methylaniline (
17) was used as a starting reagent where 2-fluoro-4-methylbenzaldehyde was not commercially available. The Heck reaction with the diazotized aniline and methyl acrylate works well, but with methyl 2-acetamidoacrylate this reaction fails completely. Aniline
17 was thus converted to aryl iodide
18 via the Sandmeyer reaction followed by Heck reaction with methyl-2-acetamidoacrylate and Pd-acetate to form amidoacrylate
19 [
4]. The reaction was done neat and gave the acrylic ester as a black tar which was purified by chromatography. Catalytic hydrogenation with (
S,S)-DuPHOS provided (
S)-amido ester
20. Reduction of ester
20 with lithium aluminium hydride gave an acetamido alcohol intermediate. Derivatisation of this intermediate as the (
R)-Mosher ester indicated, from the
1H-NMR, that we had obtained 2
S stereochemistry [
2,
3]. Deacetylation of the acetamido alcohol intermediate gave the resulting (
S)-amino alcohol
21. Analysis of the derived (
R)-Mosher amide of
21 by LC/LCMS confirmed >98% ee.
Next we examined the synthesis of chiral amines
via chiral alcohols using Noyori's chiral Ru-catalyst. The Noyori asymmetric hydrogenation catalyst is one of a family of catalysts consisting of a ruthenium based system with a phosphine and 1,2-diamine ligand. They provide highly efficient enantio- and diastereoselective hydrogenation of simple ketones. Asymmetric hydrogenation of 2-substituted cyclohexanone with the Noyori catalyst favors the
cis isomer and provides the cyclohexanol in high ee. This is attributed to the fact that from a racemic mixture, the 2
R substituted cyclohexanone undergoes hydrogenation faster and the slower 2
S substrate undergoes epimerisation under basic conditions faster than it is hydrogenated [
5].
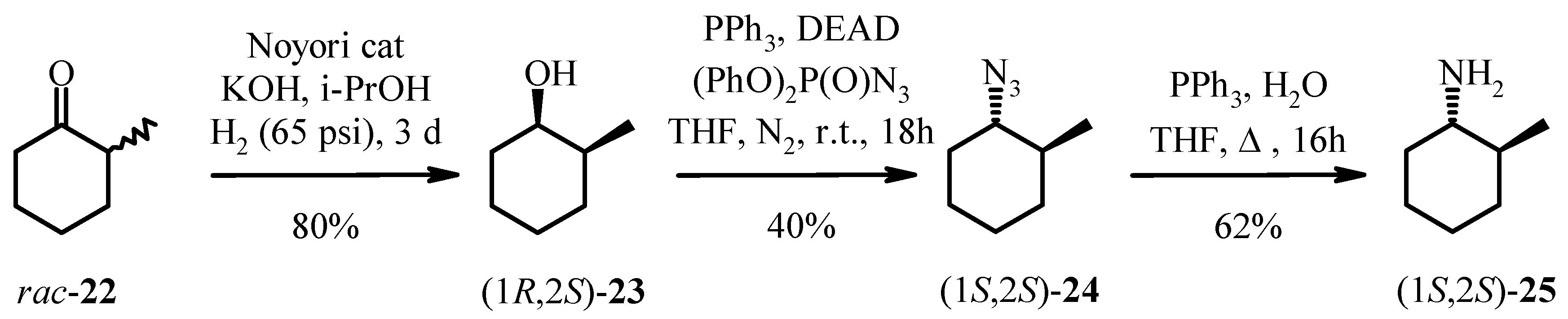
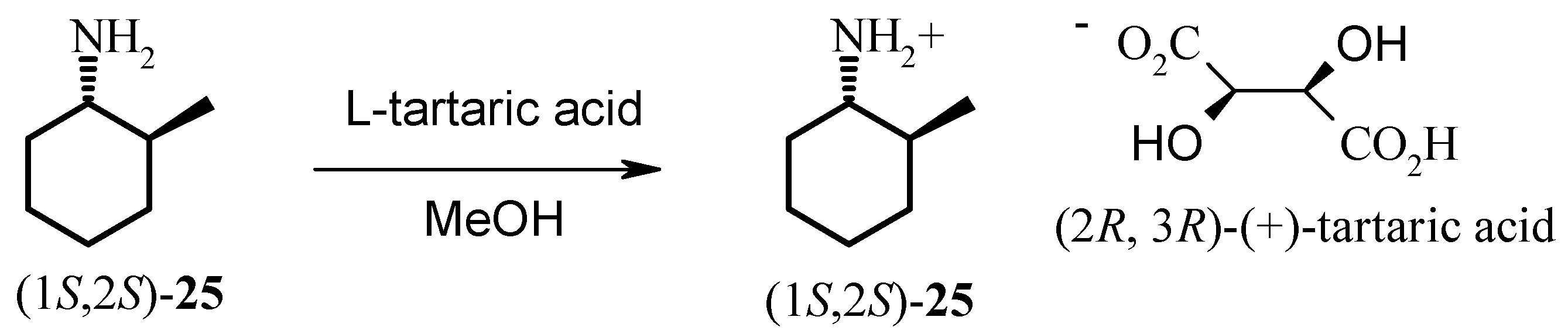
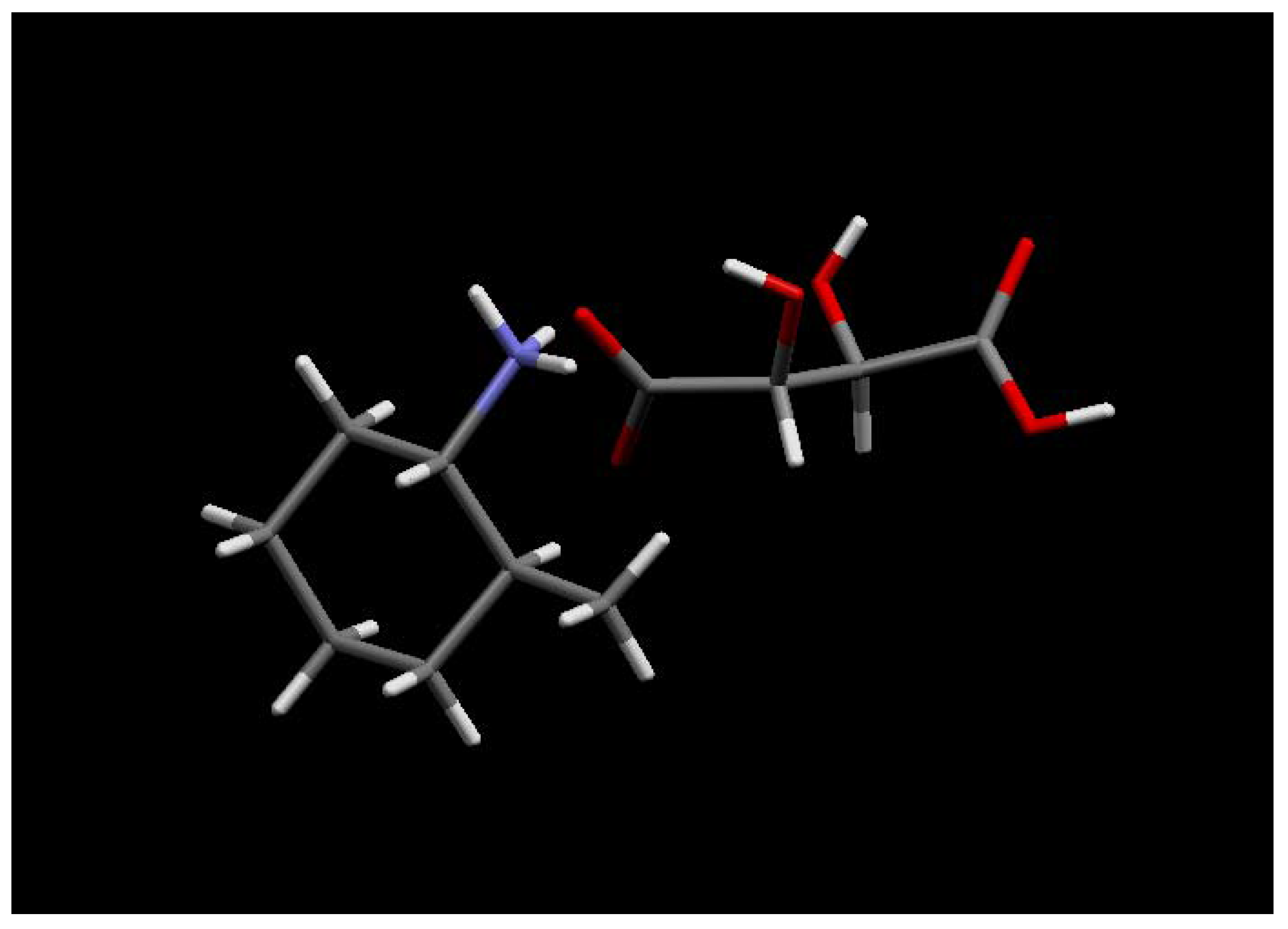
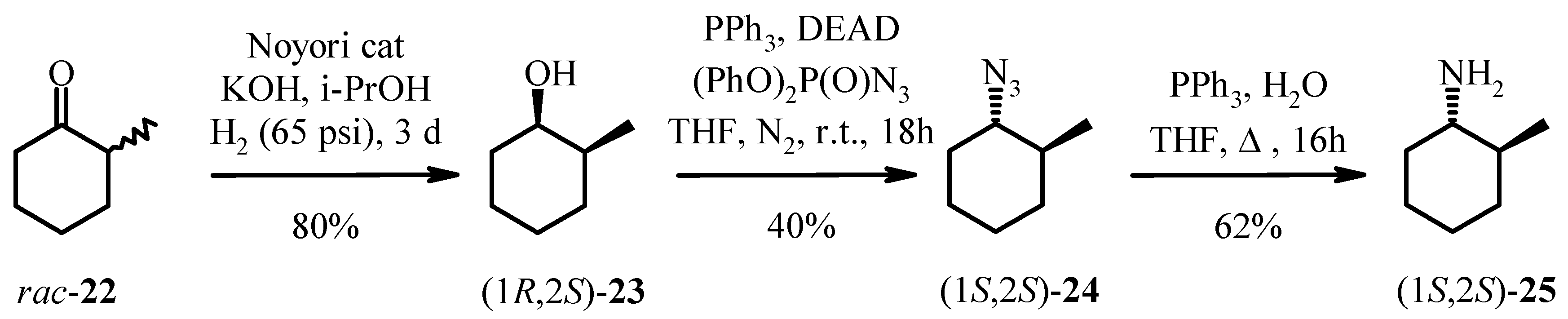
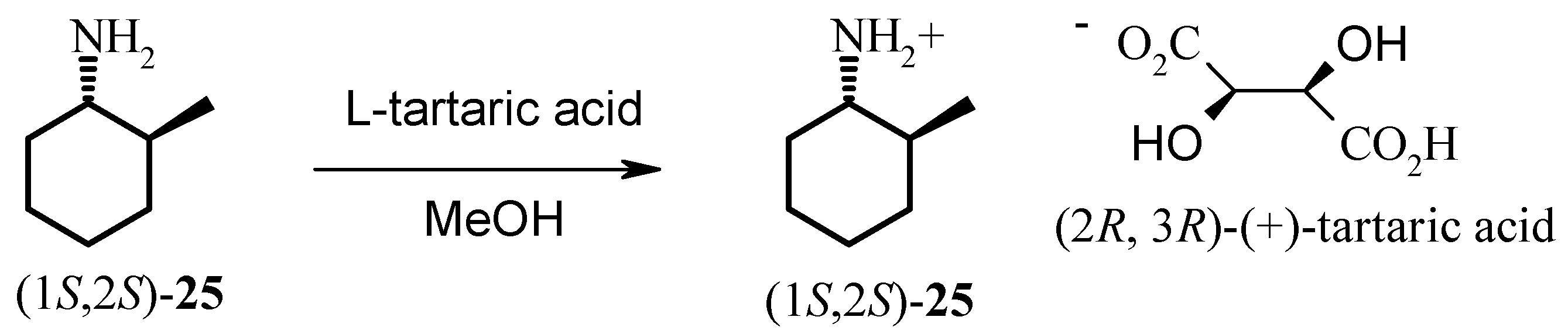
The hydrogenation of 2-methylcyclohexanone 22 with Noyori’s ruthenium catalyst, (S)-Binap-(+)- (1R, 2R)-diphenylethylenediamine, in the presence of potassium hydroxide at 65 psi affords (1R, 2S)- 2-methyl-1-cyclohexanol 23 as a 40:1 mixture of cis and trans isomers in high yield. Analysis of the Mosher ester of 23 by 19F-NMR indicated 70% ee. Importantly, the C1 stereochemistry was inverted by Mitsonobu reaction to afford the corresponding azide 24 in 100% trans form. Reduction of azide 24 with PPh3 gave the desired (1S, 2S)-methyl cyclohexylamine 25 in 70% ee as confirmed by the Mosher amide of 25. Thus, the enantiomeric excess was maintained throughout the synthesis. This was then further enhanced by resolution with tartaric acid. Amine 25 was resolved by recrystallisation with (+)- tartaric acid from methanol to give the hydrogen tartrate salt in 92% ee. The low yields in the synthesis can be attributed to the volatility of the products compounded by the poor UV absorption characteristics of these molecules which made the synthesis very difficult. Due to the high volatility of the free-base 25, the compound was prepared as the hydrogen tartrate salt. When evaporating solutions of the free-base 25, solvent should be removed at pressures greater than 600 mbar at 60 °C in order to minimise losses of the free-base through evaporation.
The absolute stereochemistry was confirmed by X-ray analysis of the L-(+)-tartaric acid salt (R, R- (+)-tartaric acid).
Figure 3.
X-ray structure of (
R,R)-(+)-tartaric acid salt of
25 [
6].
Figure 3.
X-ray structure of (
R,R)-(+)-tartaric acid salt of
25 [
6].
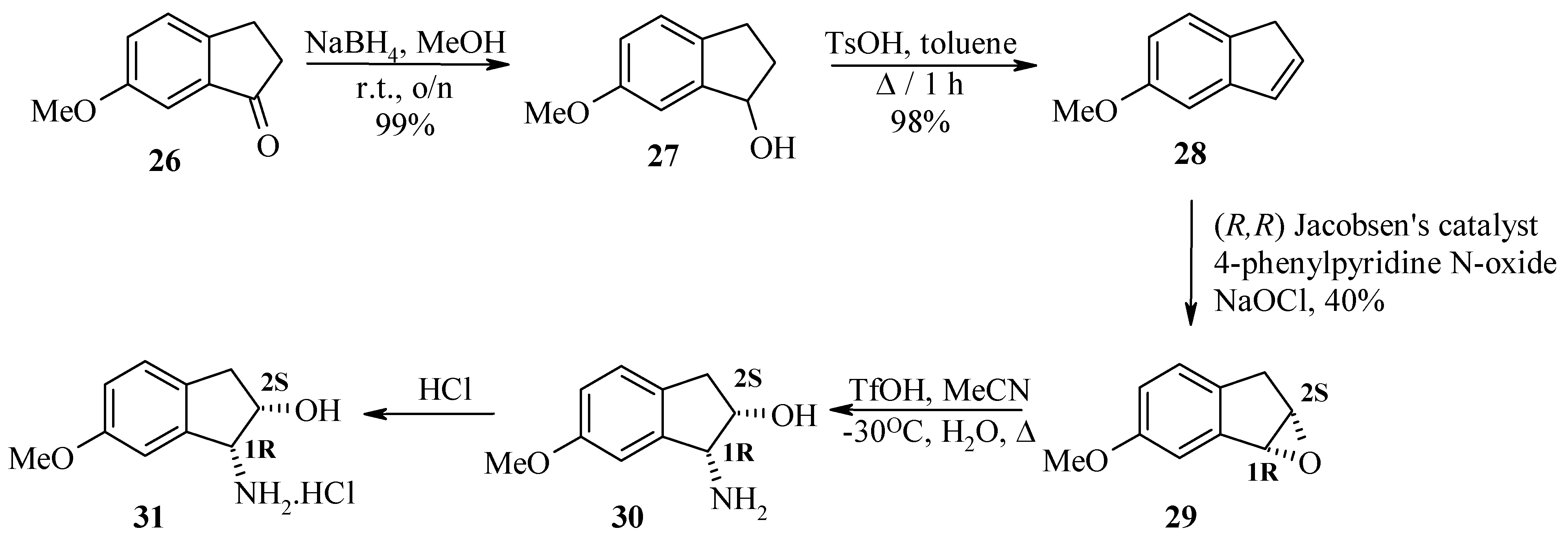
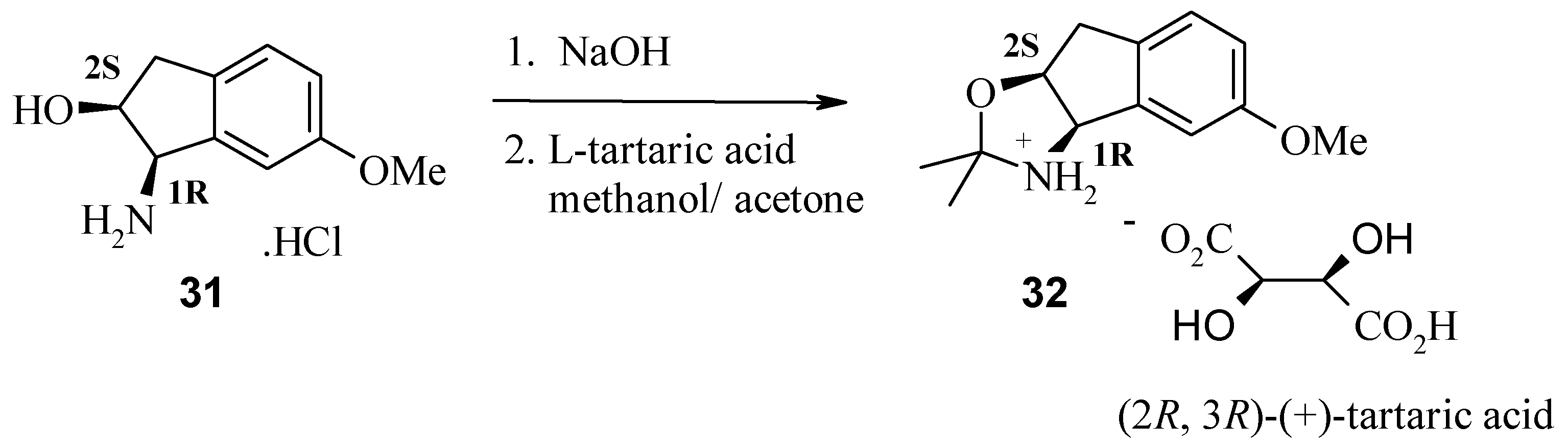
Finally, the synthesis of (1R, 2S)-cis-amino alcohol 30 is discussed. α-Amino alcohols can be prepared via asymmetric epoxidation employing Jacobsen’s catalyst. This chiral manganese epoxidation catalyst gives high ee’s with unactivated alkenes. Single enantiomer amino alcohols are then efficiently produced from the corresponding chiral epoxide by acid or base catalysed epoxide ring openings.
The synthesis of aminohydroxyindanes
via epoxidation of indenes has been reported in the literature [
7]. Sodium borohydride reduction of indanone
26 to indanol
27 followed by acid catalysed dehydration gave indene
28 in 50% yield after distillation. Chiral epoxidation of indene
28 with (
R,R)-Jacobsen’s catalyst, 4-phenylpyridine N-oxide and sodium hypochlorite provides the (
1R, 2S)-epoxide
29. Epoxide
29 then underwent a Ritter reaction with acetonitrile under acidic conditions
via a cyclic intermediate which was then hydrolysed to afford the (1
R, 2
S)-
cis-amino alcohol
30 and subsequently converted to the hydrochloride salt
31. The enantiomeric excess was determined to be >98% by LCMS and
19F-NMR analysis of the (
S)-Mosher amide derivative of
30.
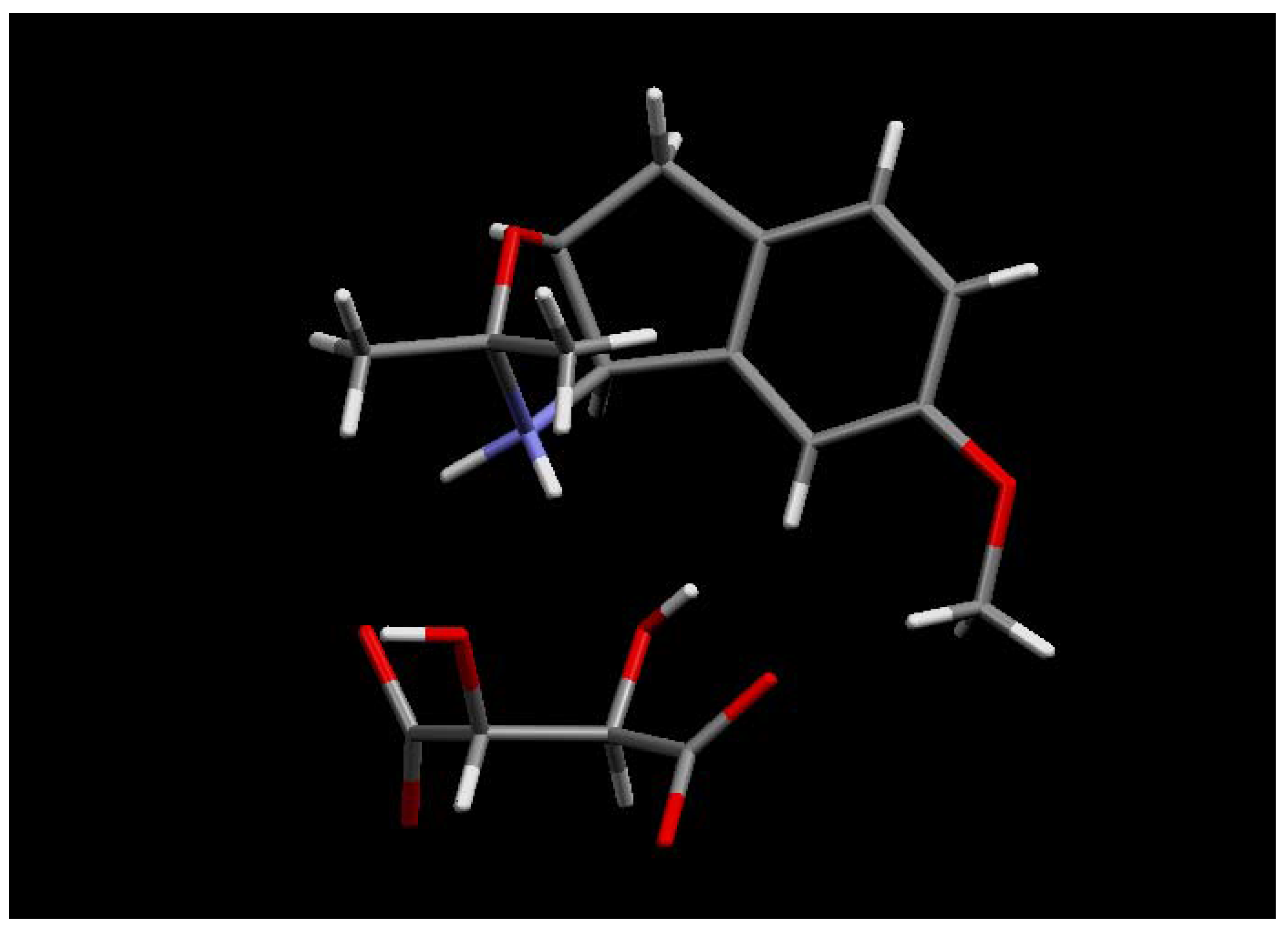
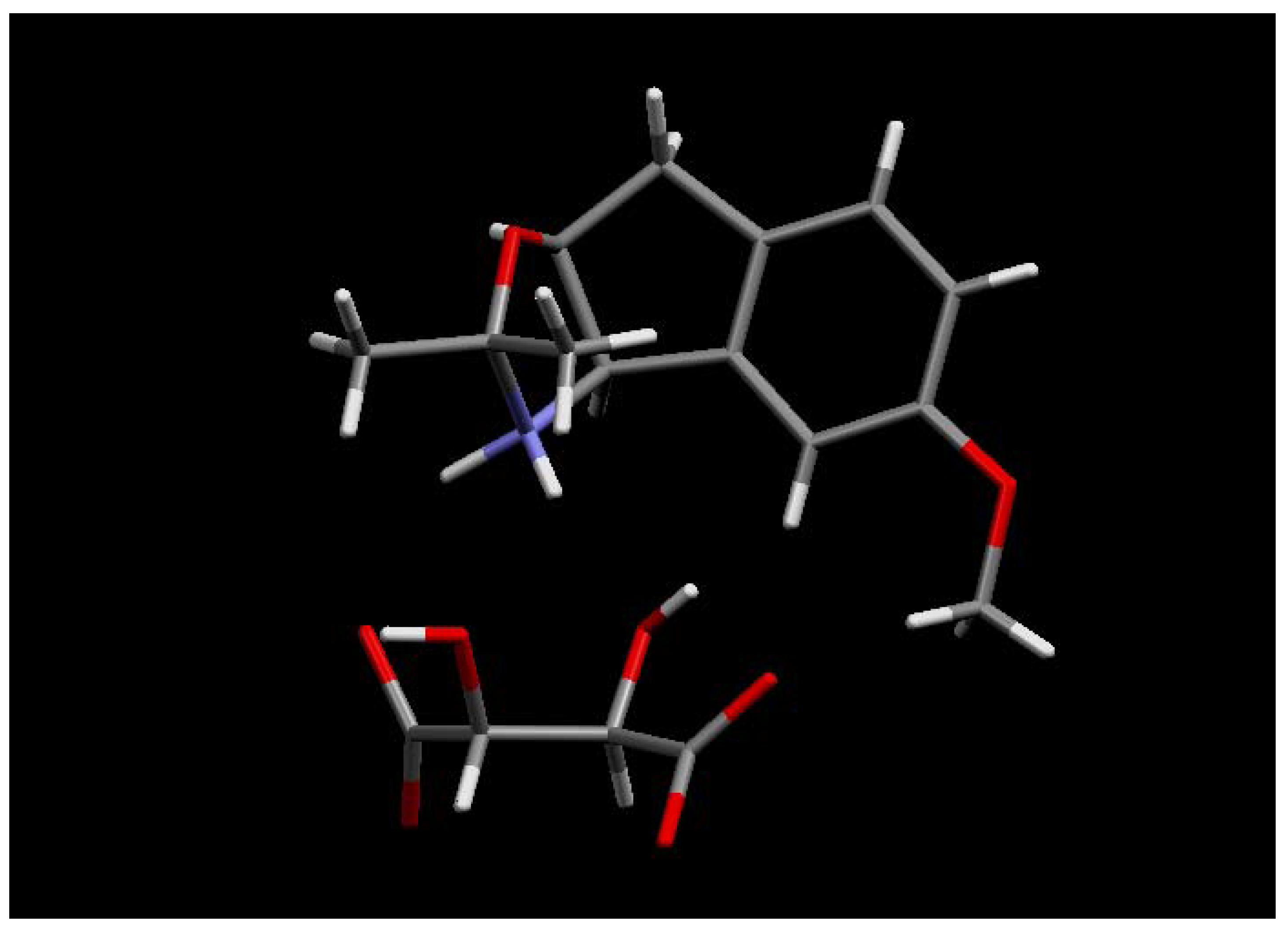
The (1R, 2S) absolute stereochemistry of 30 was confirmed by X-ray analysis of the L-(+)-tartaric acid salt (R, R-(+)-tartaric acid) of the acetonide 32, prepared in situ by recrystallisation from methanol and acetone.
Figure 4.
X-ray structure of (
R,R)-(+)-tartaric acid salt of
32 [
6].
Figure 4.
X-ray structure of (
R,R)-(+)-tartaric acid salt of
32 [
6].
Experimental
General
Reverse phase HPLC analysis was carried out using a Column Engineering Monitor C18 (5 μm), 50 x 4.6 mm. LCMS were run on a Perkin-Elmer Sciex API-100 instrument. ESMS were run on an API III LC/MS/MS from Perkin Elmer/Sciex using an electrospray inlet. 1H-, 19F- and 13C-NMR spectra were acquired on a 400 MHz Varian UNITY INOVA spectrometer and were recorded at 400 MHz, 377 MHz and 100 MHz respectively. X-ray crystallography was carried out on a Bruker Smart Apex X-ray diffractometer. The images were generated using Mercury 1.1 from .pdb files. Chiral catalysts (DuPHOS, Noyori and Jacobsen’s) were purchased from Strem. Flash chromatography was conducted on Merck Silica gel type 9385.
Synthesis of azlactone 7.
In a round bottom flask, N-acetylglycine (11.54 g, 98.51 mmol), anhydrous sodium acetate (5.77 g 70.37 mmol), 2,4-difluorobenzaldehyde (6, 20 g, 140.74 mmol) and 95% acetic anhydride (24.43 g, 239.26 mmol) were added and warmed to 40 °C until dissolved (~0.5 h). The resulting solution was refluxed for 1 h, cooled and placed in the refrigerator overnight. The light brown crystals were dissolved in ethyl acetate and washed with cold water (3 x 100 mL). The organic layer was collected, dried over sodium sulfate and the solvent removed under reduced pressure to afford azlactone 7 as a yellow powder (21.8 g, 69% yield). ECMS m/z 224.2 [M+H]+; LC/MS, tR = 8.30 min. (223.9 [M+H]+, 483.2 [2M+H]+ parent).
Synthesis of 2-acetylamino-3-(2,4-difluorophenyl)acrylic acid (8).
A solution of azlactone 7 (21.8 g, 97.68 mmol) in acetone (210 mL) and water (82 mL) was refluxed for 4 h. The reaction mixture was then cooled to room temperature and solvent removed under reduced pressure to yield a yellow solid. The crude product was dissolved in water (100 mL) and adjusted to pH 10 with potassium carbonate then the aqueous layer was extracted with ethyl acetate (3 x 100 mL). The organic layer was discarded and the aqueous layer adjusted to pH 3-4 with citric acid then extracted with ethyl acetate (5 x 100 mL). The organic layer was collected then dried over sodium sulfate and the solvent removed under reduced pressure to afford as a yellow powder (23.5 g, 88% yield). This crude material was then purified by recrystalization using boiled water to give acrylic acid 8 as a yellow solid (17.79 g, 75.6% yield). HPLC (214 nm), tR 4.76 min. (92.7%); 1H-NMR (CDCl3) δ: 2.41 (s, 3H), 3.49 (s, 1H), 6.85-6.89 (m,1H), 6.96-7.00 (m, 1H), 7.38 (s,1H), 8.71- 8.77 (m, 1H); ECMS m/z 483.1 [2M+H]+ ; LC/MS tR = 5.03 min. (242.3 [M+H]+, 483.2 [2M+H]+ parent).
Synthesis of (2S)-2-acetylamino-3-(2,4-difluorophenyl)propionic acid (9).
In an oven dried glass Parr hydrogenation flask, acrylic acid 8 (10.6 g, 41.46 mmol) was dissolved in anhydrous methanol (270 mL) along with the chiral catalyst (+)-1,2-bis((2S,5S)-2,5-diethyl phospholano)benzene(cyclooctadiene)rhodium (I) tetrafluoroborate (239 mg, 0.33 mmol). The flask was then placed in the Parr pressure reactor, which was evacuated and flushed with argon (x 5) before finally evacuating and flooding with hydrogen (x 5). The reaction was then allowed to proceed overnight at room temperature with stirring. The reaction mixture was then filtered through Celite™ and the filtrate concentrated under reduced pressure to give (S)-acetylamino acid 9 (10.0 g, 93% yield). This material was used without further purification. HPLC (214 nm), tR 5.42 min. (93.8%); 1H-NMR (CDCl3) δ: 1.83 (s, 3H), 2.87 (dd, J = 14.0, 8.8 Hz, 1H), 3.19 (dd, J = 14.0, 5.2 Hz, 1H), 4.61 (dd, J = 8.8, 5.2 Hz, 1H), 6.79-6.84 (m, 2H), 7.18-7.22 (m, 1H); ECMS m/z 244.1 [M+H]+, 285.0 [M+H+MeCN]+, 487.4 [2M+H]+; LC/MS tR = 5.24 min. (244.2 [M+H]+, 487.3 [2M+H]+ parent).
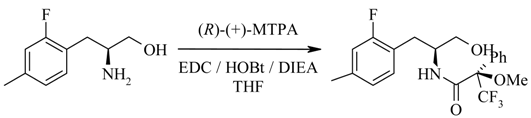
Synthesis of the (R)-Mosher amide of (2S)- 2-amino-3-(2,4-difluorophenyl) propan-1-ol (11).
To a stirred solution of (S)-amino alcohol 11 (12 mg, 64.11 mmol) in tetrahydrofuran (1 mL) at room temperature was added (R)-(+)-MTPA [(R)-(+)-α-methoxy-α-trifluoromethylphenyl acetic acid] (15 mg, 64.11 mmol), EDC (N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride) (12.3 mg, 64.11 mmol), DIEA (diisopropylethylamine) (22.3 μL, 128.2 mmol) and HOBt·H2O (1-hydroxy-benzotriazole hydrate) (9.8 mg, 66.11 mmol). The reaction was stirred at room temperature overnight then diluted with water and ethyl acetate. The organic layer was washed with citric acid, 10% sodium hydrogen carbonate and brine and dried over sodium sulfate. The organic solvent was removed under reduced pressure to yield a yellow oil. 1H-NMR, LCMS and reverse phase HPLC estimate the enantiomeric excess to be > 95%. The alternate (R)-enantiomer was also detected (0.65%). HPLC (214 nm), tR 8.92 min. (97.0%), 9.10 min. (0.65%, minor-isomer); 1H-NMR (CDCl3) δ: 2.86-2.97 (brm, 2H), 3.30 (s, 3H), 3.64 (dd, J = 11.3, 5.1 Hz, 1H), 3.69 (dd, J = 11.3, 4.5 Hz, 1H) 4.19-4.26 (brm, 1H), 6.77-6.85 (brm, 2H), 7.08 (d, J = 7.6 Hz, 1H), 7.18-7.23 (brm, 1H), 7.38-7.40 (brm, 3H), 7.49-7.50 (brm, 2H); ECMS m/z 404.1.0 [M+H]+; LC/MS tR = 8.66 min. (404.0 [M+H]+, 807.2 [2M+H]+, parention).
Synthesis of 2-benzyloxycarbonylamino-3-(2,4-dimethylphenyl)acrylic acid methyl ester (13).
To a solution of N-(benzyloxycarbonyl)-α-phosphonoglycine trimethyl ester (14.574 g, 44 mmol, 1.1 eq) in dichloromethane (80 mL) was added DBU (6.28 mL, 42 mmol, 1.05 eq). The reaction mixture was stirred for 0.5 h then 2,4-dimethylbenzaldehyde (12, 6.2 mL, 40 mmol, 1 eq) was added. The reaction was exothermic after addition of aldehyde 12. The reaction mixture was stirred at room temperature over the weekend. To this mixture was added aqueous 1N H2SO4 followed by separation of the dichloromethane layer. The aqueous phase was further extracted with an aliquot of ethyl acetate and the combined organic layers were dried over sodium sulfate, filtered and concentrated to give an oil. The crude product was purified by flash chromatography over silica gel with 20% ethyl acetate/petroleum ether as eluent followed by 50% dichloromethane/petroleum ether. The fractions containing the major band were combined and evaporated to dryness to provide amidoacrylate 13 as a colourless solid (12.19 g, 89% yield). HPLC (214nm), tR = 9.95 min. (80.8%); LC/MS tR = 9.02 min. (340.1 [M+H]+, 679.3 [2M+H]+, 696.6 [2M+NH4]+); 1H-NMR (CDCl3) δ: 2.27 (s, 3H), 2.31 (s, 3H), 3.82 (s, 3H), 5.07 (s, 2H), 6.22 (brs, 1H), 6.93 - 7.02 (m, 2H), 7.26-7.30 (m, 7H).
Synthesis of (2S)-2-benzyloxycarbonylamino-3-(2,4-dimethylphenyl)propionic acid methyl ester (14).
A mixture of amidoacrylate 13 (2.92 g, 8.59 mmol), (+)-1,2-bis((2S,5S)-2,5-diethylphosphonolano) benzene(cyclooctadiene)rhodium (I) trifluoromethanesulfonate (50 mg) dissolved in deoxygenated methanol (45 mL) was placed in a bomb. Five vacuum/N2 cycles to purge the gas lines of any oxygen followed by five vacuum/H2 cycles were carried out before the bomb was pressurised with H2 (30 psi/2 atm) and reacted at room temperature overnight. The pressure in the vessel was then released and the reaction mixture was filtered through a pad of Celite™ and concentrated to afford (S)-amido ester 14 as an oil (3.10 g, >100% yield). HPLC (214nm), tR = 9.96 min. (73.5%); LC/MS, tR = 9.06 min. (342.0 [M+H]+, 683.3 [2M+H]+); 1H-NMR (CDCl3) δ: 2.27 (s, 6H), 2.97 (dd, J = 14.0, 7.1 Hz, 1H), 3.12 (dd, J = 14.0, 6.1 Hz, 1H), 3.70 (s, 3H), 4.57-4.63 (m, 1H), 5.07 (m, 2H), 5.20 (d, J = 9.9 Hz, 1H), 6.91-6.97 (m, 3H), 7.28-7.36 (m, 5H).
Synthesis of (2S)-2-amino-3-(2,4-dimethylphenyl)propionic acid methyl ester (15).
A mixture of (S)-amido ester 14 (1.81 g, 5.31 mmol), 10% Pd / C (180 mg) in ethyl acetate (35 mL) was hydrogenated at room temperature overnight. The reaction mixture was filtered and concentrated to dryness to provide (S)-amino ester 15 as an oil (1.23 g, >100% yield). HPLC (214nm), tR = 5.57 min. (79%); LC/MS tR = 5.03 min. (208.1 [M+H]+, 415.2 [2M+H]+); 1H-NMR (CDCl3) δ: 2.28 (s, 3H), 2.31 (s, 3H), 2.74 (dd, J = 13.9, 8.8 Hz, 1H), 3.10 (dd, J = 13.9, 5.2 Hz, 1H), 3.67-3.71 (m, 4H), 6.93-7.03 (m, 3H).
(2S)-2-Amino-3-(2,4-dimethylphenyl) propan-1-ol hydrochloride (16).
In a 500 mL 3 necked round bottom flask fitted with a thermometer and flushed with nitrogen, lithium aluminium hydride (1.71 g, 45.1 mmol, 4eq) was suspended in anhydrous tetrahydrofuran (90 mL) and cooled in an ice bath to 0 °C. (S)-Amino ester 15 (2.75 g, 11.28 mmol) was solubilised in anhydrous tetrahydrofuran (140 mL) then slowly cannulated into the reaction mixture whilst maintaining temperature < 3°C. The reaction mixture was allowed to warm to room temperature, stirred for 1 h, then recooled to 0 °C and very cautiously diluted with water (15 mL), 15% sodium hydroxide (15 mL) and water (15 mL). The reaction mixture was allowed to stir for a further 0.5 h at 0°C then the aluminium salts were filtered off and the filtrate concentrated. The residue was taken up in ethyl acetate, washed with brine, extracted with ethyl acetate (x 3) and the combined organic extracts dried over sodium sulfate, filtered and concentrated. The crude material was diluted with dichloromethane, washed with 1N hydrochloric acid (x 5) and the combined aqueous layers were re-extracted with dichloromethane then basified and re-extracted with dichloromethane (x 5). The organic layer was dried over sodium sulfate, filtered and concentrated to afford a white solid (2.09 g) which was then taken up in 3 N hydrochloric acid and lyophilised to provide (S)-amino alcohol 16 as a pale yellow solid (1.99 g, 81% yield). HPLC (214nm), tR = 5.26 min. (96.4%); LC/MS tR = 4.71 min. (180.2 [M+H]+, 359.4 [2M+H]+); ESMS m/z 180.4 [M+H]+; 1H-NMR (DMSO-d6) δ: 1.22 (s, 3H), 1.24 (s, 3H), 1.80 (dd, J = 13.7, 9.9 Hz, 1H), 1.89 (dd, J = 13.7, 4.9 Hz, 1H), 2.16-2.26 (brm, 1H), 2.37 (dd, J = 11.4, 5.9 Hz, 2H), 5.93 (d, J = 7.7 Hz, 1H), 5.97 (s, 1H), 6.07 (d, J = 7.5 Hz, 1H), 7.14 (brs, 2H); 13C-NMR (DMSO-d6) δ: 19.0, 20.6, 31.8, 52.8, 59.5, 126.6, 130.0, 131.1, 131.7, 135.9, 136.2.
Synthesis of (R)-Mosher’s amide of (2S)-2-amino-3-(2,4-dimethylphenyl)propionic acid methylester (15).
Amine 15 (42 mg, 0.2 mmol) was dissoved in tetrahydrofuran (2 mL) under an atmosphere of N2, DIEA (104 mL, 0.6 mmol, 3 eq), (R)-(+)-MTPA (47 mg, 0.2 mmol), EDC (38 mg, 0.2 mmol) and HOBt.H2O (31 mg, 0.2 mmol) were added and the reaction stirred overnight. The reaction mixture was diluted with water and extracted with Et2O (2 x 25 mL). The combined organic layers were washed with brine (10 mL) then dried over anhydrous sodium sulfate, filtered and concentrated to afford the (R)-Moshers amide ester of 15 (26 mg, 30% yield) as a colourless oil. 1H-NMR, LCMS and reverse phase HPLC estimate the enantiomeric excess to be > 98% as the alternate (R, R)- diastereoisomer was undetectable. HPLC (214nm), tR = 5.76 (15, 13.8%), 10.66 (MA of 15, 60.2%) min.; LC/MS tR = 5.21 min. (15, 208.3 [M+H]+, 415.1 [2M+H]+), 9.76 min. (MA of 15, 424.2 [M+H]+, 847 [2M+H]+).
Synthesis of 2-fluoro-1-iodo-4-methyl benzene (18) via the Sandmeyer reaction.
In a round bottom flask, 2-fluoro-4-methylaniline (17, 1 g, 7.99 mmol) was suspended in water (2 mL) and concentrated hydrochloric acid (2 mL). This solution was then cooled in an ice bath with vigorous stirring. To this stirring solution was added sodium nitrite (662 mg, 9.58 mmol) dissolved in water (2 mL) dropwise over 0.5 h, keeping the temperature below 10 °C. The reaction was then stirred for a further 0.5 h. The resulting solution was then added dropwise to a solution of potassium iodide (1.99 g, 11.98 mmol) dissolved in water (2 mL) stirring in an ice bath. The reaction was then refluxed for 2 h before being allowed to stir at room temperature over night. The reaction mixture was then taken up in ethyl acetate and washed with 3N hydrochloric acid and 1M sodium hydroxide containing a small portion of sodium metabisulfite. The organic layer was then dried over sodium sulfate and the solvent removed under reduced pressure to afford a dark brown oil (1.46 g, 77% yield). On scale-up of this reaction the crude yield increased to 93%. This material was then purified via flash chromatography using a hexane running solvent and washed with 1M hydrochloric acid (x 2), 2M sodium hydroxide (x 1), brine (x 1) and dried over sodium sulfate to recover aryl iodide 18 (829 mg, 44% yield) as a colourless oil. HPLC (214 nm), tR =10.13 min. (76%); 1H-NMR (CDCl3) δ: 2.29 (s, 3H), 6.68 - 7.55 (m, 3H).
Synthesis of 2-acetylamino-3-(2-fluoro-4-methylphenyl)acrylic acid methyl ester (19) via the Heck reaction.
A mixture of aryl iodide 18 (2.00 g, 8.47 mmol), methyl-2-acetamidoacrylate (1.45 g, 10.17 mmol), Pd(OAc)2 (228 mg, 1.02 mmol), tetrabutylammonium chloride hydrate (2.83 g, 10.17 mmol), and sodium hydrogen carbonate (1.92 g, 22.88 mmol) was weighed into a thick walled oven dried glass vial, flushed with nitrogen and sealed. The vial was then heated to 80 °C for 24 h. The reaction was then cooled to room temperature and dissolved in dichloromethane. The black crude product was then absorbed onto silica gel. This crude material was then purified via flash chromatography using 45% ethyl acetate/hexane running solvent to recover amidoacrylate 19 as an off-white solid (average 90% yield). HPLC (214 nm), tR = 6.78 min. (80%); LC/MS, tR = 6.26 (252.1 [M+H]+, 503.2 [2M+H]+, parent) min.; 1H-NMR (CDCl3) δ: 2.00 (s, 3H), 2.30 (s, 3H), 3.77 (s, 3H), 6.80-6.89 (m, 2H), 7.32-7.40 (m, 2H).
Synthesis of (2S)-2-acetylamino-3-(2-fluoro-4-methylphenyl)propionic acid methyl ester (20).
In an oven dried Parr hydrogenation vial, amidoacrylate 19 (13.40 g, 53.32 mmol) was dissolved in anhydrous methanol (100 mL) along with the chiral catalyst; (+)-1,2-Bis((2S,5S)-2,5-diethyl phospholano)benzene(cyclooctadiene)rhodium (I) trifluoromethanesulfonate (38 mg, 53.32 μmol). The vial was then placed in the Parr pressure reactor, evacuated and flushed with argon (x 5) before evacuating and flooding with hydrogen (x 5). The reaction was then allowed to proceed for 48 h at room temperature with stirring. The reaction mixture was then filtered through cotton wool before removing the organic solvent under reduced pressure to yield (S)-amido ester 20 (13.80 g, >100% yield). This material was used without further purification. LC/MS tR = 6.47 min. (254.2 [M+H]+), 507.4 min. ([2M+H]+, parent); 1H-NMR (CDCl3) δ: 1.93 (s, 3H), 2.28 (s, 3H), 3.03 (dd, J = 6.0 Hz, J = 13.9 Hz, 1H), 3.12 (dd, J = 5.7 Hz, J = 13.9 Hz, 1H), 3.69 (s, 3H), 4.73-4.82 (m, 1H), 6.05 (d, J = 6.9 Hz, 1H), 6.78-6.86 (m, 2H), 6.96 (t, J = 7.8 Hz, 1H).
Reduction of methyl ester 20.
In an oven dried round bottom flask under nitrogen, lithium aluminium hydride (6.41 g, 168.88 mmol) was suspended in anhydrous tetrahydrofuran (280 mL) and cooled in an ice bath. To this stirring solution was added dropwise a solution of (S)-amido ester 20 (13.80 g, 54.47 mmol) in anhydrous tetrahydrofuran (280 mL). The reaction was then allowed to warm to room temperature and monitored via TLC (45% ethyl acetate/hexane running solvent) until completion within approximately 2 h. The reaction was then cooled in an ice bath and diluted with water and diethyl ether. To this vigorously stirring solution was added 2M sodium hydroxide, the reaction was then allowed to stir for a further 0.5 h. The aqueous layer was then extracted with diethyl ether (x 3) and the combined organic extracts dried over sodium sulfate. The organic layer was then concentrated under reduced pressure to recover the intermediate alcohol as a crude yellow solid (11.20 g, 91% yield). HPLC (214 nm), tR = 5.66 min. (56%); LC/MS, tR = 5.56 min. (226.2 [M+H]+ parent); 1H-NMR (CDCl3) δ: 1.91 (s, 3H), 2.28 (s, 3H), 2.80 (d, J = 6.6 Hz, 2H), 3.05 (bs, 1H), 3.54 (dd, J = 11.3, 4.9 Hz, 1H), 3.62 (dd, J = 11.3, 3.7 Hz, 1H), 4.10-4.13 (m, 1H), 6.15 (d, J = 7.3 Hz, 1H), 6.78-6.85 (m, 2H), 7.07 (t, J = 7.87 Hz, 1H).
Synthesis of the (R)-Mosher amide of 2-amino-3-(2-fluoro-4-methylphenyl)propan-1-ol (21).
To a solution of (S)-amino alcohol 21 (35 mg, 0.191 mmol) in tetrahydrofuran (0.5 mL) stirred at room temperature was added (R)-(+)-MTPA (45 mg, 0.191 mmol), EDC (37 mg, 0.191 mmol) and HOBt.H2O (26 mg, 0.171 mmol). The reaction mixture was the stirred at room temperature overnight then diluted with water and extracted with diethyl ether (x 3). The combined organic layers were then washed with 0.5M HCl, sodium hydrogen carbonate and brine, dried over sodium sulfate and the solvent removed under reduced pressure to yield a colourless oil. 1H-NMR, LCMS and reverse phase HPLC estimate the enantiomeric excess to be greater than 95% as the alternate (R)-enantiomer was undetectable. HPLC (214 nm), tR 9.33 min. (74%); 1H-NMR (CDCl3) δ: 2.23 (s, 3H), 2.75-2.86 (m, 2H), 2.91 (bs, 1H), 3.20 (s, 3H), 3.48 (dd, J = 11.2, 4.9 Hz, 1H), 3.54 (dd, J = 11.2, 4.0 Hz, 1H), 4.12-4.18 (m, 1H), 6.74-6.81 (m, 2H), 7.00-7.05 (m, 1H), 7.25-7.30 (m, 3H), 7.40-7.45 (m, 2H); LC/MS tR = 8.51 min. (400.2 [M+H]+, 799.3 [2M+H]+, parent).
(1R, 2S)-2-Methyl-1-cyclohexanol (23).
A pellet of potassium hydroxide (105 mg, 1.87 mmol) was fused under an atmosphere of argon and allowed to cool under a stream of argon. Anhydrous 2-propanol (25 mL) was added and the mixture stirred with heating to dissolve the potassium hydroxide. A solution of 2-methyl-1-cyclohexanone (22, 5.652 g, 50.4 mmol) in anhydrous 2-propanol (25 mL) was prepared under argon. The solution was sonicated under a stream of argon for 15 min. Dichloro[(S)-2,2’-bis(diphenylphosphino)-1,1’-binaphthyl][(1R,2R)-1,2-diphenylethylenediamine]ruthenium(II) (50 mg, 50 µmol) was purged with argon and drawn into a syringe as a suspension in anhydrous 2-propanol (10 mL). The potassium hydroxide solution was added to the 2-methyl-1-cyclohexanone solution via syringe followed by the ruthenium catalyst suspension. A hydrogenation flask was heated with a heat gun and allowed to cool under a stream of argon. The cyclohexanone/catalyst/KOH mixture was added to a hydrogenation flask via syringe. The argon atmosphere was exchanged with hydrogen with five evacuation/hydrogen purge cycles. The mixture was stirred at room temperature under an atmosphere of hydrogen (65 psi) for 65 h. Water (250 mL) was added to the reaction mixture and the aqueous phase extracted with ether (3 × 100 mL). The combined ether extracts were washed with water (250 mL) and brine (250 mL), dried over anhydrous sodium sulfate and filtered.The filtrate was evaporated to dryness (60 °C, 600 mbar) to give a pale brown oil which was purified by flash chromatography over silica gel with dichloromethane as eluent. Fractions which contained the cis isomer (Rf 0.46) were combined and evaporated to dryness (60 °C, 850 mbar) to give (1R, 2S)-2-methyl-1-cyclohexanol (23) as a clear oil (5.40 g, 94%). By comparison of the integrals in the 1H-NMR spectrum of the resonance due to dichloromethane and the resonance at δ 3.77, the purity of the sample was 81 mol% or 85 wt%. (Corrected yield after taking dichloromethane into account: 4.58 g, 80% yield). 1H-NMR (CDCl3) δ: 0.94 (d, J = 6.8 Hz, 3H), 1.20-1.78 (m, 10H), 3.77 (br s, 1H); 13C-NMR (CDCl3) δ: 17.1, 20.9, 24.7, 30.0, 32.7, 36.0, 71.3.
(1S, 2S)-1-Azido-2-methylcyclohexane (24).
Solutions of triphenylphosphine (4.13 g, 15.8 mmol) in anhydrous tetrahydrofuran (60 mL) and (1R, 2S)-2-methyl-1-cyclohexanol (23, 1.50 g, 13.1 mmol) in anhydrous tetrahydrofuran (40 mL) were prepared under nitrogen. Diethyl azodicarboxylate (2.50 mL, 15.9 mmol) was added dropwise to the triphenylphosphine solution under nitrogen and the mixture stirred at room temperature for 10 min. Diphenylphosphoryl azide (3.40 mL, 15.7 mmol) was added dropwise to this mixture under nitrogen. The (1R, 2S)-2-methyl-1-cyclohexanol solution was transferred via cannula to the mixture. The mixture was stirred at room temperature for 18 h. The reaction mixture was evaporated to dryness (60 °C, 285 mbar). The residue was dissolved in a 1:1 dichloromethane/n-hexane mixture (80 mL) and purified by flash chromatography over gel with 1:1 dichloromethane/n-pentane as eluent. Fractions eluting prior to the first fraction that could be visualised under a 254 nm lamp (Rf 0.60, eluent: dichloromethane) were combined and evaporated to dryness (60 °C, 600 mbar) to give a clear oil (647 mg, 35% yield). 1H-NMR analysis of the first fraction that could be visualised under a 254 nm lamp showed that it contained (1S,2S)-1-azido-2-methylcyclohexane (24) in addition to triphenyl- phosphine oxide and tetrahydrofuran. This fraction was dissolved in 1:2 dichloromethane/n-pentane (20 mL) and purified by flash chromatography over silica gel with 1:2 dichloromethane/n-pentane (23 × 40 mL), 1:1 dichloromethane/n-pentane (17 × 40 mL), as eluent. The fractions containing the (1S,2S)-1-azido-2-methylcyclohexane 24 were evaporated to dryness (60 °C, 600 mbar). The samples of 24 obtained from the two columns were combined and evaporated to dryness (60 °C, 600 mbar) to give the title compound as a clear oil (865 mg, 47% yield). By comparison of the integrals in the 1H-NMR spectrum of the resonance due to dichloromethane and the resonance at δ 2.78, the purity of the sample was 78 mol% or 85 wt%. (corrected yield after taking dichloromethane into account: 736 mg, 40% yield). HPLC (214 nm), tR = 6.61 min. (9%), 10.28 min. (85%); 1H-NMR (CDCl3) δ: 1.01 (d, J = 6.8 Hz, 3H), 1.15-1.45 (m, 5H), 1.61-1.68 (m, 1H), 1.71-1.84 (m, 2H), 2.01-2.08 (m, 1H), 2.78 (td, J = 10.6, 4.0 Hz, 1H); 13C-NMR (CDCl3) δ: 19.6, 25.3, 25.5, 31.7, 34.1, 37.9, 67.3; ESMS m/z No molecular ion observed; LC/MS tR = 5.85 min. (no molecular ion observed), 9.78 min. (no molecular ion observed).
(1S, 2S)-2-Methyl-1-cyclohexylamine (25).
A mixture of (1S, 2S)-1-azido-2-methylcyclohexane (24, 865 mg, 6.21 mmol), triphenylphosphine (3.282 g, 12.5 mmol), water (225 µL, 12.5 mmol) and tetrahydrofuran (28 mL) was heated at reflux for 16 h. The reaction mixture was evaporated to dryness (60 °C, 285 mbar).Ether (50 mL) and hydrochloric acid (0.1 M, 100 mL) were added to the crude product. The phases were separated and the aqueous phase washed with ether (2 × 50 mL). A 10% sodium carbonate solution (25 mL) was added and the aqueous phase extracted with dichloromethane (3 × 50 mL). The combined dichloromethane extracts were dried over anhydrous sodium sulfate and filtered. The filtrate was evaporated to dryness (60 °C, 500 mbar) to give (1S, 2S)-2-methyl-1-cyclohexylamine (25) as a pale yellow oil (crude: 680 mg, 97% yield). By comparison of the integrals in the 1H-NMR spectrum of the resonance due to dichloromethane and the multiplet at δ 2.20, the purity of the sample was 57 mol% or 64 wt% (corrected yield after taking dichloromethane into account: 435 mg, 62% yield). 1H-NMR (CDCl3) δ: 0.97 (d, J = 6.4 Hz, 3H), 0.99-1.34 (m, 7H), 1.60-1.75 (m, 3H), 1.77-1.84 (m, 1H), 2.20 (ddd, J = 10.9, 9.5, 4.0 Hz, 1H); 13C-NMR (CDCl3) δ: 19.3, 26.0, 26.4, 34.5, 36.6, 41.1, 56.9.
Resolution of 25: (1S, 2S)-2-Methyl-1-cyclohexylammonium hydrogen (+)-tartrate.
(+)-Tartaric acid (790 mg, 5.26 mmol) was dissolved in methanol (2.0 mL) with heating and allowed to cool. 2-Methyl-1-cyclohexylamine (25, 680 mg, 5.32 mmol) was dissolved in methanol (3.0 mL). The tartaric acid solution was added to the 2-methyl-1-cyclohexylamine solution. The volume of the 2-methyl-1-cyclohexylamine / tartaric acid mixture was reduced to ca. 4 mL by heating. The mixture was allowed to cool to room temperature and then placed in the freezer overnight. The mixture was removed from the freezer and allowed to warm to room temperature then filtered and the residue dried at the pump to give (1S, 2S)-2-methyl-1-cyclohexylammonium hydrogen (+)-tartrate as a white solid (390 mg, 28% yield). 1H-NMR (D2O) δ: 1.07 (d, J = 6.4 Hz, 3H), 1.11-1.20 (m, 1H), 1.24- 1.48 (m, 3H), 1.52-1.64 (m, 1H), 1.69-1.76 (m, 1H), 1.79-1.88 (m, 2H), 2.02-2.08 (m, 1H), 2.87 (td, J = 10.7, 3.2 Hz, 1H), 4.58 (s, 2H); 13C-NMR (D2O) δ: δ 17.8, 24.4, 24.9, 30.8, 33.4, 36.0, 56.7, 73.0, 176.4. (n.b. 13C-NMR chemical shifts are relative as there was no internal reference present).
Determination of the enantiomeric excess of 25: preparation of the Mosher amide.
(1S,2S)-2-methyl-1-cyclohexylammonium hydrogen (+)-tartrate (20 mg) was dissolved in deuterium oxide (0.7 mL). Water (1.0 mL) and a 10% sodium carbonate solution (2.0 mL) were added to the NMR sample and the aqueous phase extracted with dichloromethane (3 × 2 mL). The organic extracts were dried over anhydrous sodium sulfate and filtered. The filtrate was evaporated to dryness (60 °C, 600 mbar) to give (1S,2S)-2-methyl-1-cyclohexylamine 25 as a white solid. A mixture of EDC (36.4 mg, 0.19 mmol), (S)-α-methoxy-α-trifluoro-methylphenylacetic acid (35.3 mg, 0.15 mmol), 1-hydroxybenzotriazole hydrate (29.8 mg, 0.19 mmol) and diisopropylethylamine (62 µL, 0.36 mmol) in tetrahydrofuran (2.0 mL) was stirred at room temperature for 0.5 h. 2-Methyl-1-cyclohexyl-amine (25) was dissolved in tetrahydrofuran (1.0 mL) and added to the mixture. The mixture was stirred at room temperature for 15.5 h. Water (10 mL) was added and the mixture extracted with ethyl acetate (3 × 10 mL). The ethyl acetate extracts were washed with hydrochloric acid (1 M, 2 × 10 mL), a 10% sodium carbonate solution (2 × 10 mL) and brine (10 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was evaporated to dryness. The residue was dissolved in dichloromethane and filtered through a plug of silica gel with dichloromethane as eluent. The filtrate was evaporated to dryness to give a white solid (crude: 20 mg, 80% yield overall for the neutralisation and Mosher amide formation). 19F-NMR analysis of the Mosher amide of 25 showed two resonances. The integral of these two peaks was 96.1:3.9, which is equivalent to an enantiomeric excess of 92%. 19F-NMR (CDCl3) δ: -70.2 (s, 96.1% of total integral, (1S, 2S, S-MTPA)), -70.5 (s, 3.9% of total integral, (1R, 2R, S-MTPA)). (n.b. 19F-NMR chemical shifts are relative as there was no internal reference present).
Preparation of 6-methoxy -1-indanol (27).
6-Methoxy-1-indanone (26, 92.5 mmol) was dissolved in methanol (150 mL) at room temperature. Sodium borohydride powder (107.7 mmol) was added portionwise over several minutes with stirring. After overnight stirring at room temperature, the reaction mixture was diluted with distilled water (100 mL) and acidified with 2M hydrochloric acid (100 mL) then extracted with ether (3 x 150 mL). The combined ether extracts were washed with brine (150 mL), dried over anhydrous sodium sulfate and concentrated to give 6-methoxy-1-indanol 27 (15.5 g, 98.6% yield) as a clear oil. HPLC (214 nm), tR = 6.13 min. (96.4%); 1H-NMR (CDCl3) δ: 1.95-1.99 (m, 1H), 2.41-2.57 (m, 1H), 2.65-2.79 (m, 1H), 2.90-3.00 (m, 1H), 3.78 (s, 3H), 5.17 (t, J = 6.0 Hz, 1H), 6.80 (dd, J = 8.4, 2.4 Hz, 1H), 6.93 (d, J = 2.4 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H); 13C-NMR (CDCl3) δ: 29.1, 36.8, 55.7, 76.9, 109.0, 115.2, 125.7, 135.3, 146.5, 159.2; LC/MS tR = 5.73 min. (147.3 [M+H-18]+).
Preparation of 5-methoxy-1H-indene (28).
The crude 6-methoxy-1-indanol 27 (92.5 mmol) and 4-toluenesulfonic acid (5.81 mmol) were dissolved in toluene (200 mL). The mixture was boiled under a Dean-Stark water collector for 5 h then washed with 5% sodium sulfate (3 x 80 mL) and brine (1 x 50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and the solvent evaporated to give crude product as a brown oil. The crude oil was distilled under vacuum to give pure 5-methoxy-1H-indene 28 as a clear pale oil (11.2 g, 83.3% yield). HPLC (214 nm), tR = 8.86 min. (98.4%); 1H-NMR (CDCl3) δ: 3.33 (s, 2H), 3.82 (s, 3H), 6.56-6.58 (m, 1H), 6.75 (dd, J = 8.4, 2.4 Hz, 1H), 6.81-6.83 (m, 1H), 6.96 (d, J = 4 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H); 13C-NMR (CDCl3) δ: 38.5, 55.7, 106.7, 110.9, 124.3, 132.2, 135.8, 136.0, 146.5, 159.1; LC/MS tR = 8.48 min.
Chiral epoxidation of 5-methoxy-1H-indene (28) to produce epoxide 29.
A stirred mixture of 5-methoxy indene (28, 13.9 mmol), (R,R)-Jacobsen’s catalyst (1.39 mmol) and 4-phenylpyridine N-oxide (1.39 mmol) in dichloromethane (1.6 mL) was cooled to 5 °C. A cold aqueous solution of sodium hypochlorite (10.7 mL) was added in slowly with vigorous stirring while maintaining the reaction temperature between 0 – 2 °C. Upon complete addition of the bleach, the reaction mixture was stirred for another one hour at 0 °C. At this point, hexane (11 mL) was added in one portion with stirring and the reaction mixture was filtered through a pad of Celite™ on a large buchner funnel. The filtrate brown organic layer was washed with brine (2 x 50 mL), dried over anhydrous sodium sulfate, filtered and concentrated to give crude epoxide 29 as a brown liquid (990 mg, 40% yield). HPLC (214 nm), tR = 4.81 min. (65.4%); 1H-NMR (CDCl3) δ: 2.91 (dd, J = 17.6, 2.8 Hz, 1H), 3.14 (d, J = 17.6 Hz, 1H), 3.8 (s, 3H), 4.12 (t, J = 2.8 Hz, 1H), 4.21 (m, 1H), 6.80 (dd, J = 8.2, 2.4 Hz, 1H), 7.07 (d, J = 2.4 Hz, 1H), 7.12 (d, J = 8.2 Hz, 1H); 13C-NMR (CDCl3) δ: 34.0, 55.7, 58.5, 59.3, 111.4, 114.4, 126.8, 135.3, 142.3, 158.6; ESMS m/z 163.2 [M+H]+, 180.9 [M+H+H2O]+, 204.5 [M+H+MeCN]+, 325.5 [2M+H]+; LC/MS tR = 4.51 min. (204.1 [M+H+MeCN]+).
Preparation of (1R, 2S)-1-amino-6-methoxy-indan-2-ol (30).
A three-necked flask under nitrogen atmosphere was charged with indene oxide 29 (4.0 mmol), acetonitrile (5 mL) then stirred and cooled to –40 °C. To this slurry was added trifluoromethanesulfonic acid (8.0 mmol) while maintaining the reaction temperature at –30 °C. The reaction mixture was warmed to room temperature and stirred for 1 h. Water (10 mL) was added and aged for 15 min. After removal of acetonitrile under reduced pressure, the reaction mixture was heated at reflux for 5 h. After cooling to room temperature, dichloromethane (5 mL) was added and stirred for 10 min. The two phases were separated and the aqueous layer containing the amino indanol was collected. The aqueous solution was basified with 3M sodium hydroxide (5 mL) and extracted with ethyl acetate (3 x 30 mL). The organic layer was concentrated under reduced pressure to about 10 mL. To this solution, cold hydrochloric acid in ethyl acetate was added slowly with stirring. The resulting solid salt was collected and washed with cold petroleum spirit to give the desired (1R, 2S)-cis-amino alcohol as the hydrochloride salt 31. HPLC (214 nm) tR = 4.10 min. (92.4%); 1H-NMR (CDCl3) δ: 2.86 (dd, J = 16.0, 2.7 Hz, 1H), 3.03 (dd, J = 16.0, 5.6 Hz, 1H), 3.8 (s, 3H), 4.28 (brs,1H), 4.37 (m, 1H), 6.78 (dd, J = 8.4, 2.4 Hz, 1H), 6.85 (d, J = 2.4 Hz, 1H), 7.12 (d, J = 8.2 Hz, 1H); 13C-NMR (CDCl3) δ: 38.8, 55.7, 59.0, 73.6, 109.6, 114.1, 126.2, 132.8, 145.61, 159.43; ESMS m/z 180.4 [M+H]+, 221.5 [M+H+MeCN]+; LC/MS tR = 3.80 (180.3 [M+H]+) min.
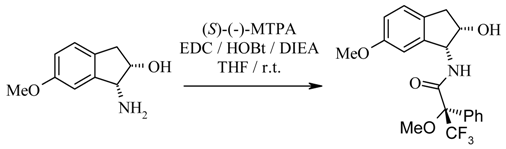
Determination of the chiral purity via (S)-Mosher amide of (30).
A mixture of EDC (0.06 mmol, 1 eq), (S)-(-)-MTPA [(S)-α-methoxy-α-trifluoromethylphenylacetic acid, 0.06 mmol, 1 eq], HOBt·H2O (0.06 mmol, 1 eq) and DIEA (0.12 mmol, 2 eq) in tetrahydrofuran (300 µL) was stirred at room temperature for 15 min. A solution of (1R, 2S)-cis-amino alcohol 30 (10 mg, 0.06 mmol) in tetrahydrofuran (200 µL) was added and the mixture stirred at room temperature for two days. Ethyl acetate (20 mL) was added and the solution was washed with 5% citric acid (2 x 10 mL), 10% sodium hydrogen carbonate solution (2 × 10 mL) and brine (2 x 10 mL), then dried over anhydrous sodium sulfate and filtered. The filtrate was evaporated to dryness to give the (S)-Mosher amide derivative of 30; HPLC (214 nm), tR = 9.01 min. (73.7%); 19F-NMR (CDCl3) δ: - 69.1; LC/MS tR = 8.53 min. (396.2 [M+H]+). An extracted ion chromatogram (mass = 396.0 – 397.1) showed only 1 peak corresponding with the (S)-Mosher amide derivative of 30. The chiral purity of the amide therefore was over 99%. This agreed with 19F NMR data, showing one line for one isomer only. Thus, the enantiomeric excess of 1-amino-6-methoxy-indan-2-ol (30) was determined to be >98%.

{kind=link}
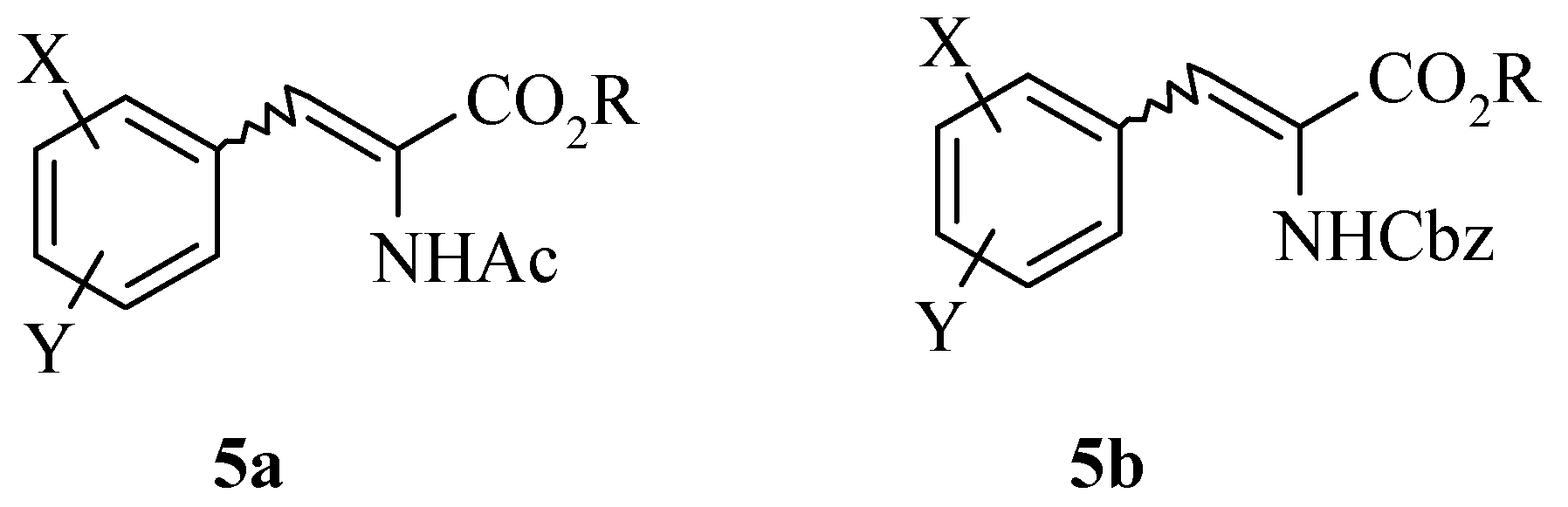{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}