Solid State Deprotection of Acetals and Thioacetals Using Benzyltriphenylphosphonium Peroxymonosulfate

Abstract
:Introduction
Results and Discussion


{kind=link}
| Substrate | R1 | R2 | n, X | Subs./AlCl3/Oxidant | Time (min) | Yield (%)b |
|---|---|---|---|---|---|---|
| 2a | Ph | Me | 1, S | 1/1/2 | 12 | 96 |
| 2b | 4-Cl-C6H4 | Me | 1, S | 1/1/2 | 15 | 90 |
| 2c | 2-O2N-C6H4 | H | 2, S | 1/1/2 | 20 | 87 |
| 2d | 3-O2N-C6H4 | H | 2, S | 1/1/2 | 20 | 88 |
| 2e | 4-O2N-C6H4 | H | 2,S | 1/1/2 | 20 | 91 |
| 2f | 4-Cl-C6H4 | H | 2, S | 1/1/2 | 16 | 93 |
| 2g | 4-Br-C6H4 | CH2Br | 2, S | 1/1/2 | 15 | 86 |
2h | 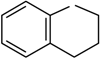 | __ | 2, S | 1/1/2 | 10 | 97 |
| 2i | Ph | H | 2, S | 1/1/2 | 5 | 99 |
| 2j | 2-MeOC6H4 | H | 2, S | 1/1/2 | 12 | 90 |
| 2k | Ph | Me | 1, O | 1/1/1 | 5 | 91 |
| 2l | 2-MeOC6H4 | H | 1, O | 1/1/1 | 10 | 88 |
| 2m | 2-O2N-C6H4 | H | 1, O | 1/1/1 | 12 | 80 |
| 2n | 2-Cl-C6H4 | Me | 1, O | 1/1/1 | 13 | 85 |
2o | | __ | 1, O | 1/1/1 | 5 | 99 |
| 2p | 4-Ph-C6H4 | Me | 1, O | 1/1/1 | 10 | 92 |
| R1 | R2 | n | Yield (%)/Time (min) | |||
|---|---|---|---|---|---|---|
| Ia | IIb | IIIc | IVd | |||
| Ph | H | 2 | 99/5 | 96/90 | 70/50 | __ |
| 4-Cl-C6H4 | H | 2 | 93/16 | 92/120 | __ | __ |
| Ph | Me | 1 | 96/12 | __ | 91/120 | 98/25 |
Conclusions
Acknowledgments
Experimental
General
Typical Procedure for Solid Phase Deprotection of Acetals and Thioacetals with Reagent 1: Reaction of 2-Methyl-2-(4-chlorophenyl)-1,3-dithiolane (2b).
References and Notes
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis; Wiley: New York, 1991. [Google Scholar] Komatsu, N.; Uda, M.; Suzuki, H. Synlett 1995, 984.
- Guanti, G.; Banfi, L.; Brusco, S.; Riva, R. Tetrahedron Lett. 1993, 34, 8549.
- Firouzabadi, H.; Iranpoor, N.; Karimi, B. Synthesis 1999, 58. Seebach, D.; Corey, E.J. J. Org. Chem. 1975, 40, 231. Groblel, B.T.; Seebach, D. Synthesis 1977, 357. Hatch, R.P.; Shringarpure, J.; Weinreb, S.M. J. Org. Chem. 1978, 43, 4172. Marshall, J.A.; Belletire, J.L. Tetrahedron Lett. 1971, 871.
- Lebouc, A.; Simonet, J.; Gelas, J.; Dehbi, A. Synthesis 1987, 320.
- Firouzabadi, H.; Iranpoor, N.; Zolfigol, M.A. Bull. Chem. Soc. Jpn. 1998, 71, 2169. Varma, R.S.; Saini, R.K. Tetrahedron Lett. 1997, 38, 2633. Meshram, H.M.; Reddy, G.S.; Yadav, J.S. Tetrahedron Lett. 1997, 38, 8891. Curini, M.; Marcotullio, M.C.; Pisani, E.; Rosati, O. Synlett 1997, 769. Curini, M.; Ceccherelli, P.; Marcotullio, M.C.; Epifano, F.; Rosati, O. Synlett 1996, 767. Komatsu, N.; Taniguchi, A.; Uda, M.; Suzuki, H. Chem. Commun. 1996, 1847. Schmittel, M.; Levis, M. Synlett 1996, 315. Haroutounian, S.A. Synthesis 1995, 39. Hirano, M.; Ukawa, K.; Yakabe, S.; Clark, J.H.; Morimoto, T. Synthesis 1997, 858. Lee, J.G.; Hwang, J.P. Chem. Lett. 1995, 507. Meshram, H.M.; Reddy, G.S.; Sumitra, G.; Yadav, J.S. Synth. Commun. 1999, 29, 1113.
- Firouzabadi, H.; Hazarkhani, H.; Karimi, B.; Niroumand, U.; Ghassamipour, S. Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), September 1-30, 2000.
- Mohammadpoor-Baltork, I.; Nourozi, A.R. Synthesis 1999, 487. Meskens, F.A.J. Synthesis 1981, 501. Gros, P.; Perchec, P.L.; Senet, J.P. J. Chem. Res. (S) 1995, 196. Marcantoni, E.; Nobili, F. J. Org. Chem. 1997, 62, 4183.
- For some recent work using Oxone® as an oxidant, see: Hajipour, A.R. Indian J. Chem. 1997, 36B, 1069. Hajipour, A.R.; Mahboubkhah, N. Org. Prep. Proced. Int. 1999, 31, 112. Meunier, B. New J. Chem. 1992, 16, 203.
- Hajipour, A.R.; Mallakpour, S.E.; Adibi, H. Phosphorus, Sulfur and Silicon 2000, 167, 71. Hajipour, A.R.; Mallakpour, S.E.; Adibi, H. Chem. Lett. 2000, 460. Hajipour, A.R.; Mallakpour, S.E.; Mohammadpoor-Baltork, I.; Adibi, H. Phosphorus, Sulfur and Silicon 2000, 165, 155. Hajipour, A.R.; Mallakpour, S.E.; Mohammadpoor-Baltork, I.; Adibi, H. Synth. Commun. 2001, 31, 3401. Hajipour, A.R.; Mallakpour, S.E.; Adibi, H. Chem. Lett. 2001, 164. Hajipour, A.R.; Mallakpour, S.E.; Adibi, H. Phosphorus, Sulfur and Silicon 2002, in press.
- Toda, F. Acc. Chem. Res. 1995, 28, 480. Toda, F.; Tanaka, K. Chem. Rev. 2000, 100, 1025.
- Hajipour, A.R.; Mallakpour, S.E.; Khoee, S. Chem. Lett. 2000, 120. Hajipour, A.R.; Mallakpour, S.E.; Imanzadeh, Gh. Indian J. Chem. 2001, 40B, 237. Hajipour, A.R.; Mallakpour, S.E.; Afrousheh, A. Phosphorus, Sulfur and Silicon 2000, 160, 67. Hajipour, A.R.; Mallakpour, S.E.; Khoee, S. Synlett 2000, 740. Hajipour, A.R.; Mallakpour, S.E.; Afrousheh, A. Tetrahedron 1999, 55, 2311. Hajipour, A.R.; Mallakpour, S.E.; Mohammadpoor-Baltork, I.; Khoee, S. Synth. Commun. 2001, 31, 1138. Hajipour, A.R.; Mallakpour, S.E. Mol. Cryst. and Liq. Cryst. 2001, 55, 356.
- Trost, B.M.; Braslau, R. J. Org. Chem. 1988, 53, 532. [CrossRef]
- Dictionary of Organic Compounds, 6th ed.; Chapman and Hall: London, 1982.
- Sample availability: Samples are available from the authors
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Hajipour, A.R.; Mallakpour, S.E.; Mohammadpoor-Baltork, I.; Adibi, H. Solid State Deprotection of Acetals and Thioacetals Using Benzyltriphenylphosphonium Peroxymonosulfate. Molecules 2002, 7, 674-680. https://doi.org/10.3390/70800674
Hajipour AR, Mallakpour SE, Mohammadpoor-Baltork I, Adibi H. Solid State Deprotection of Acetals and Thioacetals Using Benzyltriphenylphosphonium Peroxymonosulfate. Molecules. 2002; 7(8):674-680. https://doi.org/10.3390/70800674
Chicago/Turabian StyleHajipour, Abdol Reza, Shadpour E. Mallakpour, Iraj Mohammadpoor-Baltork, and Hadi Adibi. 2002. "Solid State Deprotection of Acetals and Thioacetals Using Benzyltriphenylphosphonium Peroxymonosulfate" Molecules 7, no. 8: 674-680. https://doi.org/10.3390/70800674
APA StyleHajipour, A. R., Mallakpour, S. E., Mohammadpoor-Baltork, I., & Adibi, H. (2002). Solid State Deprotection of Acetals and Thioacetals Using Benzyltriphenylphosphonium Peroxymonosulfate. Molecules, 7(8), 674-680. https://doi.org/10.3390/70800674