DivCalc: A Utility for Diversity Analysis and Compound Sampling

{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
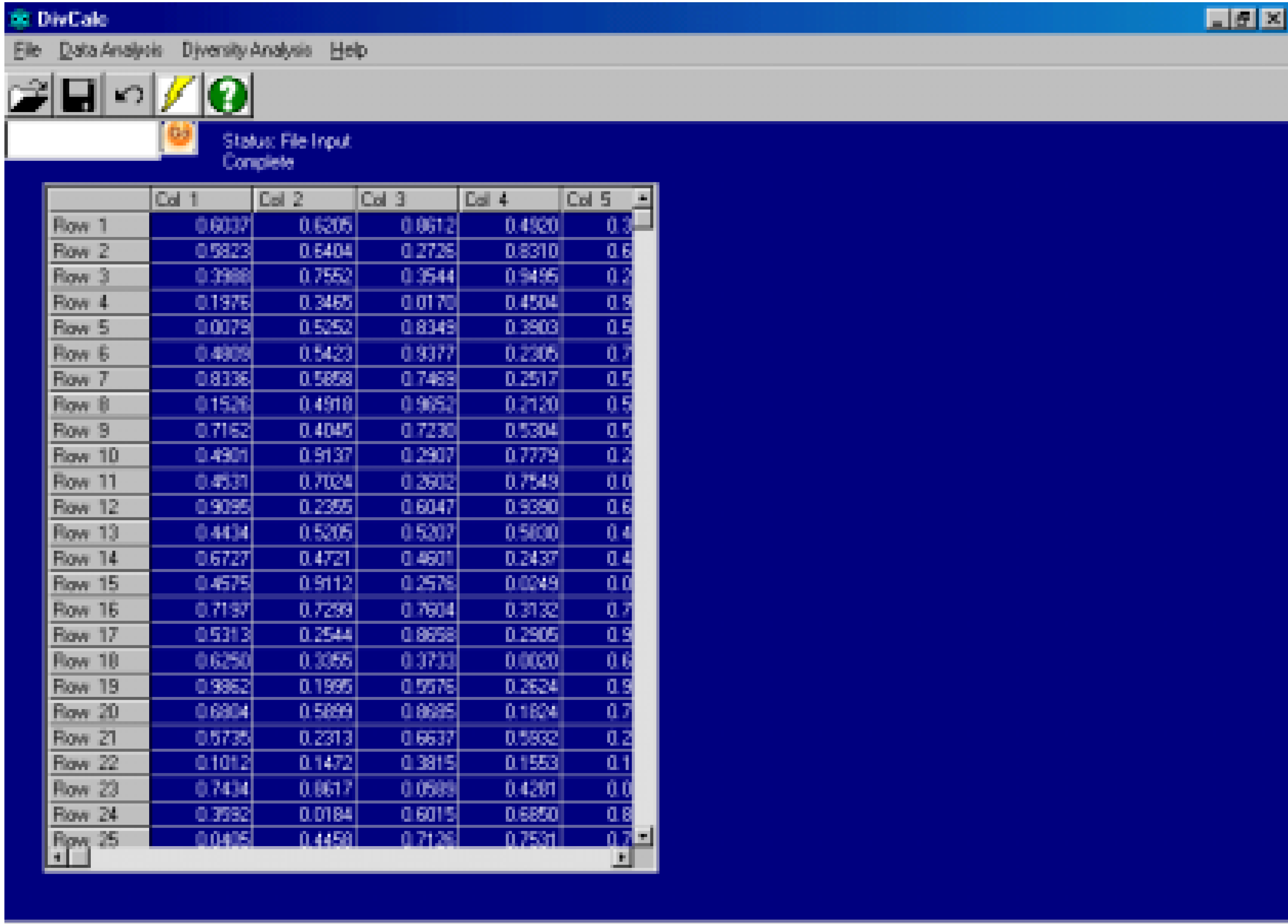
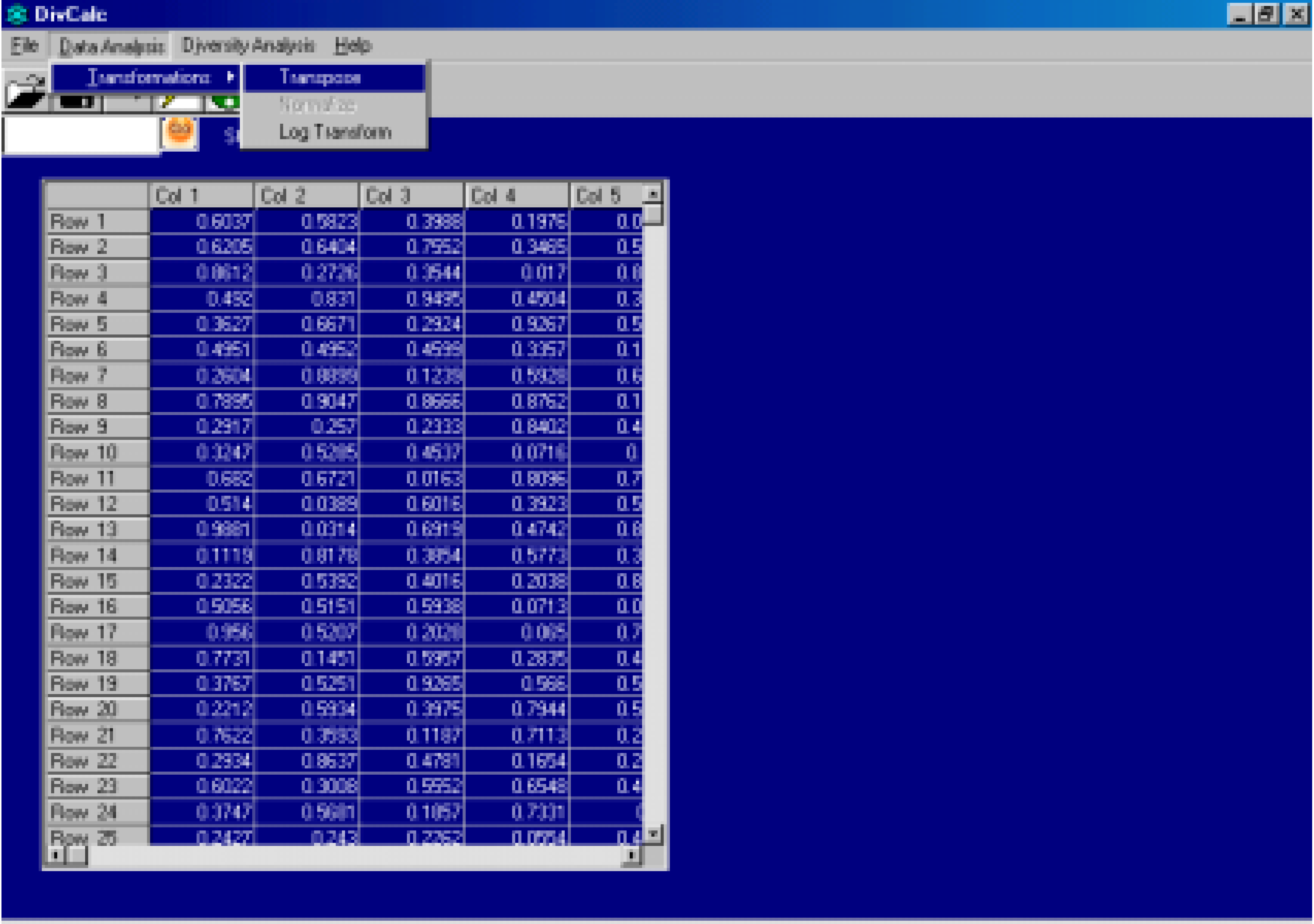
Features And Implementation
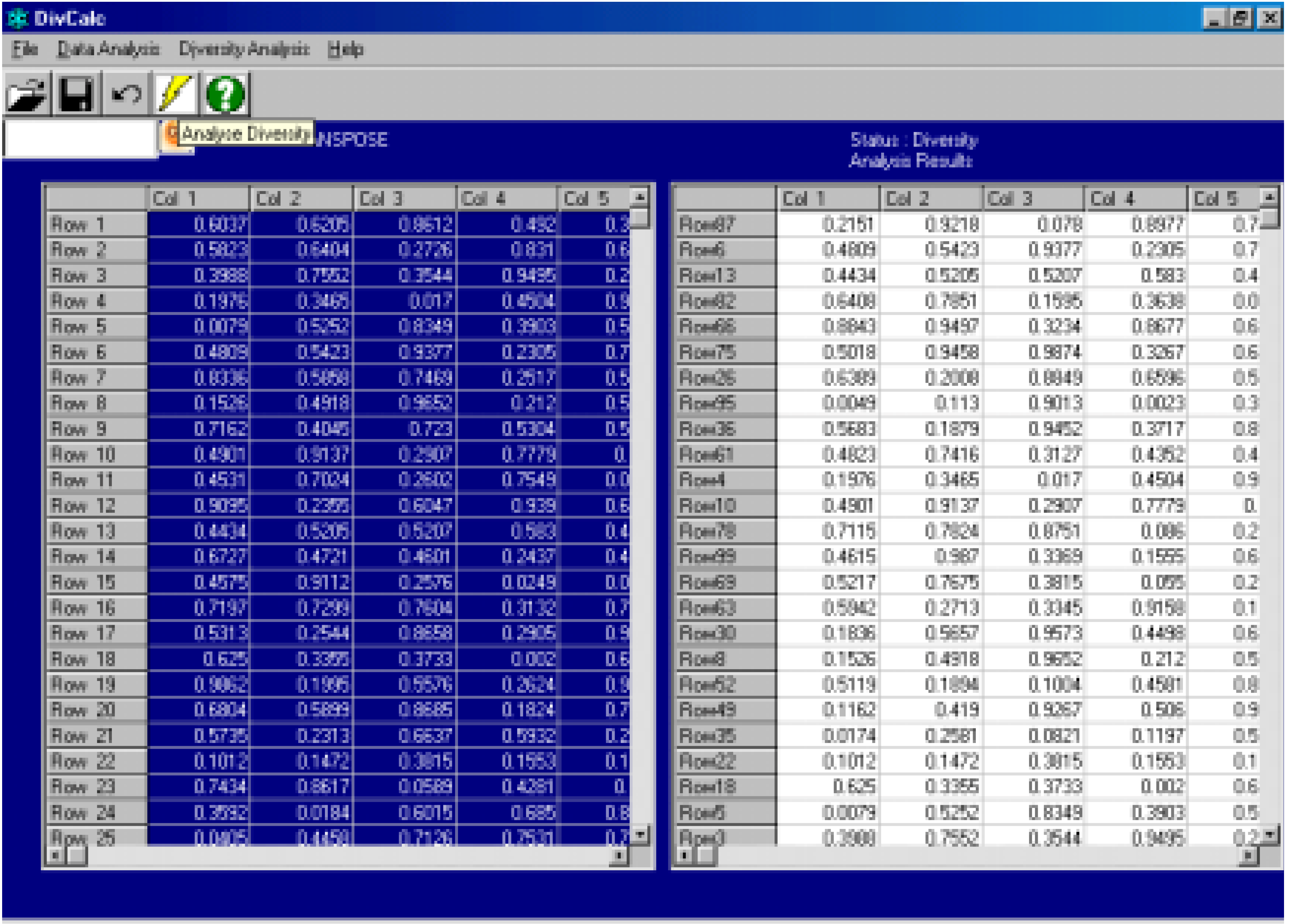
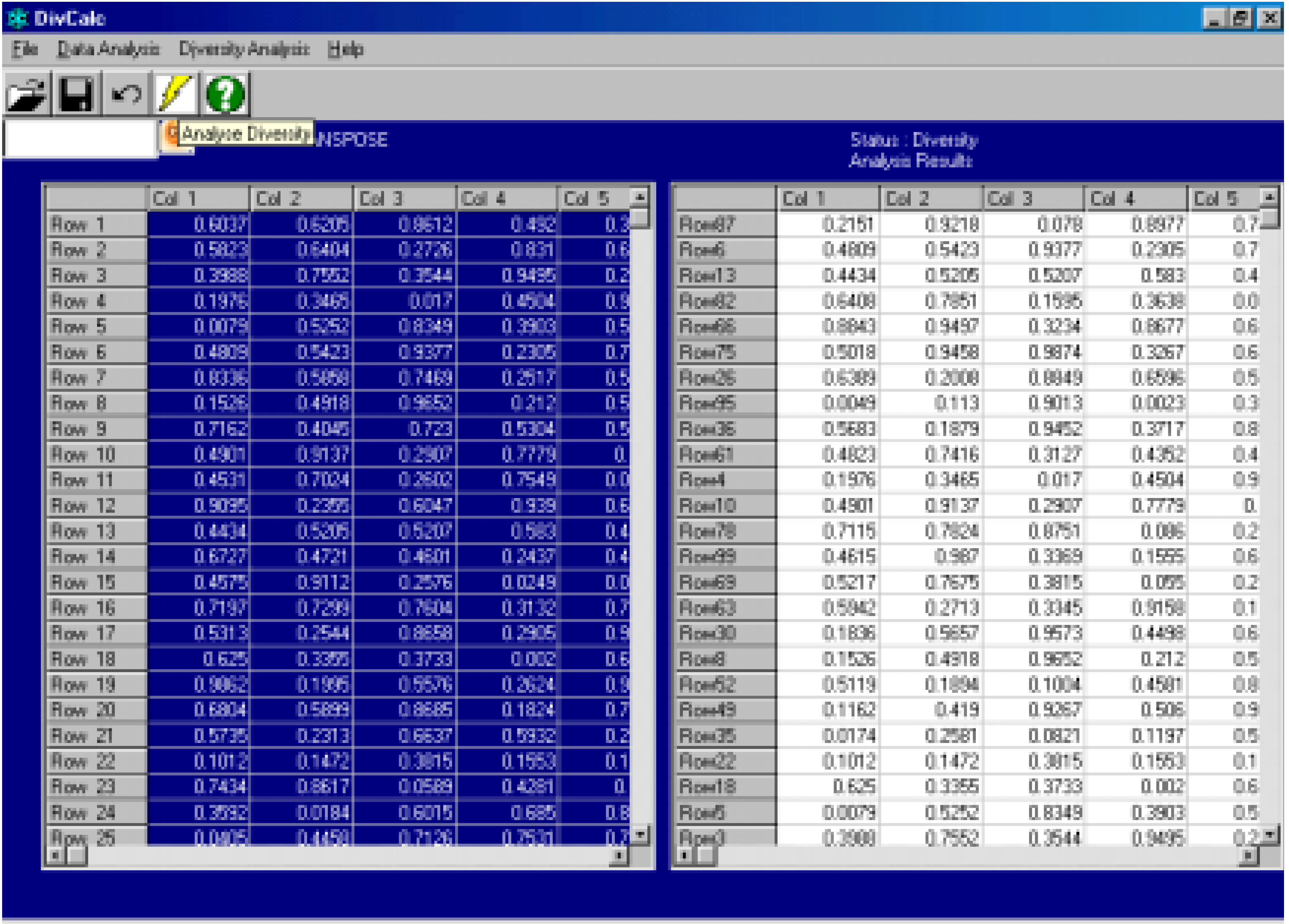
Algorithm
- The centroid of the input data is calculated.
- The compound most distant from the centroid is the first selected compound. Thus the compound with the maximum Euclidean distance from the centroid is the first selected compound.
- The compound most distant from the 1st compound is selected next.
- Hereafter the algorithm increases in complexity. At each iteration the procedure is to find the compound from the list of unselected compounds whose minimum distance to selected compounds is a maximum amongst all unselected compounds.
- Step 4 is repeated till the required number of compounds are selected.
Discussion
References
- Flower, D. DISSIM: A program for the analysis of chemical diversity. J. Mol. Graphics Mod. 1998, 16, 239–253. [Google Scholar] [CrossRef]
- Van Drie, J. H.; Lajiness, M. S. Approaches to virtual library design. DDT 1998, 3, 274–283. [Google Scholar]
- Linusson, A.; Gottfries, J.; Lindgren, F.; Wold, S. Statistical Molecular Design of Building Blocks for Combinatorial Chemistry. J. Med. Chem. 2000, 43, 1320–1328. [Google Scholar] [CrossRef]
- Leach, A. R.; Hann, M. M. The in silico world of virtual libraries. DDT 2000, 5, 326–336. [Google Scholar] [CrossRef]
- Gorse, D.; Rees, A.; Kaczorek, M; Lahana, R. Molecular Diversity and its analysis. DDT 1999, 4, 257–385. [Google Scholar] [CrossRef]
- Todeschini, R; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim (Germany), 2000; vol. 11; p. 667. [Google Scholar]
- Sample Availability: Not applicable
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Gangal, R. DivCalc: A Utility for Diversity Analysis and Compound Sampling. Molecules 2002, 7, 657-661. https://doi.org/10.3390/70800657
Gangal R. DivCalc: A Utility for Diversity Analysis and Compound Sampling. Molecules. 2002; 7(8):657-661. https://doi.org/10.3390/70800657
Chicago/Turabian StyleGangal, Rajeev. 2002. "DivCalc: A Utility for Diversity Analysis and Compound Sampling" Molecules 7, no. 8: 657-661. https://doi.org/10.3390/70800657
APA StyleGangal, R. (2002). DivCalc: A Utility for Diversity Analysis and Compound Sampling. Molecules, 7(8), 657-661. https://doi.org/10.3390/70800657