Niobium Pentachloride Catalysed Ring Opening of Epoxides

Abstract
:Introduction
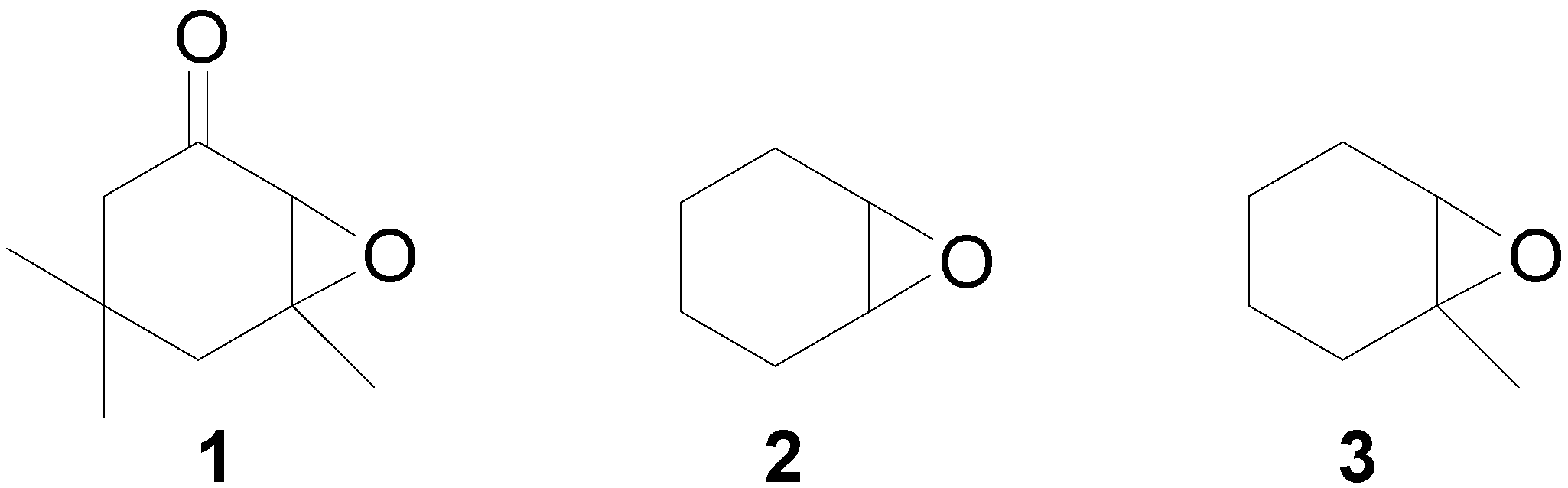
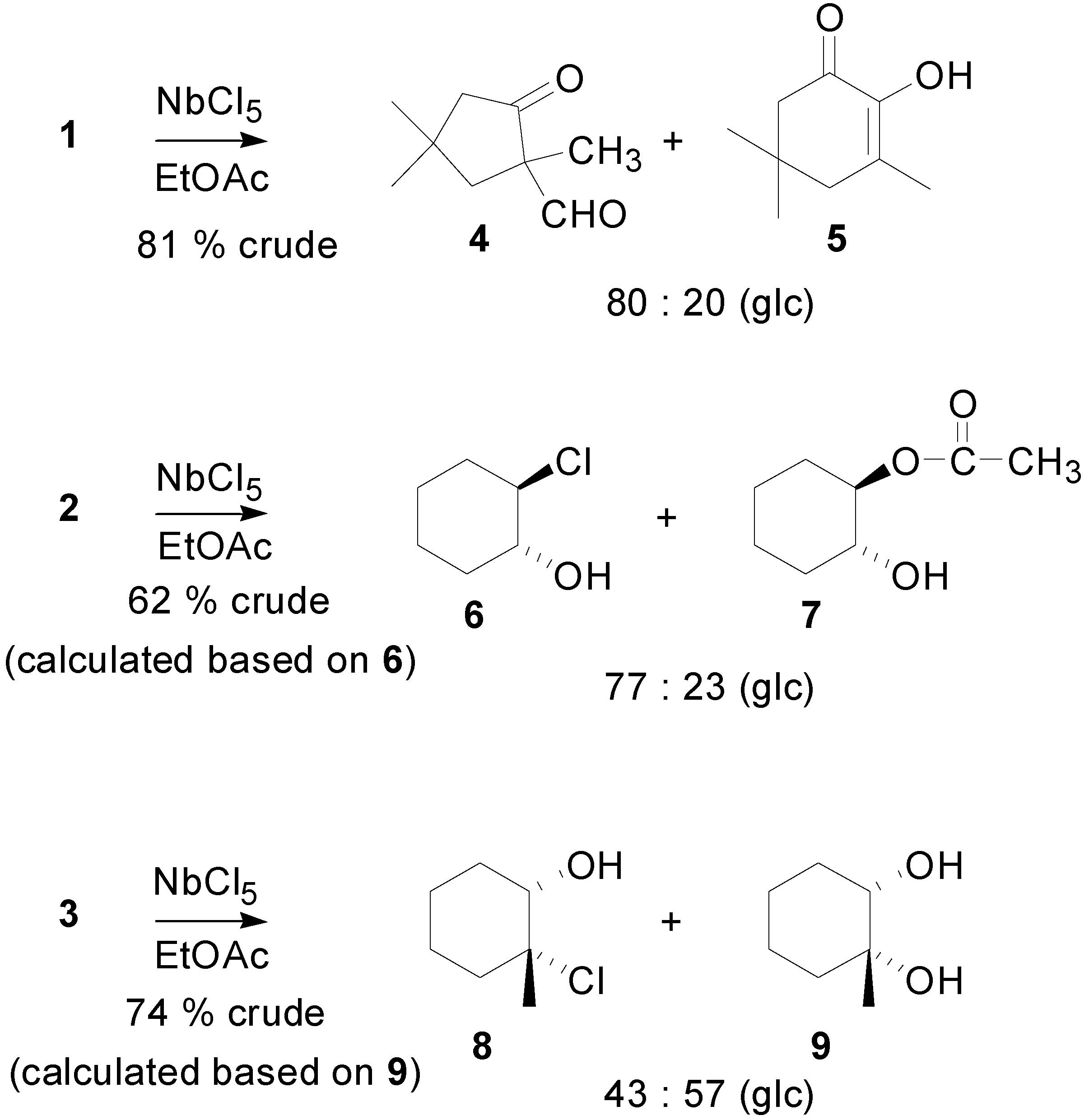
Results and Discussion
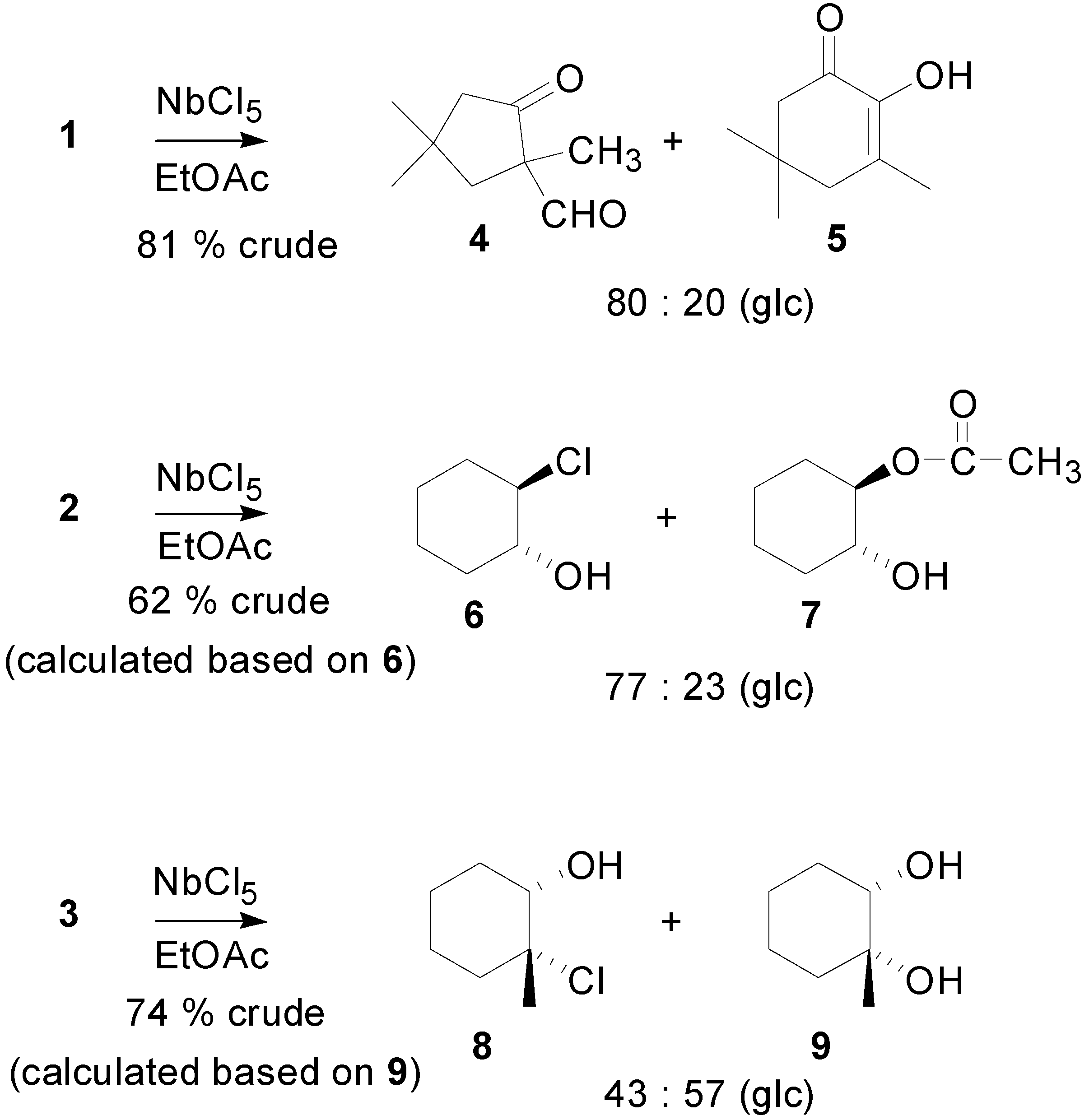
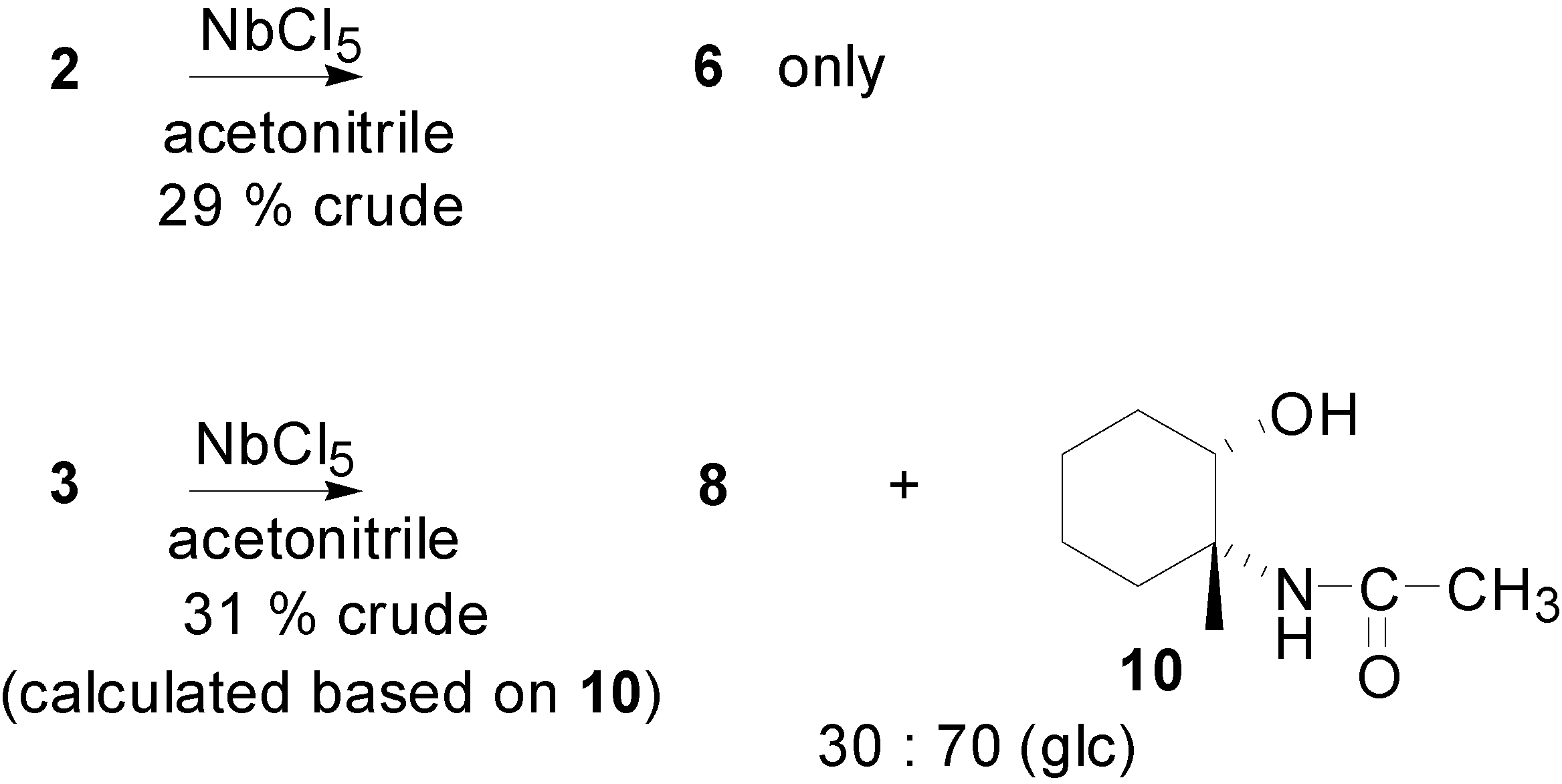

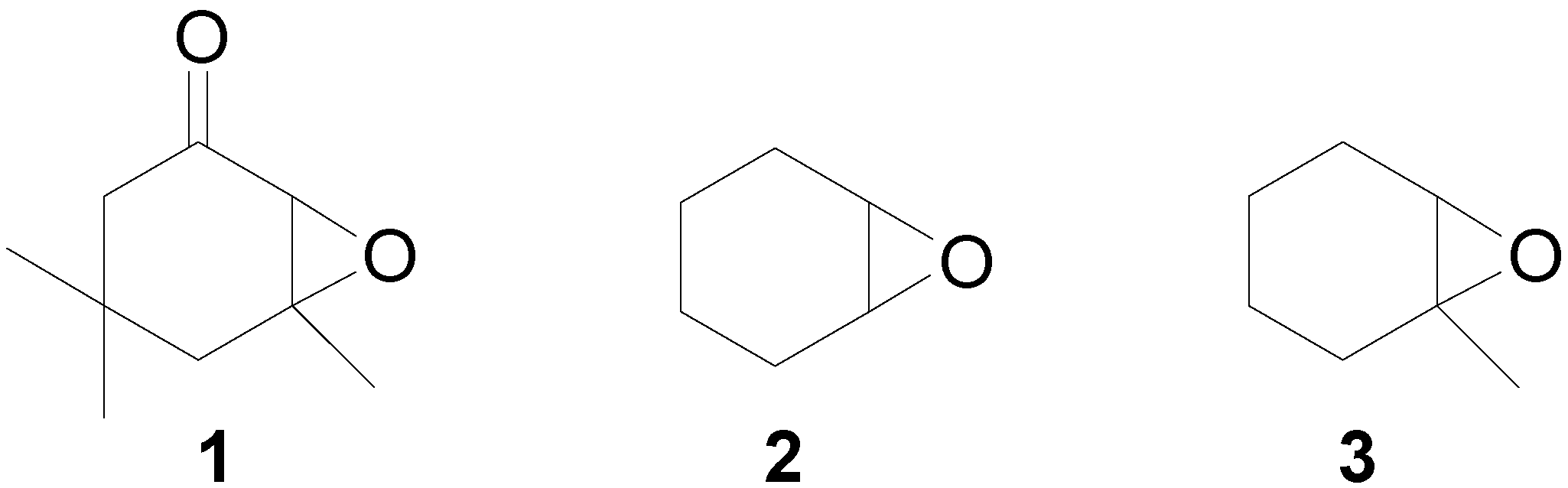{kind=link}
{kind=link}
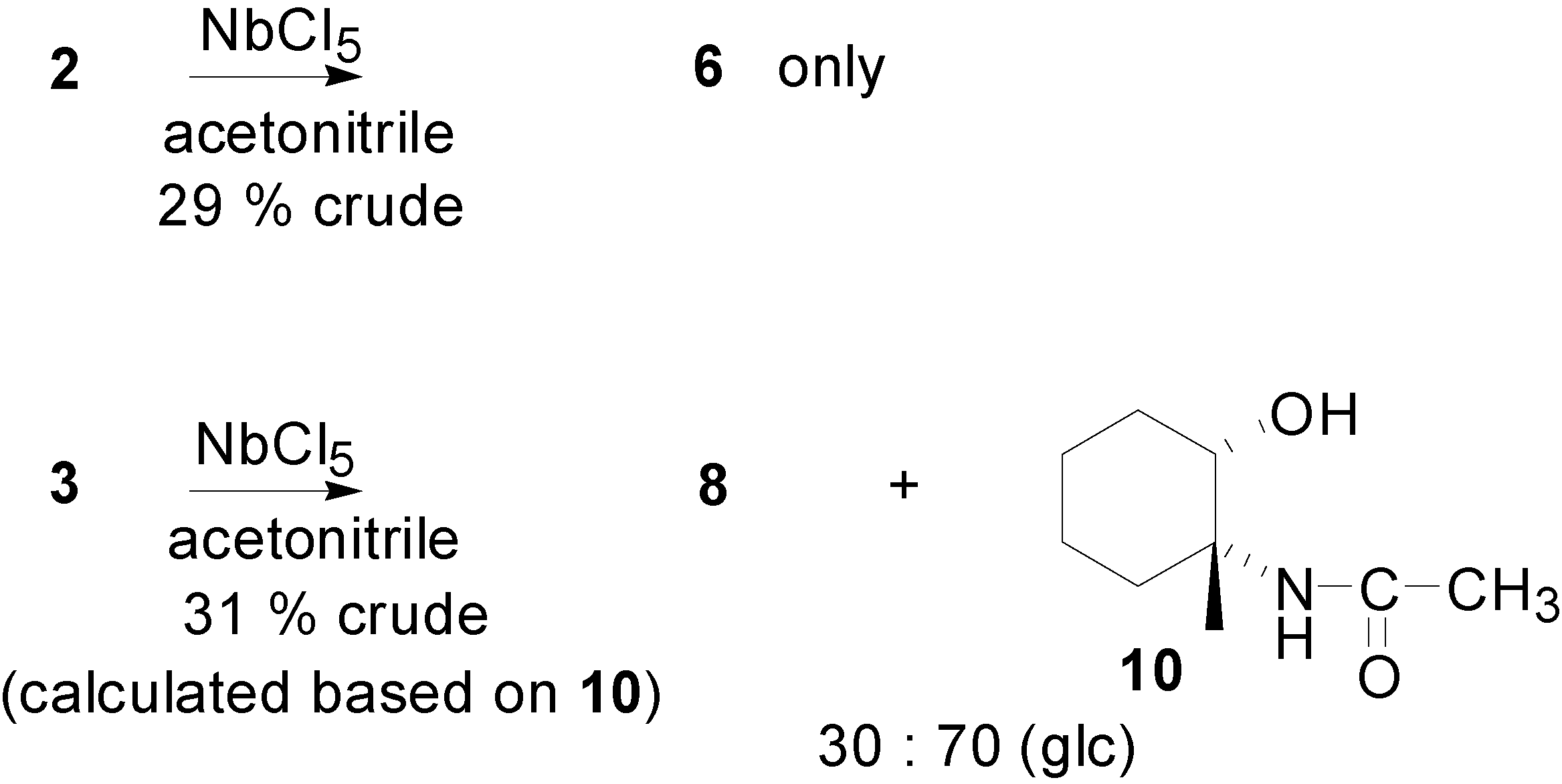{kind=link}
| Compound | Compound 8 | Compound 10 | ||
| Coupling constant | J1 (Hz) | J2 (Hz) | J1 (Hz) | J2 (Hz) |
| Calculated for trans isomer: | 2.7 | 4.3 | 3.0 | 6.4 |
| Calculated for cis isomer: | 10.6 | 4.4 | 11.0 | 4.4 |
| Experimental values: | 9.5 | 4.3 | 10.7 | 4.8 |
Experimental
General
Preparation of substrates
(±) – 4,4,6-Trimethyl-7-oxabicyclo[4.1.0]heptan-2-one (1).
7-Oxa-bicyclo[4.1.0]heptane (2) [8].
(±)-1-Methyl-7-oxa-bicyclo[4.1.0]heptane (3).
General Procedure for the Reactions of Epoxides with NbCl5.
Characterization of the products
Acknowledgments
References and Notes
- Constantino, M.G.; Losco, P.; Castellano, E.E. J. Org. Chem. 1989, 54, 681–683. Constantino, M.G.; Prado, M.C. Química Nova 1991, 14, 22–25. Losco, P.; Ferraz, H.M.C.; Constantino, M.G. Química Nova 1992, 15, 18–20. Constantino, M.G.; Donate, P.M.; Frederico, D.; Carvalho, T.V.; Cardoso, L.E. Synth. Commun. 2000, 30, 3327–3340.
- Howarth, J.; Gillespie, K. Tetrahedron Lett. 1996, 37, 6011–6012. Furstner, A.; Hupperts, A.; Ptock, A.; Janssen, E. J. Org. Chem. 1994, 59, 5215–5229. Sato, M.; Oshima, K. Chem. Lett. 1982, 157–160. Roskamp, E.J.; Pedersen, S.F. J. Am. Chem. Soc. 1987, 109, 6551–6553. Hartung, J.B.; Pedersen, S.F. J. Am. Chem. Soc. 1989, 111, 5468–5469. Maeta, H.; Nagasawa, T.; Handa, Y.; Takei, T. Tetrahedron Lett. 1995, 36, 899–902. Howarth, J.; Gillespie, K. Molecules 2000, 5, 993–997. Stohrer, I.; Hoffmann, H.M.R. Tetrahedron 1992, 48, 6021–6032. Segi, M.; Nakajima, T.; Suga, S. Bull. Chem. Soc. Jpn. 1980, 53, 1465–1466.
- In tests carried out with each of the three substrates, TLCs of reaction mixtures quenched after 1.0 min, 2.0 min and 5.0 min were identical
- The formation of 5 from 1 could also be explained by a hydrogen migration. In this case, however, we should expect the formation of a corresponding compound from 3.
- Garret, C. E.; Fu, G. C. J. Org. Chem. 1997, 62, 4534–4535. Sarangi, C.; Das, N. B.; Nanda, B.; Nayak, A.; Sharma, R. P. J. Chem. Research (S) 1997, 18. Archer, I. V. J.; Leak, D. J.; Widdowson, D. A. Tetrahedron Lett. 1996, 37, 8819–8822.
- PCMODEL, version 7.0, Serena Software, P. O. Box 3076, Bloomington, IN 474-23076; GMMX, version 1.5, Serena Software, P. O. Box 3076, Bloomington, IN 474-23076.
- Wasson, R.L.; House, H.O. Organic Synthesis; Wiley: New York, 1963; Collet. Vol. IV, pp. 552–553. [Google Scholar] Constantino, M.G.; Matias, L.G.O.; da Silva, G.V.J.; Heleno, V.C.G.; Gambardella, M.T.P. Synth. Commun. 1997, 27, 4285–4295.
- This compound is commercially available. See also the literature NMR data in The Aldrich in Pouchert, C.J. Library of NMR Spectra, 2nd ed.; Aldrich Chemical Co.: Wisconsin, 1983; Vol. I, p. 196. [Google Scholar]
- Sample availability: Samples of compounds 4-10 are available from MDPI. Additional samples of compound 4 are available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Constantino, M.G.; Lacerda, V., Jr.; Aragão, V. Niobium Pentachloride Catalysed Ring Opening of Epoxides. Molecules 2001, 6, 770-776. https://doi.org/10.3390/60900770
Constantino MG, Lacerda V Jr., Aragão V. Niobium Pentachloride Catalysed Ring Opening of Epoxides. Molecules. 2001; 6(9):770-776. https://doi.org/10.3390/60900770
Chicago/Turabian StyleConstantino, Mauricio Gomes, Valdemar Lacerda, Jr., and Valquiria Aragão. 2001. "Niobium Pentachloride Catalysed Ring Opening of Epoxides" Molecules 6, no. 9: 770-776. https://doi.org/10.3390/60900770
APA StyleConstantino, M. G., Lacerda, V., Jr., & Aragão, V. (2001). Niobium Pentachloride Catalysed Ring Opening of Epoxides. Molecules, 6(9), 770-776. https://doi.org/10.3390/60900770