General
Melting points were taken on a STUART apparatus and are uncorrected. The IR spectra (KBr disks) were measured with a Jasco FT/IR 5300 spectrometer. The 1H-NMR spectra were obtained on a Varian Gemini 200 instrument at 200 MHz using DMSO-d6 as a solvent and TMS as internal standard. Mass spectra were performed by Shimadzu-GC MS QP 1000 EX using the direct inlet system. Thin layer chromatography (TLC) was carried out on silica gel (MN-Kieselgel G., 0.2 mm thickness) with ethylacetate:n-hexane (E/H) as the solvent and the plates were scanned under 254 nm ultraviolet light. Microanalyses were performed by the Microanalytical Unit at Cairo University. All the compounds gave satisfactory elemental analyses. Antimicrobial and antifungal activity tests were carried out by the Microbiology Lab., Faculty of Science, Al-Azhar University, Cairo, Egypt.
General procedure for synthesis of tosyl aminoacids 1a-f.- The tosyl amino acid derivatives were prepared according to McChesney et al [
12] whereby the aminoacid (0.026 mol) was dissolved in 1N sodium hydroxide (25 mL) and over a period of thirteen minutes a solution of p-toluenesulphonyl chloride (0.027 mol) in ether (30 mL) was added in portions. The mixture was stirred at room temperature for 3hrs. The excess p-toluenesulphonyl chloride was filtered off and the solution treated with 2N HCl until acidic to Congo Red (pH 5). After cooling acidification caused the product to precipitate. The crude product was filtered, washed with water and dried. The crude materials were recrystallized to give
1a-f.
General procedure for synthesis of the amides 2a-f.- To a mixture of tosyl aminoacid (0.01 mol) and methyl anthranilate (0.01 mol) in xylene (20 mL), phosphorous trichloride (2-3 mL) was added. The mixture was heated under reflux for 3-4 hrs. After cooling and addition of pet.ether (bp 40-60°C; 10 mL) the precipitate was filtered and washed with two 5 mL portions of pet.ether, dried and recrystallized from the appropriate solvent to give 2a-f.
Methyl N-[phenylsulphonyl-glycyl]anthranilate (2a).- From phenylglycyl chloride as colourless crystals (65%), m.p. 125°C; IR νmax/cm-1 (2NH), 1703 (ester CO), 1680 (CO).
Methyl N-[4'-methylphenylsulphonyl-2-phenylglycyl]anthranilate (2b).- From tosyl 2-phenylglycyl acid chloride as colourless crystals (71%), m.p. 155°C; IR νmax/cm-1 3190 (2NH), 1700 (ester CO), 1681 (CO).
Methyl N-[4'-methylphenylsulphonyl-phenylalaninyl]anthranilate (2c).- From tosyl phenylalaninyl acid chloride as colourless crystals (68%), m.p. 120°C; IR νmax/cm-1 3195 (2NH), 1715 (ester CO), 1662 (CO).
Methyl N-[4'-methylphenylsulphonyl-β-alaninyl]anthranilate (2d).- From tosyl-β-alaninyl acid chloride as colourless crystals (73%), m.p. 130°C; IR νmax/cm-1 3190 (2NH), 1695 (ester CO), 1660 (CO).
Methyl N-[4'-methylphenylsulphonyl-DL-valinyl]anthranilate (2e).- From tosyl-DL-valinyl acid chloride as colourless crystals (75%), m.p. 170°C; IR νmax/cm-1 3220 (2NH), 1721 (ester CO), 1676 (CO); 1H-NMR δ 0.85-0.87 (d, 3H, isopropyl CH3), 0.90-0.93 (d, 3H, isopropyl CH3), 2.02-2.13 (m, 1H, CH), 2.37 (d, 1H, CH), 2.45 (s, 3H, CH3), 3.98 (s, 3H, OCH3), 7.26-8.55 (m, 8H, ArH’s), 11.12 ppm (s, 2H, 2NH).
Methyl N-[4'-methylphenylsulphonyl-DL-leucinyl]anthranilate (2f).- From tosyl-DL-leucine acid chloride as colourless crystals (68%), m.p. 165°C; IR νmax/cm-1 3180 (2NH), 1698 (ester CO), 1663 (CO).
General procedure for synthesis of N-amino quinazolin-4(3H)-one derivatives (3a-f).- The corresponding amide 2a-f (0.01 mol) and 95% hydrazine hydrate (0.05 mol) were dissolved in nbutanol (30 mL) and refluxed for 6-8 hrs. Cooling in ice gave the crude product which was filtered off and recrystallized to give compounds 3a-f.
3-Amino-2-(phenylsulphonamidomethyl)quinazolin-4(3H)-one (3a).- Colourless crystals (74%); m.p. 180°C; IR νmax/cm-1 3320 (NH2), 3210 (NH), 3060 (CH-arom.), 2960 (CH-aliph.), 1660 (CO).
3-Amino-2-(1`(4-methylphenylsulphonyl)-1`-(phenyl)methyl]quinazolin-4(3H)-one (3b).- Colourless crystals (65%), m.p. 135°C; IR νmax/cm-1 3325 (NH2), 3298 (NH), 3060 (CH-arom.), 2910 (CH-aliph.), 1669 (CO), 1598 (C=N).
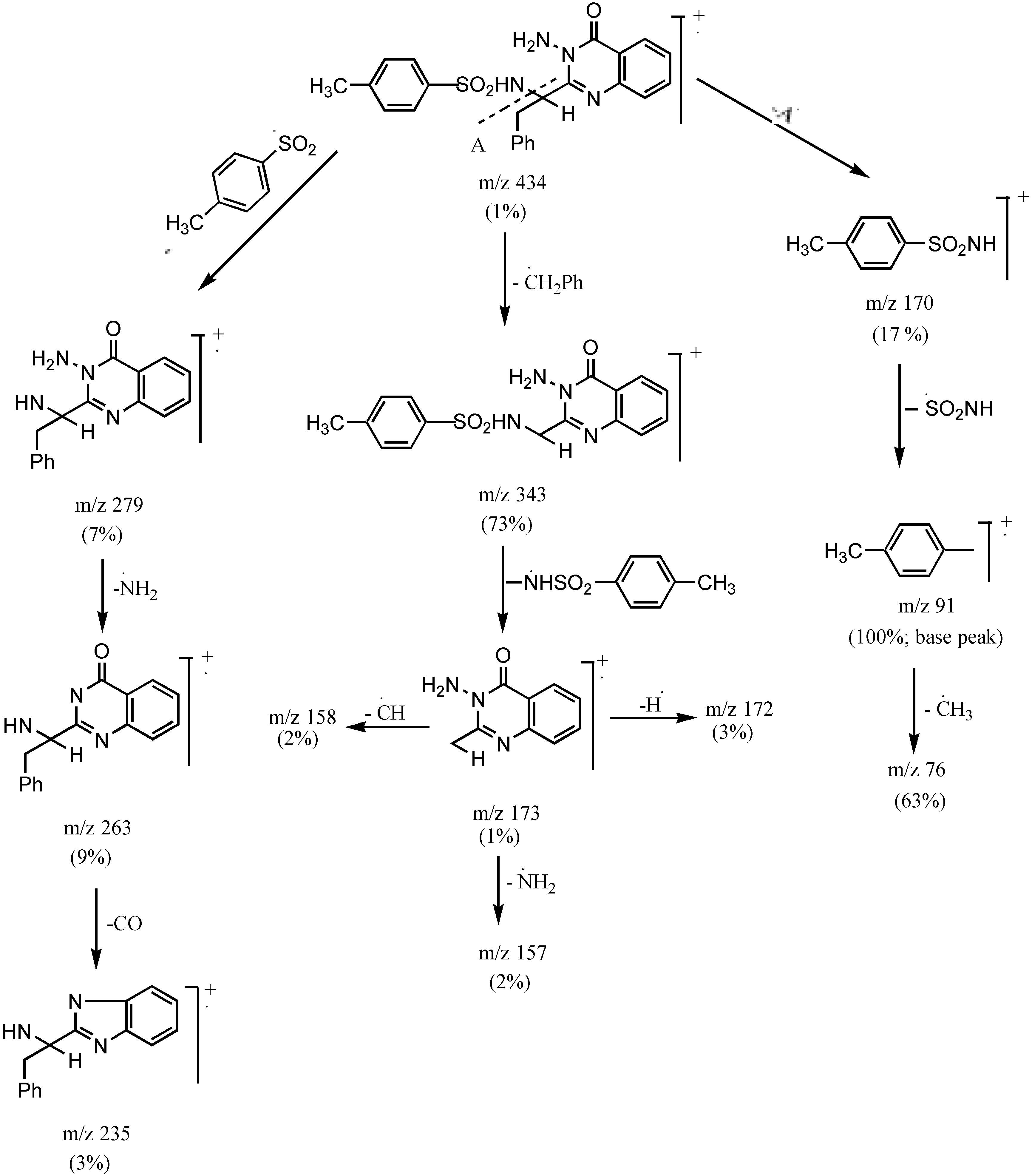
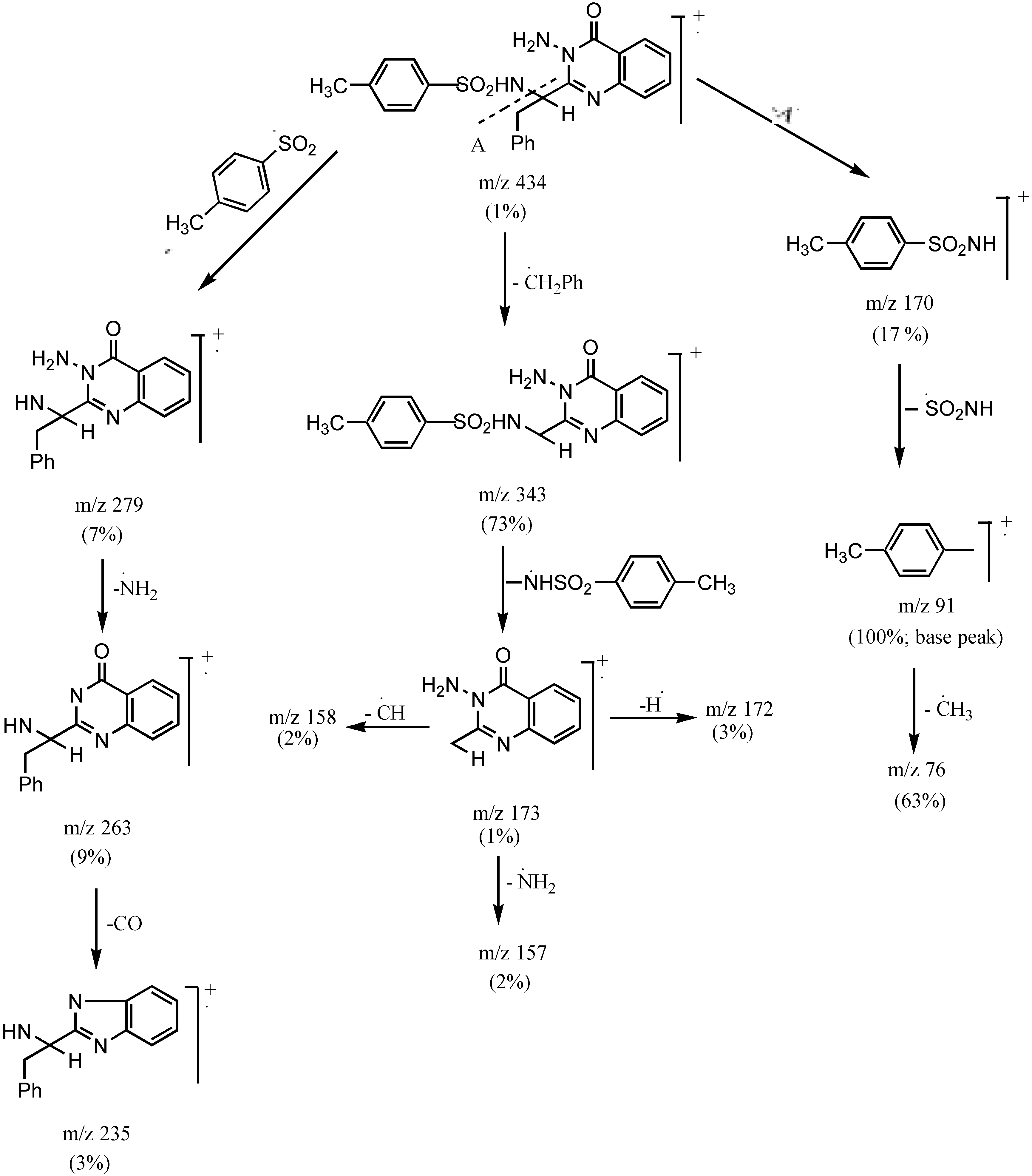
3-Amino 2-[1`(4-methyl phenylsulphonamido)-1`-(benzyl)methyl]quinazo-line-4-(3H)-one (3c).- Colourless crystals (68%), m.p. 170°C; IR νmax/cm-1 3316 (NH2), 3241 (NH), 3055 (CH-arom.), 2972 (CH-aliph.), 1667 (CO); MS (C23H22N4O3S): m/e 434 (M+, 1%), 343 (73.3%), 328 (10.6%), 235 (2.7%), 188 (4.6%), 172 (2.7%), 145 (4.2%), 91 (100%, base peak), 65 (22.6%).
3-Amino-2-[4-methyl phenylsulphonamidoethyl]quinazolin-4-(3H)-one (3d).- Colourless crystals (70%), m.p. 185°C, IR νmax/cm-1 3322, 3294 (NH2), 3260 (NH), 1666 (CO.), 1614 (C=N).
3-Amino-2-[1`-(4-methylphenylsulphonamido)-1`-(iso-propyl)methyl]quinazolin-4-(3H)-one (3e).- Colourless crystals (76%), m.p. 190°C; IR νmax/cm-1 3320, 3290 (NH2), 3250 (NH), 1665 (CO), 1611 (C=N); MS (C19H22N4O3S): m/e 387(M+1, 4.5%), 353 (5.6%), 343 (5%), 275 (11%), 226(39.6%), 185 (15.3%), 155 (51.6%), 91 (100%, base peak), 65 (32%); 1H-NMR δ 0.85-0.92 (d, 6H, 2CH3), 1.88 (s, 3H, CH3), 2.15-2.24 (m, 1H, CH), 2.50-2.51 (d, 1H, CH +DMSO), 5.51 (s, 2H, NH2; exchangeable with D2O), 6.82-8.02 (m, 8H, Ar-H’s), 10.6 (s, 1H, NH; exchangeable with D2O).
3-Amino 2-[1`-(4-methylphenylsulphonamido)-1`-(iso-butyl)methyl]quina-zolin-4-(3H)-one (3f).- Colourless crystals (71%), m.p. 170°C; 1H-NMR δ 0.88-0.94 (d, 6H, 2CH3), 1.52-1.59 (t, 2H, CH2), 1.82-1.90 (m, 1H, CH), 2.50-2.52 (d, 1H, CH +DMSO), 5.57 (s, 2H, NH2; exchangeable with D2O), 6.86-8.04 (m, 8H, Ar-H’s), 10.2 (s, 1H, NH; exchangeable with D2O).
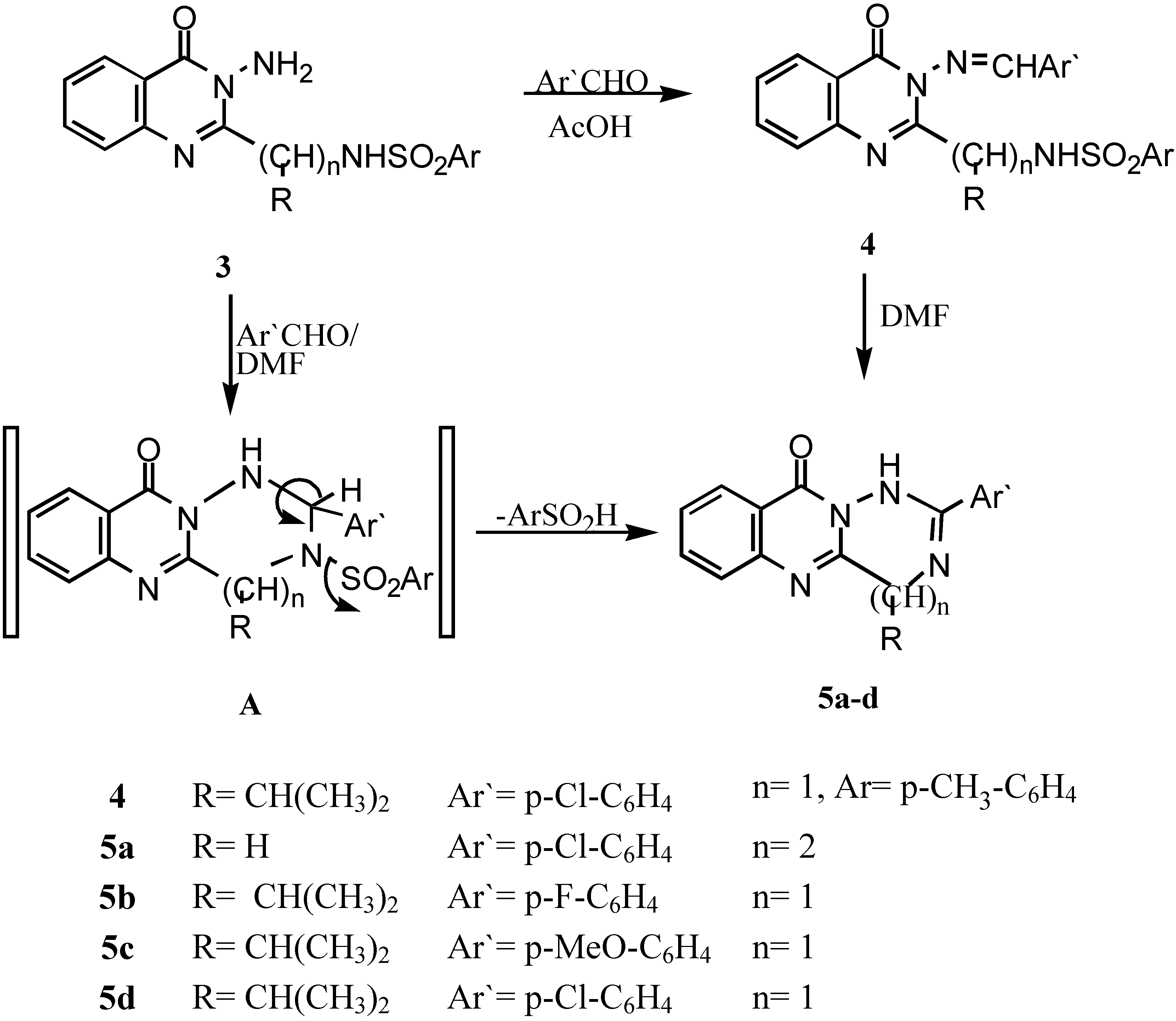
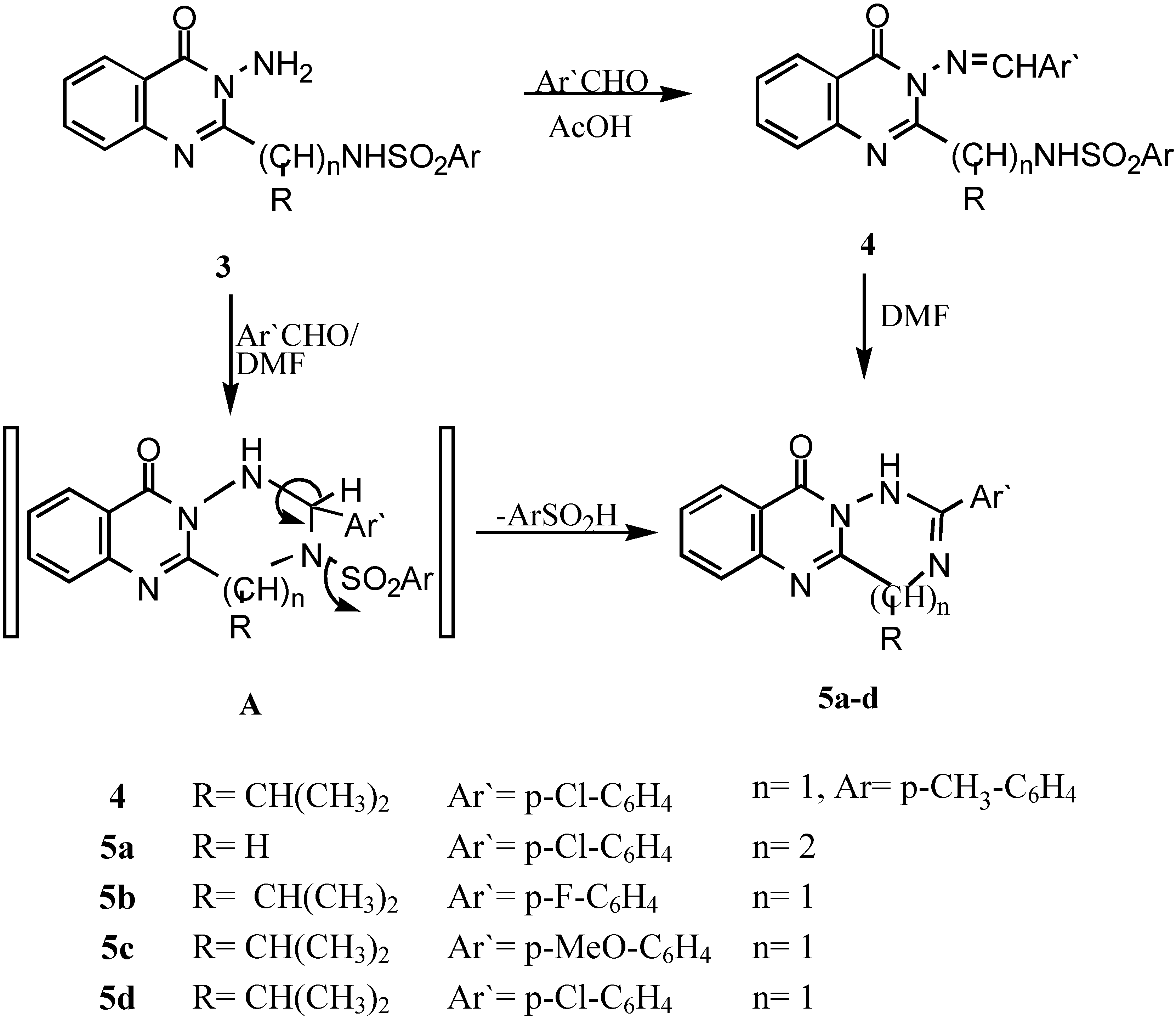
The Schiff’s base of 3-amino-2-[1`-(4-methyl phenyl sulphonamido)-1`-(iso-propyl)methyl]-quinazolin-4-(3H)-one (4).- Prepared from 3e (0.01 mol) and 4-chlorobenzaldehyde (0.01 mol) in acetic acid (20 mL). The mixture was refluxed for 4 hrs., cooled and colected by filteration as pale brown crystals (69%); m.p. 160°C; IR νmax/cm-1 3220 (NH), 3050 (CH-arom.), 2985 (CH-aliph.), 1660 (CO).
General procedure for the synthesis of 5a-c.- Compounds 3d,e (0.01 mol) and the corresponding aldehyde derivative (0.01 mol) in DMF (20 mL) containing 2 drops of triethylamine was refluxed for 6-8 hrs., after cooling and acidification with dil. HCl, a precipitate was formed, which was collected by filtration, washed with water and recrystallized to give 5a-c.
2-(4-Chlorophenyl)-4,5,11-trihydro-1H[1,2,4]triazepino[7,1-b]quinazolin-11-one (5a).- Prepared from 3d and 4-chlorobenzaldehyde as brown crystals (67%), m.p. 260°C; IR νmax/cm-1 3210 (NH), 1664 (CO), 1590 (C=N); 1H-NMR δ 4.23, 4.6 (2s, 4H, 2CH2), 7.2-8.4 (m, 8H, Ar-H’s), 9.2 (s, 1H, NH; exchangeable with D2O).
2-(4-Fluorophenyl)-4-isopropyl-4,10-dihydro-1H[1,2,4]triazino[6,1-b]-quinazolin-10-one (5b).- Prepared from 3e and 4-fluorobenzaldehyde as pale brown crystals (62%), m.p. 180°C; IR νmax/cm-1 3195 (NH), 2995, 2890 (CH-aliph), 1658 (CO).
2-(4-Methoxyphenyl-4-isopropyl-4,10-dihydro-1H-[1,2,4]triazino[6,1-b] quinazolin-10-one (5c).- Prepared from 3e and 4-methoxybenzaldehyde as brown crystals (60%), m.p. 170°C; IR νmax/cm-1 3210 (NH), 2990, 2885 (CH-aliph.), 1665 (CO).
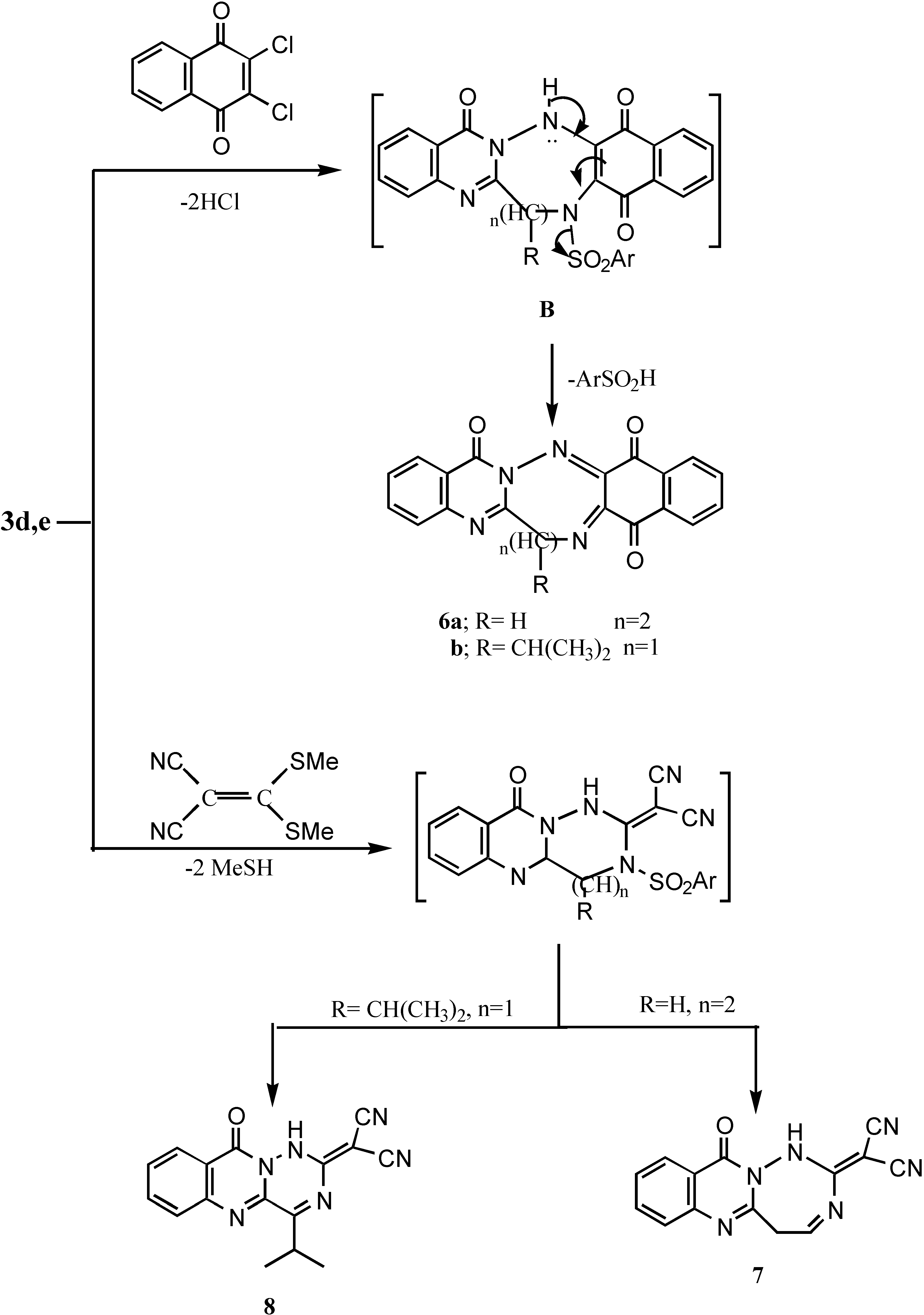
6,7,9,14,17-Pentahydronaphtho[2`,3`:3,4][1,2,5]triazocino[8,1-b]qinozolin-9,14,17-trione (6a).- Prepared in a similar fashion as 6a from 3d and 2,3-dichloro-1,4-naphthoquinone as deep brown crystals (63%), m.p. > 300°C; IR νmax/cm-1 3060 (CH-arom.), 1670, 1660 (3CO).
6-Isopropyl-6,8,13,16-tetrahydronaphtho[2`,3`:3,4][1,2,5]triazepino[7,1-b]-quinazolin-8,13,16 trione (6b).- Prepared from 3e (0.01 mol); 2,3-dichloro-1,4-naphthoquinone (0.01 mol) in DMF (20 mL) and TEA (0.05 mL), the reaction mixture was refluxed for 6 hrs., cooled and acidified with HCl, the precipitate was filtered, washed with water and recrystallized to give 6a as dark brown crystals (60%); m.p. > 300°C; MS (C22H16N4O3) m/e 384 (5.9%), 328 (8.1%), 278 (12.5%), 224 (14.8%), 183 (21.7%), 137 (24.3%)), 97 (100%), 1H-NMR δ 0.87-0.89 (dd, 3H, isopropyl CH3), 0.91-0.94 (dd, 3H, isopropyl CH3), 2.04-2.15 (m, 1H, isopropyl CH), 7.02-8.34 (8H,m,Ar-H’s).
2-[11-Oxo5,11-dihydro-1H-[1,2,4]triazepino[7,1-b]quinazolin-2-ylidine]-malononitrile (7).- From 3d and [bis(methylthio)methylene]malononitrile as yellow crystals (61%); m.p. 250°C; MS (C14H8N6O) m/e 276 (3%), 229 (3.8%), 203 (26.3%), 186 (17.6%), 160 (77.4%), 91 (100%, base peak), 65 (46.8%), 1H-NMR δ 4.3 (d, 2H, CH2), 4.5 (dd, 1H, CH), 7.2-8.3(m, 4H, Ar-H’s + 1H, NH; exchangeable with D2O).
2[4-isopropyl-10-oxo-1,10-dihydro-[1,2,4]-triazino-[6,1-b]quinazolin-2-ylidine) malononitrile (8) Prepared from 3e (0.01 mol), [bis(methylthio)methylene)malononitrile (0.02 mol) in DMF (20 mL) and TEA (0.5 mL). The reaction mixture was refluxed for 5 hrs. until the odour of methyl mercaptan disappeared. Cooling and acidification caused the precipitation of the product which was then collected and recrystallized to give 7 as pale yellow crystals (66%), m.p. > 300°C; IR νmax/cm-1 3210 (NH), 2210 (2CN), 1659 (CO).
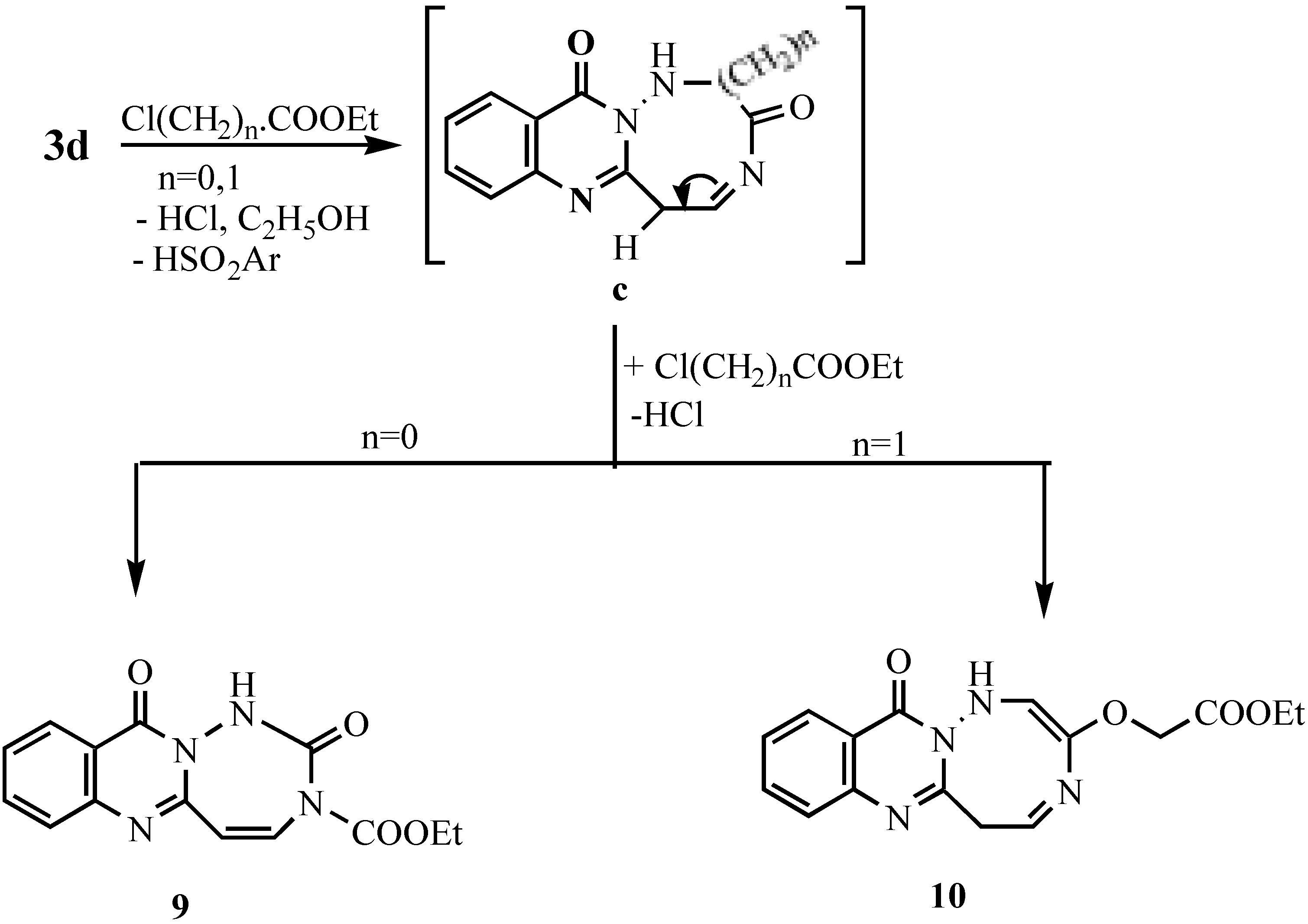
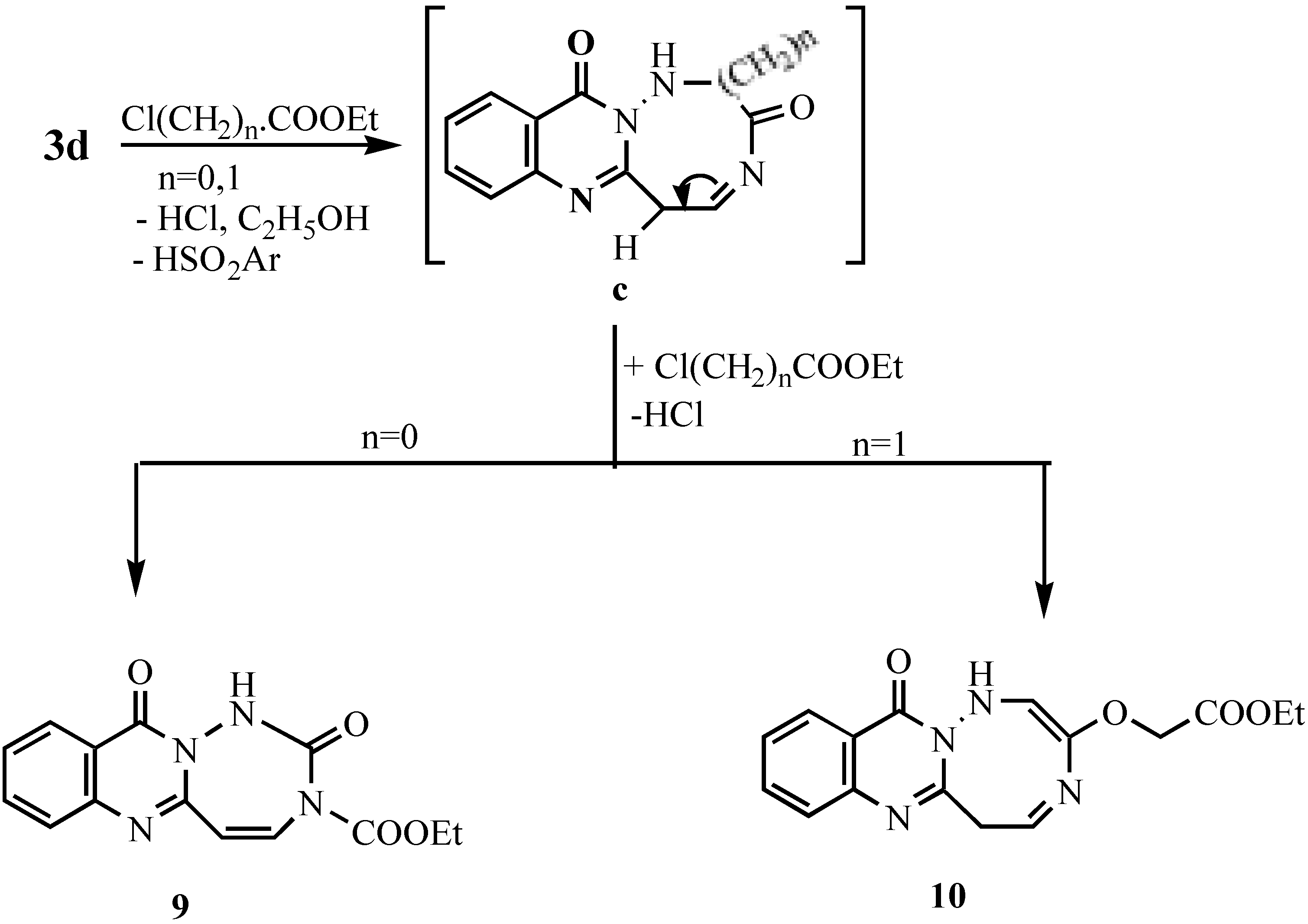
Ethyl 1-[11-oxo-5,11-dihydro-[1,2,4]triazepino[7,1-b]quinazolinyl]formate (9).- Prepared from 3d (0.01 mol), ethyl chloroformate (0.03 mol) and TEA (0.5 mL) in DMF (20 mL). The mixture was refluxed for 8 hrs., cooled and acidified with dil. HCl. The solid obtained was collected and crystallized to give 9 as pale brown crystals (60%), m.p. 265°C; MS (C14H12N4O4) m/e 300 (M+1, 0.5%), 284 (2.9%), 256 (7%), 213 (4.9%), 185 (6.2%), 149 (16.6%), 129 (10.8%), 91 (22.3%), 65 (8%); 1H-NMR δ 1.28-1.33 (t, 3H, CH3), 4.23-4.30 (q, 2H, CH2), 4.41, 4.5 (2s, 2H, 2CH), 7.29-8.29 (m, 4H, Ar-H’s + 1H, NH; exchangeable with D2O).
Ethyl-1-[12-oxo-6,12-dihydro[1,2,5]triazocino[8,1-b]quinazolinyl]acetate (10).- Prepared like 9 from 3d and ethyl chloroacetate as pale brown crystals (63%), m.p. > 300°C, MS (C16H16N4O4) m/e 328 (0.5%), 299 (0.7%), 284 (0.8%), 256 (7.3%), 213 (4.3%), 185 (5.2%), 149 (15.8%), 69 (100%; base peak); 1H-NMR δ 1.24-1.32 (t,3H, CH3 -ethyl), 1.80 (s, 2H, CH2), 4.09-4.15(q, 2H, CH2 ethyl), 7.2-8.3 (m, 4H, Ar-H’s + 2H, 2NH; exchangeable with D2O).

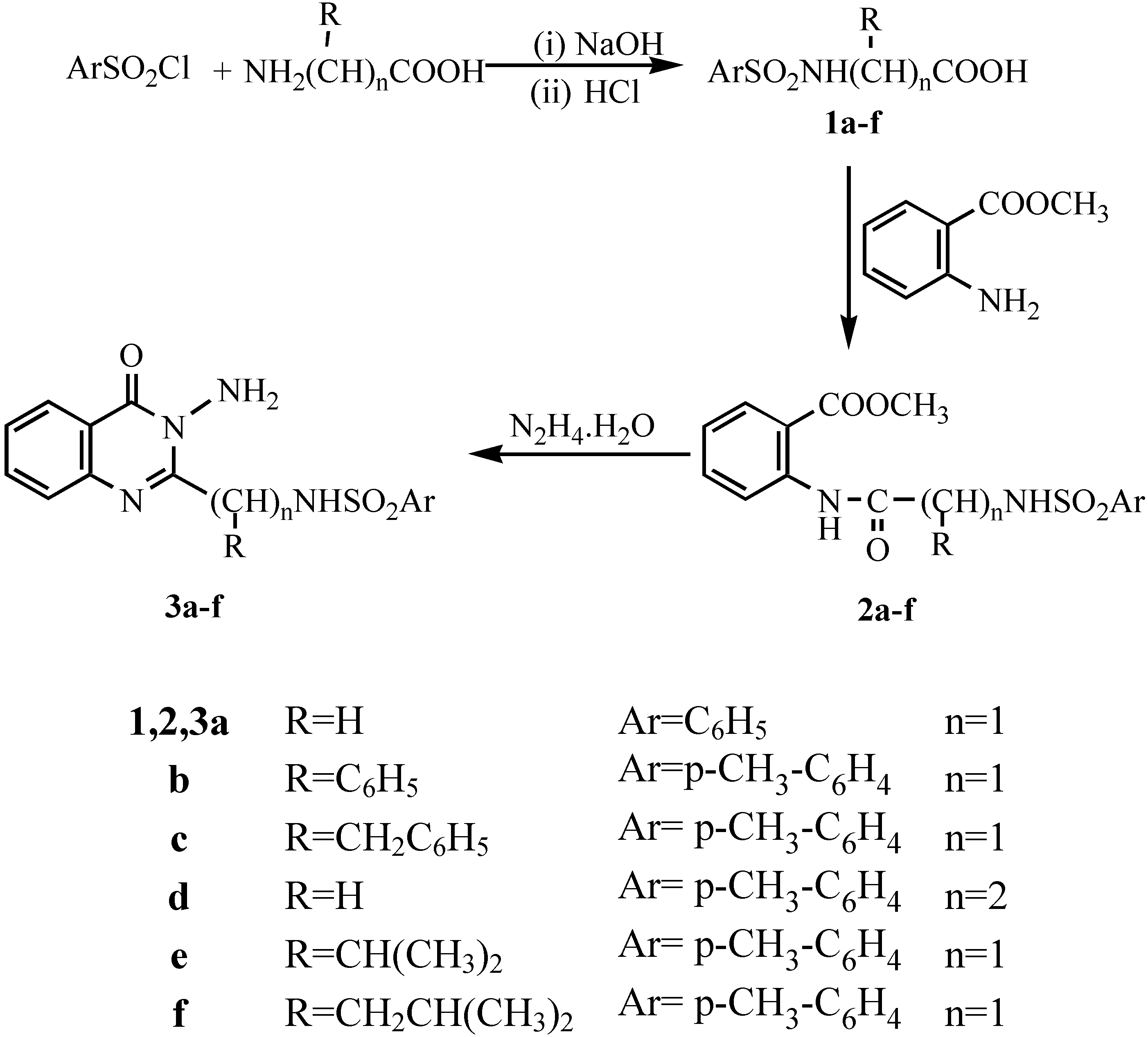

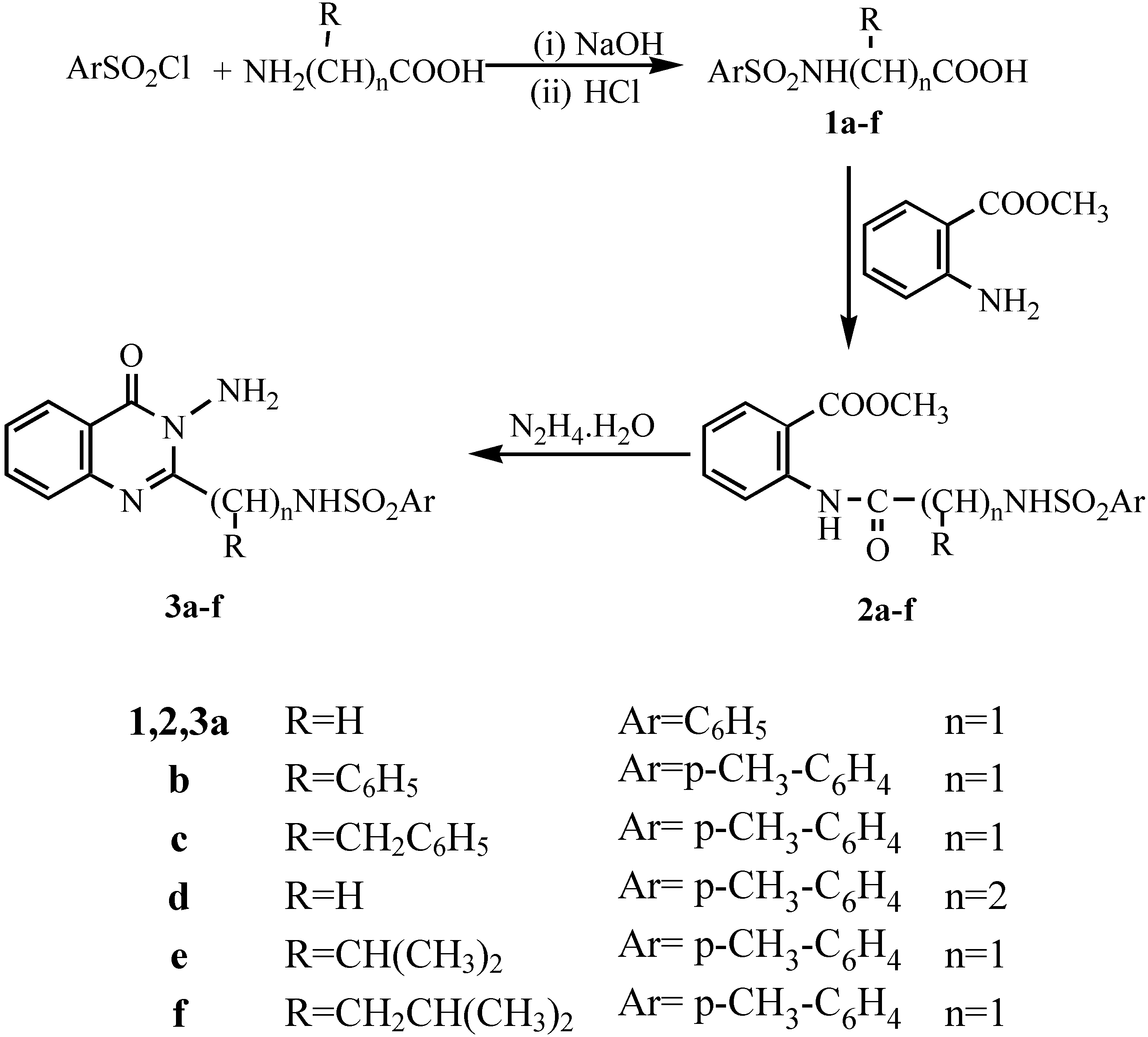{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}