Synthetic methods
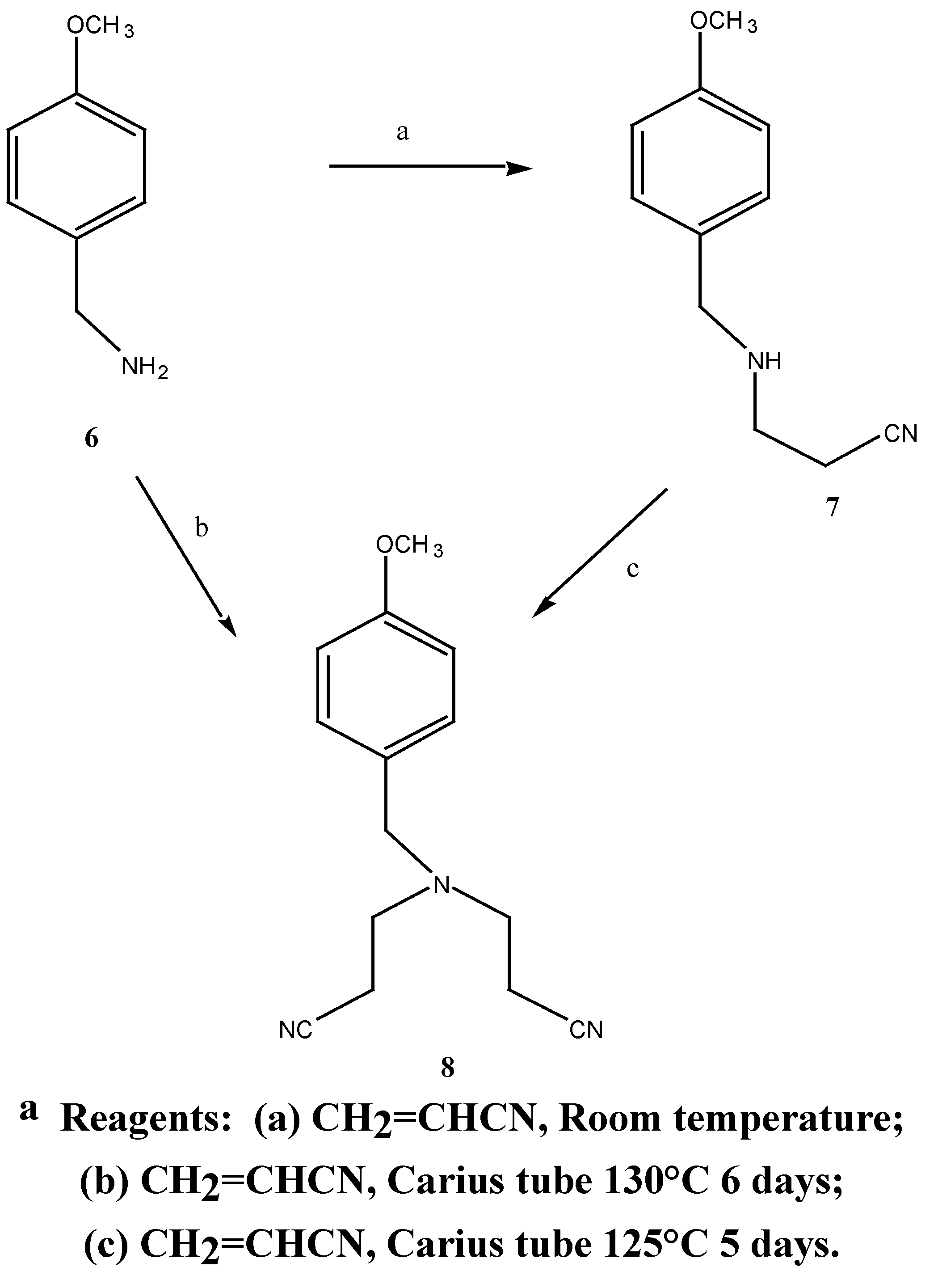
N-(2-cyanoethyl)-4-methoxybenzylamine (7). A mixture of 4-methoxybenzylamine (6) (11.02 g,
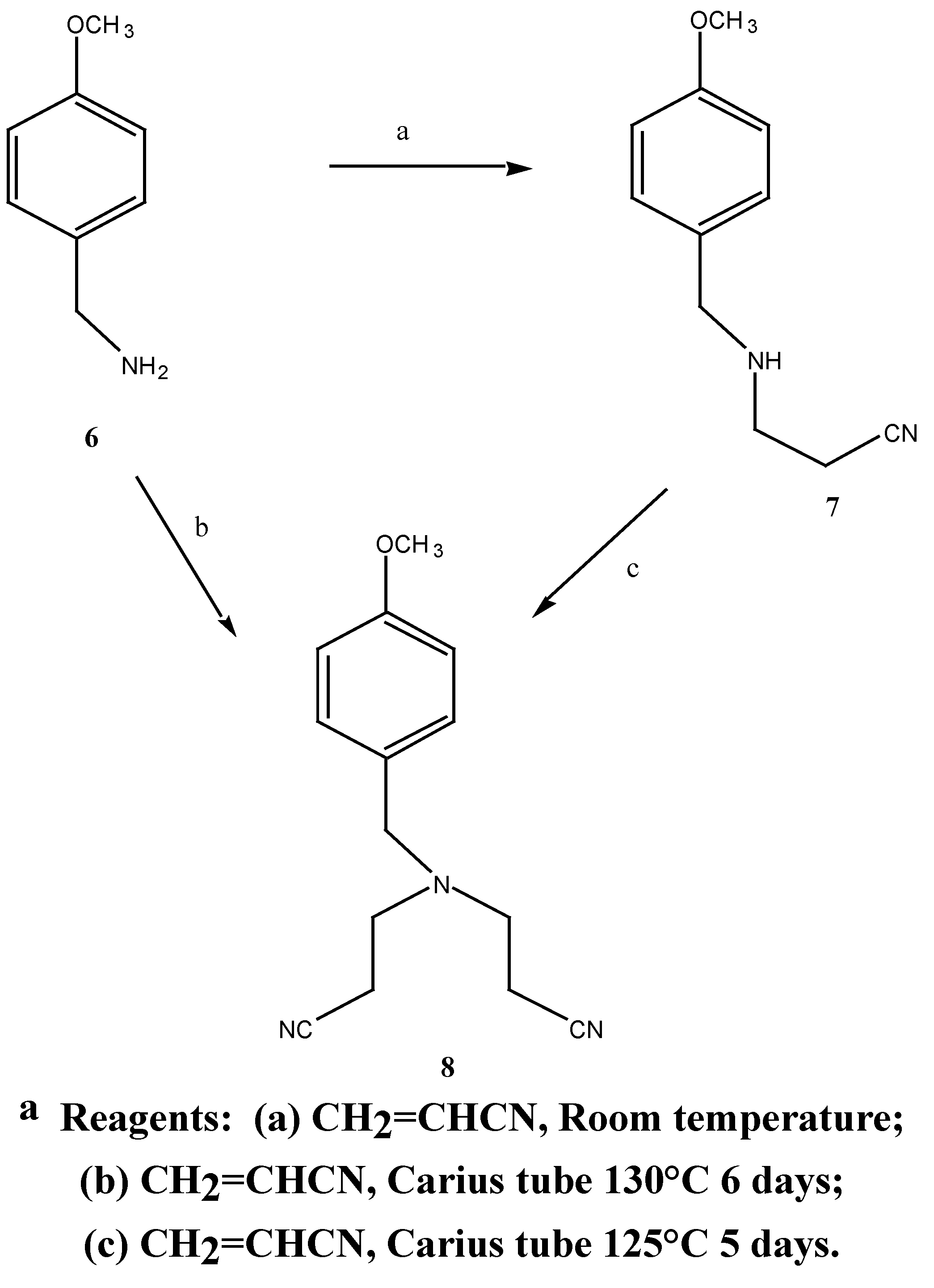
80.0 mmol) and acrylonitrile (4.97g, 93.0 mmol) was stirred for 45 hours at room temperature. Fractional distillation of the reaction mixture gave N-(2-cyanoethyl)-4-methoxybenzylamine (7) (12.2 g, 81 %) as a clear oil b.p. = 245°C at 0.5 mmHg. MS m/z (M+) 190 1H-NMR δ 1.6, s, NH; 2.44, t, J = 5.6 Hz, CH2CN; 2.88, t, J = 6.5 Hz, NHCH2; 3.70, s, PhCH2; 3.78, s, PhCH2; 3.78, s, OCH3; 7.04, d, J3,5 9.0 Hz, H-3, H-5; 7.26, d, J2,6 9.0 Hz, H-2, H-6. 13C-NMR δ 131.5 (C-1), 129.2 (C-2, C-6), 113.8 (C-3, C-5), 158.0 (C-4), 55.2 (C-7), 52.5 (C-8), 44.2 (C-9), 18.7 (C-10), 118.7 (C-11). Anal. found C 69.3, H 7.5; C11H14N2O requires C 69.5, H 7.4 %.
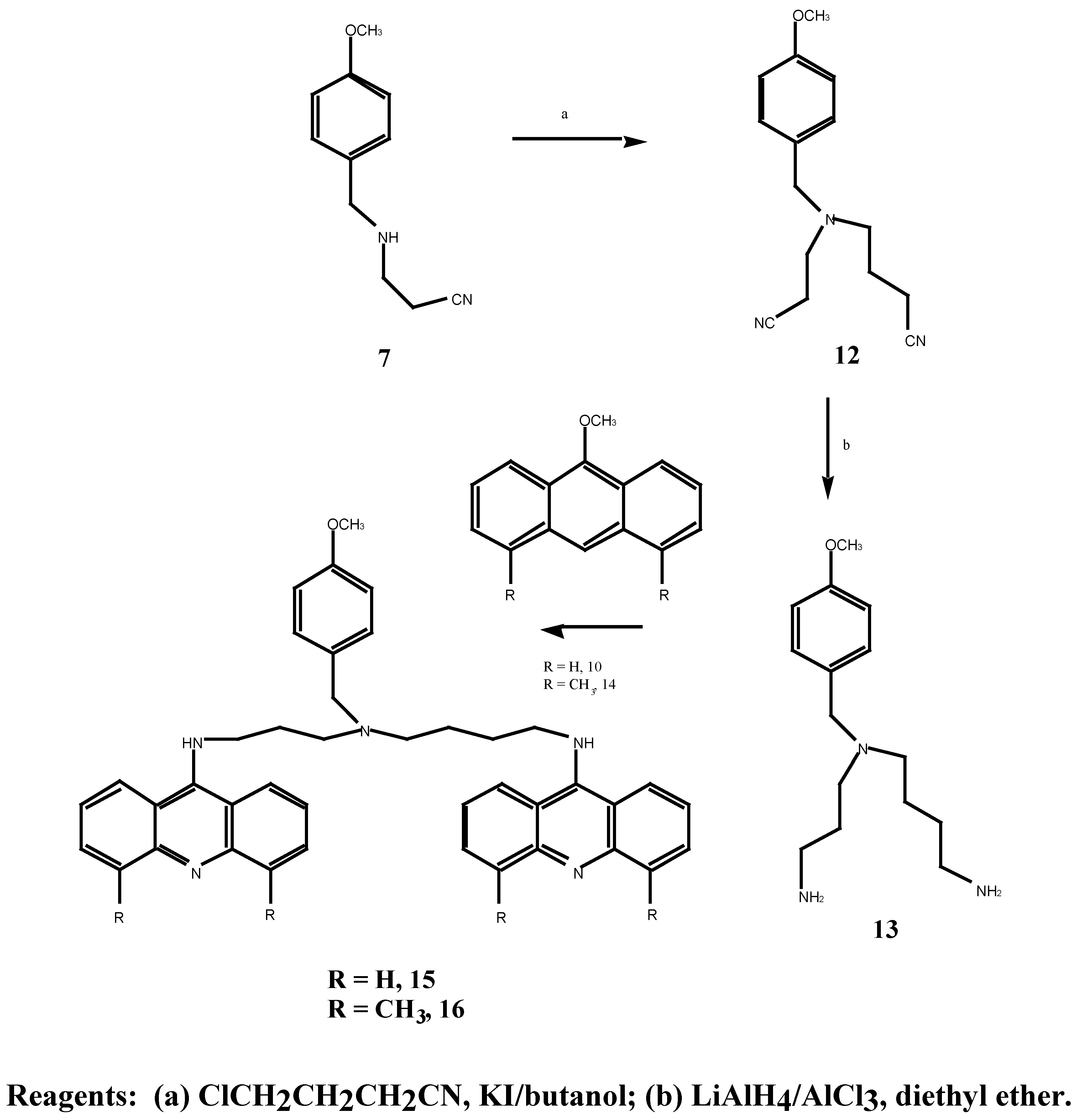
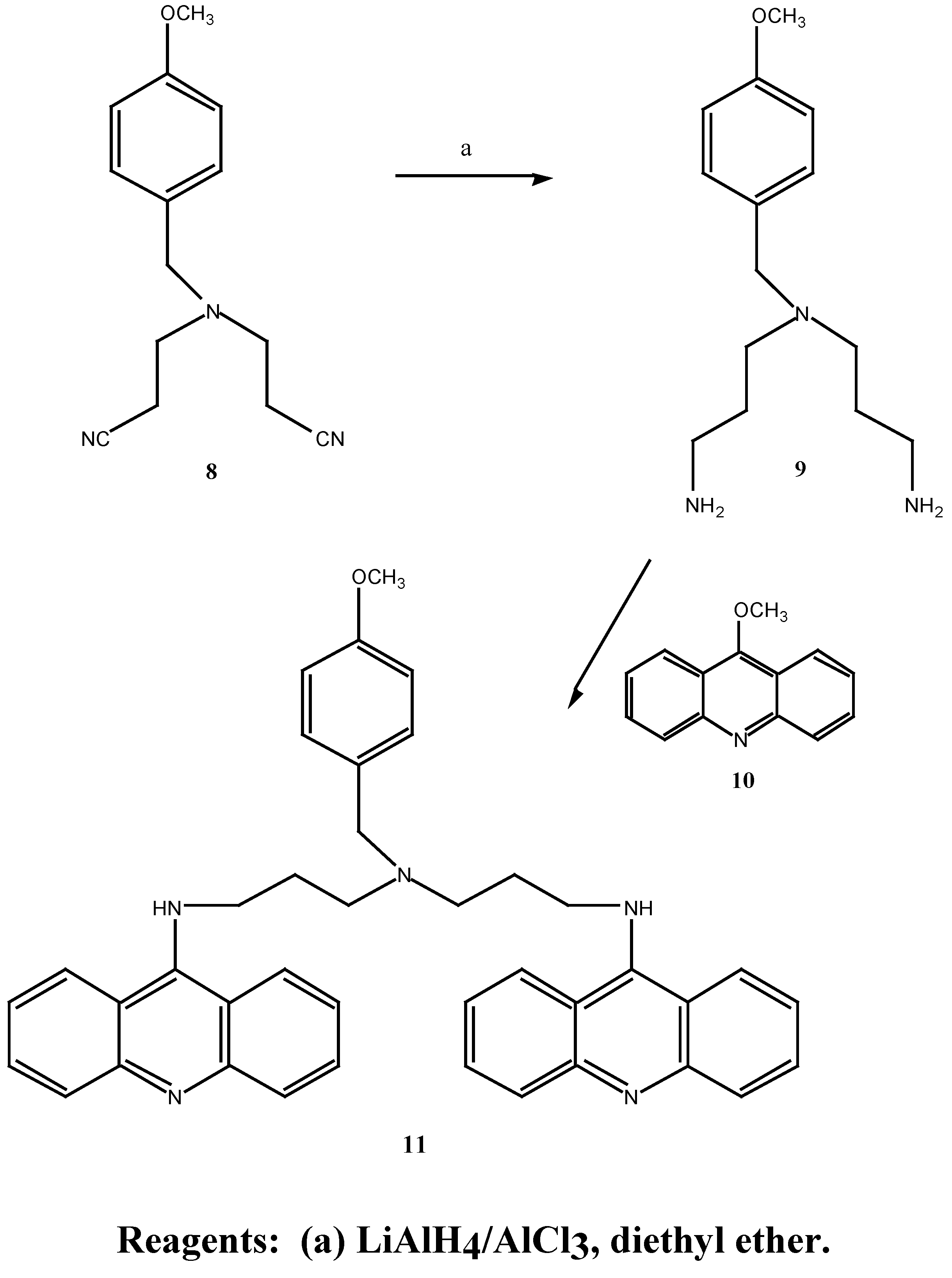
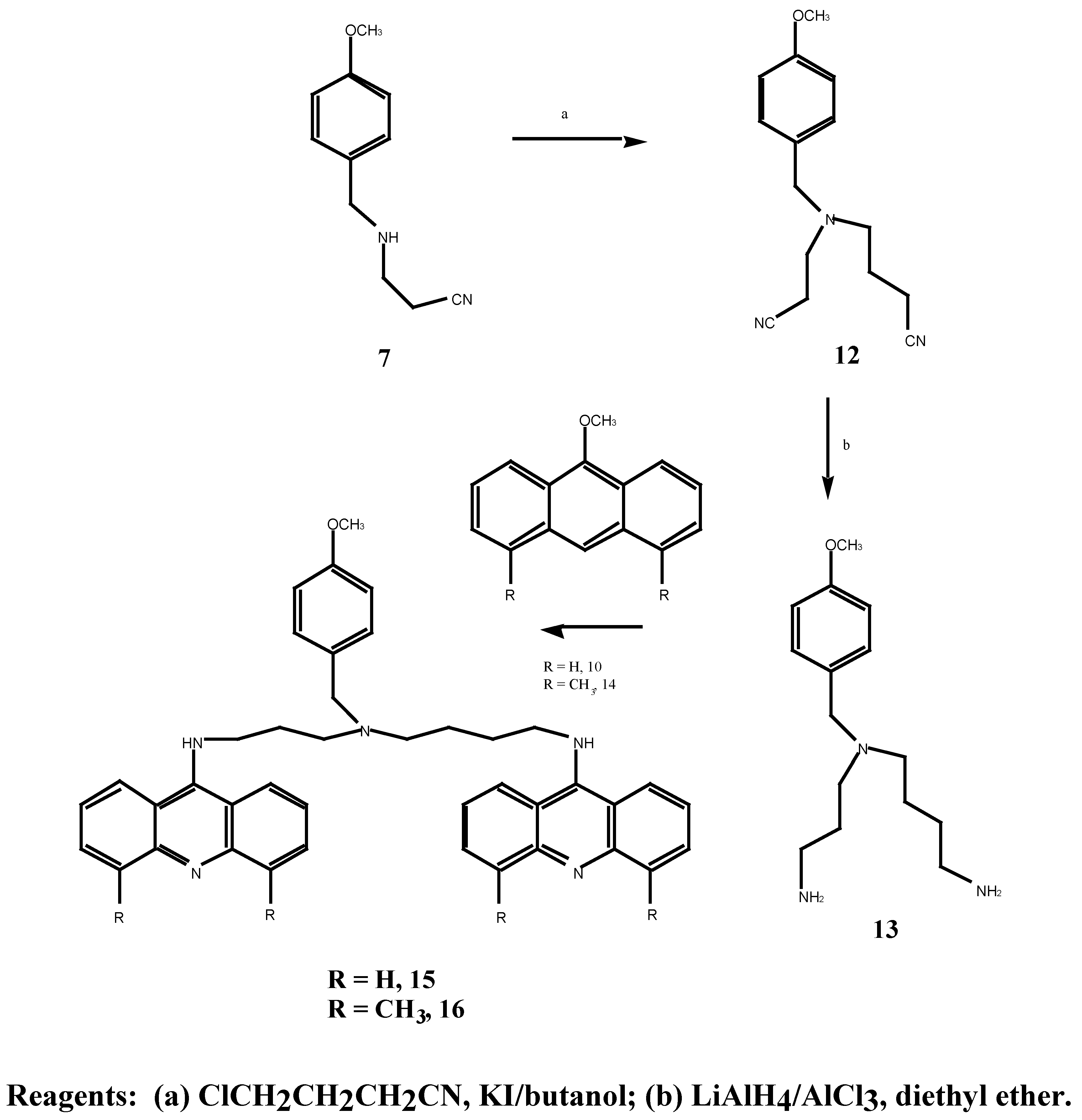
N-(2-Cyanoethyl)-N-(3-cyanopropyl)-4-methoxybenzylamined (12) A solution of 4-chloro-butyronitrile (2.46 g, 25.0 mmol) in anhydrous n-butanol (9.1 mL) was added over 2 hours to a mixture of N-(2-cyanoethyl)-4-methoxybenzylamine (5.07 g, 40.0 mmol), anhydrous sodium carbonate (4.22g, 40.0 mmol) and potassium iodide (0.64 g, 4.0 mmol) at 117°C. The mixture was stirred for an additional 20.5 hours at 117°C. After cooling to room temperature, the mixture was filtered and the solid washed with diethyl ether. The combined filtrate and washing were extracted with 3N HCl (3 x 40 mL) and the resulting acid solution was washed with ether (2 x 40 mL), made basic with potassium carbonate and extracted with ether (3 x 70 mL). The final ethereal extracts were dried over sodium sulphate, filtered and evaporated to give an oil (4.45 g, 50.0 %). Subsequent distillation afforded 3.24 g (43.3 %) of the desired N-(2-cyanoethyl)-N-(3-cyanopropyl)-4-methoxybenzylamine (12) as a light yellow oil b.p. = 235° at 0.06 mmHg. MS m/z 257 (M+); 1H-NMR δ 1.79, q, , J 6.0 Hz, CH2CH2CH2, 2.46, m, 2 x NCH2, 2 x CH2CN, 3.54, s, PhCH2, 3.79, s, OCH3, 6.87, d, , J 9.0 Hz, H-3, H-5, 7.23, d, , J 9.0 Hz, H-2, H-6. 13C-NMR δ 113.7 (C-1), 129.7 (C-2, C-6), 113.9 (C-3, C-5), 158.9 (C-4), 57.8 (C-7), 55.2 (C-8), 49.2 (C-9), 14.6 (C-10), 119.7 (C-11), 51.8 (C-12), 23.4 (C-13), 118.9 (C-15). Anal. found C 69.7, H 7.3, N 16.2; C15H19N3O requires C 70.0, H 7.3, N 16.3 %.
N-(3-Aminopropyl)-N-(4-aminobutyl)-4-methoxybenzylamine (13). A solution of anhydrous aluminium chloride (2.04 g, 15.3 mmol) in anhydrous diethyl ether (21 mL) was rapidly added to a suspension of lithium aluminium hydride (0.6 g, 15.8 mmol) in anhydrous ether (30 mL). After vigorous stirring of the mixture, a solution of N-(cyanoethyl)-N-(cyanopropyl)-4-methoxy benzylamine (12) (1.47 g, 6.0 mmol) in anhydrous diethyl ether (21.0 mL) was added over 2 hours and the reaction was stirred at room temperature for 29 hours. The solution was cooled to 0°C, quenched with 30 % aqueous potassium hydroxide (w/v; 30 mL) and the ethereal layer decanted. The combined ethereal extracts were washed with cold saturated aqueous sodium chloride (2 x 25 mL). Evaporation of the diethyl ether gave a light brown oil which was taken up in chloroform (60 mL), dried over anhydrous magnesium sulphate, filtered and the chloroform evaporated under reduced pressure. A crude oil was obtained (0.73 g) which upon fractional distillation afforded 0.52 g (34 %) of the desired N-(3-aminopropyl)-N-(4-aminobutyl)-4-methoxybenzylamine (13) as a light yellow oil. MS m/z 265 (M+); 1H-NMR δ 1.12, s, 2 x NH2, 1.49, m, 2 x (H-10, H-11, H-14), 2.50, m, 2 x (H-9, H-12, H-13, H-15), 3.47, s, PhCH2, 3.79, s, OCH3, 7.20, d, , J 10 Hz, H-2, H-6. Anal. found C 67.9, H 10.0, N 16.2; C15H27N3O requires C 67.9, H 10.2, N 15.8 %.
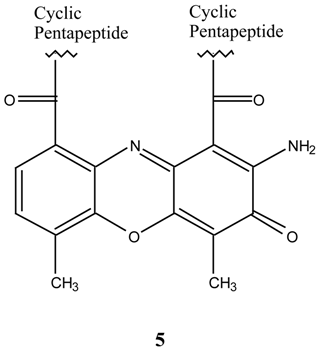
N1,N8-Bis-(9-acridinyl)-N4-(4-methoxybenzyl)spermidine (15). N-(3-aminopropyl)-N-(4-amino-butyl)-4-methoxybenzylamine 13 (0.34 g, 128 mmol) was dissolved in methanol (2.0 mL) and added to 9-methoxyacridine 10 (0.54 g, 2.39 mmol). The mixture was stirred for 70 hours at 30°C, the methanol was evaporated under reduced pressure followed by gel filtration to afford 250 mg (30 %) of the desired bis-aminoacridine 15 as an orange gum. 1H-NMR δ 1.5-3.3, m, 7 x CH2, 2.22, s, 2 x NH, 3.55, s, PhCH2, 3.71, s, OCH3), 7.02-7.96, m, H-2, H-6, H-3, H-5, and acridine protons, 20H. Anal. found C 70.1, H 6.7; C41H41N5O requires C 69.5, H 5.9 %.
N1,N8-Bis-(4,5-dimethyl-9-acridinyl)-N4-(4-methoxy benzyl)spermidine (16). N-(3-aminopropyl)-N-(4-aminobutyl)-4-methoxybenzylamine 13 (60 mg, 0.23 mmol) was dissolved in tetrahydrofuran (1.0 mL) and added to 4,5-dimethyl-9-methoxyacridine (14) (85 mg, 0.18 mmol). The mixture was stirred for 48 hours at room temperature, then stirred at 35°C for 8 hours. Evaporation of the tetra-hydrofuran under reduced pressure followed by gel filtration afforded the desired N1,N8-bis-(4,5-dimethyl-9-acridinyl)-N4-(4-methoxy benzyl)spermidine (16) as an orange gum. 1H-NMR δ 1.1-3.2, m, 8 x CH2, 2.21, s, 2 x PhCH3, 3.42, s, OCH3, 7.02-7.81, m, H-2, H-6, H-3, H-5 and acridine protons. UV (log E), 226, 266, 398 (4.02, 3.86, 3.0). vmax 3400, 2950, 2150, 1570, 1410, 1250, 1180, 1025 cm-1.
N,N-Bis-(2-cyanoethyl)-4-methoxybenzylamine (8): Method 1:-A mixture of 4-methoxybenzyl-amine (0.406 G, 2.9 mmol), an excess of acrylonitrile (0.85 g, 16.0 mmol) and a trace of hydro-quinone was added to a Carius tube [
15]. The tube was sealed under vacuum (0.1 mmHg) and the tube was heated at 45° for 8 hours, then 130°C for 140 hours. The resulting amber brown liquid was distilled under vacuum to afford 0.53 g (72 %) of the desired
N,
N-bis-(2-cyanoethyl)-4-methoxy-benzylamine (
8) as a colourless oil. b.p. = 225°C at 0.21 mmHg, which formed a white solid, m.p. = 48°C on cooling to room temperature.
Method 2:- A mixture of N-(2-cyanoethyl)-4-methoxybenzylamine 7 (1.63 g, 8.6 mmol), an excess of acrylonitrile (1.69 g, 31.8 mmol) and a trace of hydroquinone was added to a Carius tube. The tube was sealed under vacuum (0.3 mmHg) and heated at 125° for 144 hours. The resulting brown oil was distilled under vacuum to afford 1.93 g (92.4 %) of the desired N,N-bis-(2-cyanoethyl)-4-methoxybenzylamine (8) as a light yellow oil. b.p. = 220°C at 0.2 mmHg. MS m/z 243 (M+); 1H-NMR δ 2.41, t, 2 x (H-10, H-13), J 6.6 Hz, 2.84, t, 2 x (H-9, H-12), J 6.0 Hz, 3.6, s, PhCH2), 3.77, s, OCH3, 6.85, d, , J 9.0 Hz, H-2, H-6, 6.89, d, , J 8.6 Hz, H-3, H-5, 7.20, d, , J 10 Hz, H-2, H-6. 13C-NMR δ 116.0 (C-1), 129.8 (C-2, C-6), 113.9 (C-3, C-5), 159.1 (C-4), 57.4 (C-7), 55.2 (C-8), 49.2 (C-9), 16.7 (C-10), 118.8 (C-11), 49.2 (C-12), 16.7 (C-13). Found M+. = 243.135; C14H17N3O requires M+. = 243.137.
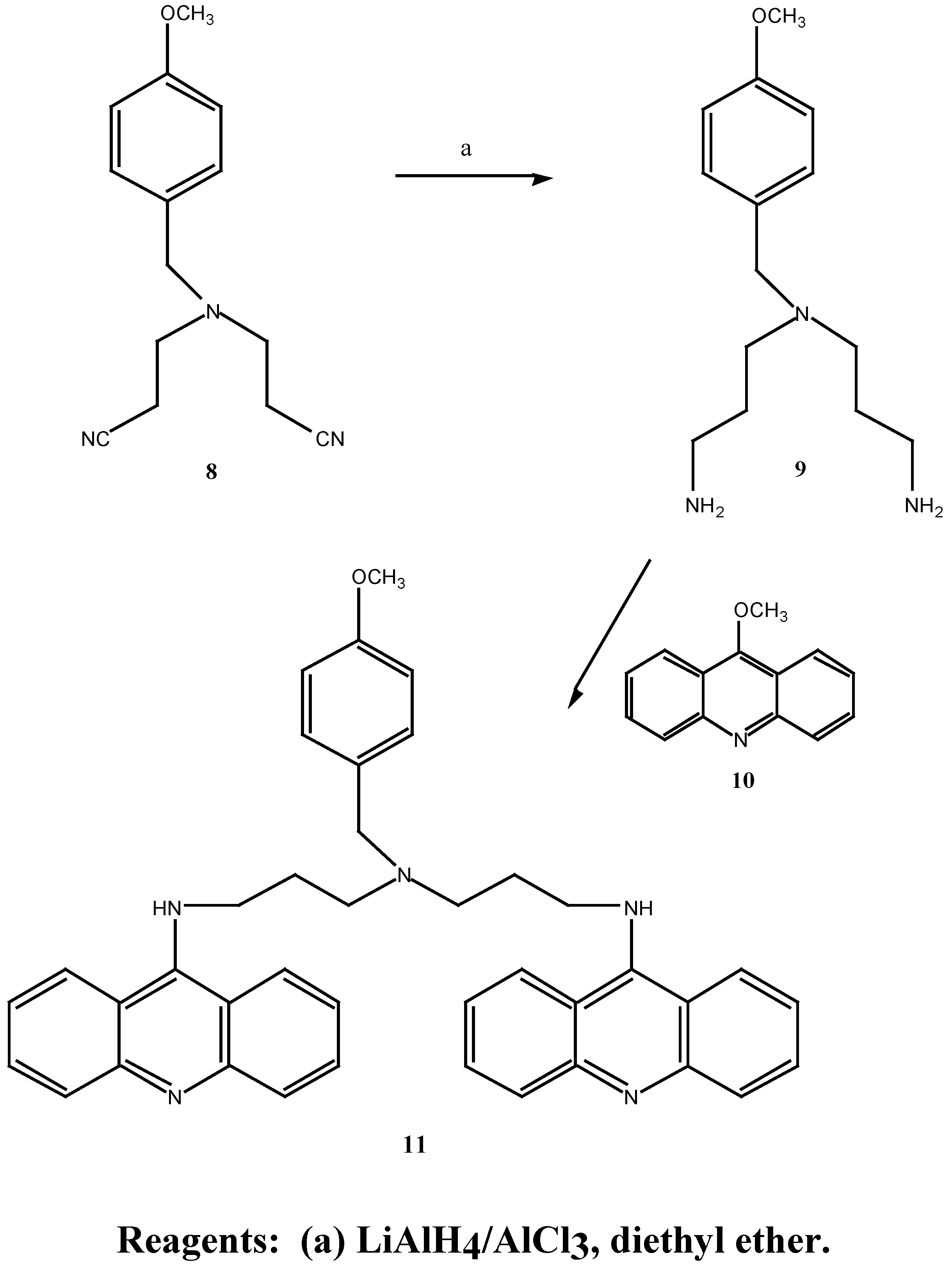
N,N-Bis-(3-aminopropyl)-4-methoxybenzylamine (9):- A solution of aluminium chloride (2.66 g, 19.8 mmol) in anhydrous diethyl ether (25 mL) was rapidly added to a suspension of lithium aluminium hydride (0.75 g, 19.8 mmol) in anhydrous diethyl ether (35 mL). The mixture was vigorously stirred and a solution of N,N-bis-(2-cyanoethyl)-4-methoxybenzylamine (1.52 g, 6.27 mmol) in anhydrous diethyl ether (25 mL) was added over 1.5 hours. After stirring at room temperature for 50 hours the mixture was cooled to 0°, quenched with aqueous 30 % potassium hydroxide (w/v; 30 mL) and the ethereal layer decanted. The remaining emulsion was extracted with diethyl ether (4 x 100 mL) and the combined diethyl ether extracts washed with saturated aqueous sodium chloride (2 x 25 mL). Evaporation of the diethyl ether under reduced pressure gave a brown oil which was taken up in chloroform (50 mL), dried over anhydrous magnesium sulphate, filtered and the chloroform evaporated under reduced pressure. Fractional distillation of the resulting oil afford 0.391 g (25 %) of the desired N,N-bis-(3-aminopropyl)-4-methoxybenzylamine (9) as a light yellow oil. b.p. = 190° at 0.4 mmHg. MS m/z 251 (M+); 1H-NMR δ 1.57, s, 2 x NH2, 2.37-2.68, m, 6 x CH2, 3.72, s, PhCH2, 3.79, s, OCH3, 6.85, d, , J 9.0 Hz, H-2, H-6, 7.22, d, , J 9.0 Hz, H-3, H-5. UV (log E) 271 nm (3.20). Found M+. = 251.19975; C14H25N3O requires M+. = 251.19975.
N1,N7-Bis-(9-acrinyl)-N4-(4-methoxybenzyl)-bis-(3-aminopropyl) amine (11). N,N-Bis-(3-amino-propyl)-4-methoxybenzylamine (9) (40.0 mg, 0.16 mmol) was dissolved in methanol (1.5 mL), added to 9-methoxyacridine (10) (66.0 mg, 0.32 mmol) and the mixture was stirred at room temperature for 63 hours. Evaporation of the methanol under reduced pressure, followed by gel filtration afforded 88.0 mg (91 %) of the desired N1,N7-bis-(9-acrinyl)-N4-(4-methoxybenzyl)-bis-(3-aminopropyl) amine (11) as an orange-red gum. 1H-NMR δ 2.17, s, 2 x NH, 2.31-3.65, m, 6 x CH2), 3.74, s, PhCH2, 3.80, s, OCH3, 6.82-7.61, m, H-3, H-5, H-2, H-6 and acridine protons, 20 H; UV (log E) 225, 267, 414 nm. 4.3, 4.45, 4.36. Found M+. = 605.31543; C40H39N5O requires M+. = 605.31543.
9-Chloroacridine (21). Prepared from 2-carboxydiphenylamine (1.0 g, 4.6 mmol) and freshly distilled phosphorus oxychloride 3.0 mL, 32.7 mmol) according to Albert [
19] and obtained as pale yellow crystals (0.37 g, 37 %). M.p. = 120°C (lit [
27] = 120°C). MS m/z 215 (M
+).
9-Methoxyacridine (10). Prepared from 9-chloroacridine (0.5 g, 2.4 mmol) and a sodium methoxide solution generated from sodium (0.1 g, 4.4 mmol) in methanol (5.8 mL) by the method of Albert [
19] and obtained as dark yellow crystals. Recrystallisation from light petroleum (60-80°C) afforded 0.31 g (63 %) of the desired 9-methoxyacridine (
10) as pale yellow crystals, m.p. = 99-101°C (lit [
19] = 103°C).
1H-NMR δ4.26, s, OCH
3, 8.0, m, acridine protons.
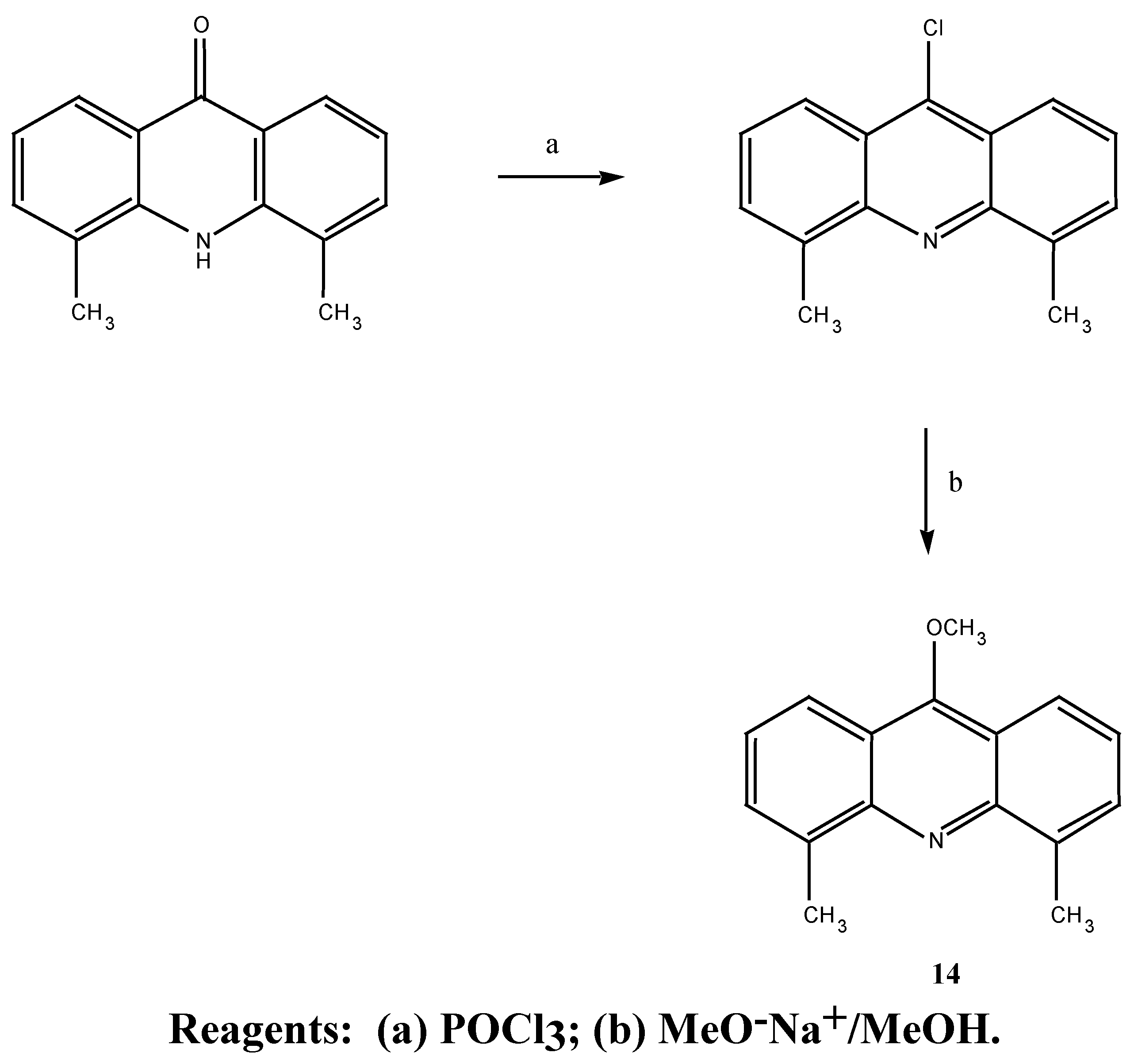
4,5-dimethyl-9-chloroacridine (18). 4,5-dimethylacridone (
20) (1.0 g, 4.5 mmol) and freshly distilled phosphorus oxychloride (7.0 mL) were refluxed at 130°C for 1.5 hours. Unused phosphorus oxychloride was then distilled off under reduced pressure. The residue was allowed to cool and then thinned with chloroform (10.0 mL). The solution was then poured into a mixture of ice (40.0 g) and concentrated aqueous ammonia (33 %) (10.0 mL) with heat being liberated. The solution was stirred, the chloroform layer separated and the aqueous phase extracted with chloroform (2 x 30 mL). The combined chloroform extracts were dried over anhydrous magnesium sulphate, filtered and the chloroform evaporated under reduced pressure to give a yellow powder which upon recrystallisation from petroleum ether (b.p. 60-80°C) afforded 0.83 g (84 %) of the desired 4,5-dimethyl-9-chloro-acridine (
21) as yellow-green needles m.p. = 150-151°C (lit [
29] 151-152°C). MS m/z 241 (M
+);
1H-NMR δ 2.94, s, 2 x CH
3, 7.42-8.31, m, acridine protons.
4,5-Dimethyl-9-methoxyacridine (14). 4,5-Dimethyl-9-chloroacridine (220 mg, 0.91 mmol) was added to a methoxide solution generated from sodium (160 mg, 6.95 mmol) in methanol (15.0 mL) and tetrahydrofuran (0.5 mL). The solution was gently refluxed for 40 hours and allowed to cool and the methanol reduced under vacuum. The residue was taken up in benzene, filtered from the salt and the benzene evaporated under reduced pressure to give a yellow powder which was recrystallised from heptane to afford 145 mg (67 %) of the desired 4,5-dimethyl-9-methoxyacridine (14) as yellow crystals m.p. = 90-91°C. MS m/z 237 (M+); 1H-NMR δ2.93 (6H, s, 2 x CH3), 4.19 (3H, s, OCH3), 7.32-8.17 (6H, m, acridine protons). Anal. Found C 80.0, H 6.4, N 5.9%; C16H15NO requires C 80.6, H 6.4, N 5.9 %. UV (log E) 225, 357, 373, 392 nm. (4.63, 3.81, 3.80, 3.66).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}