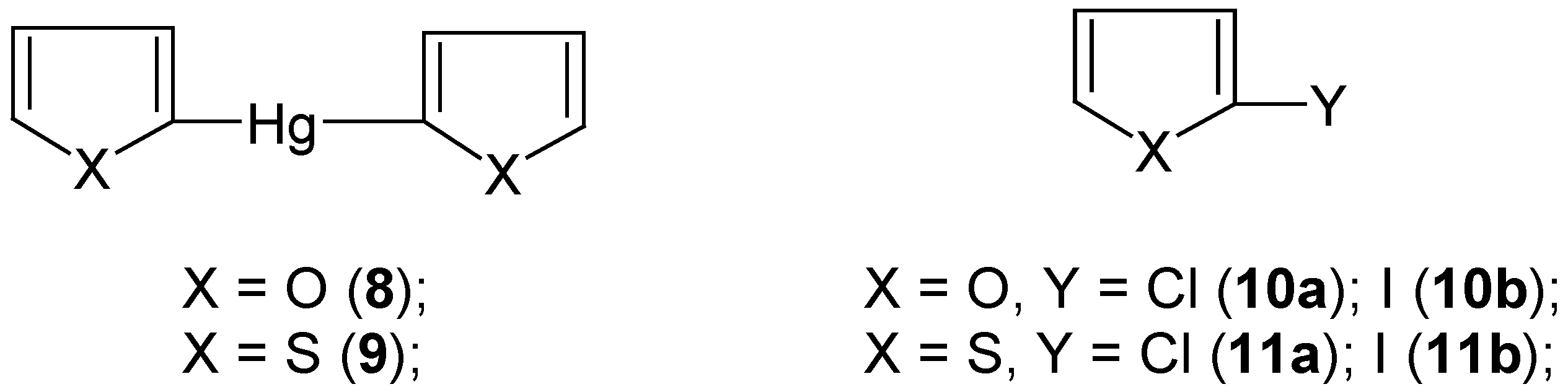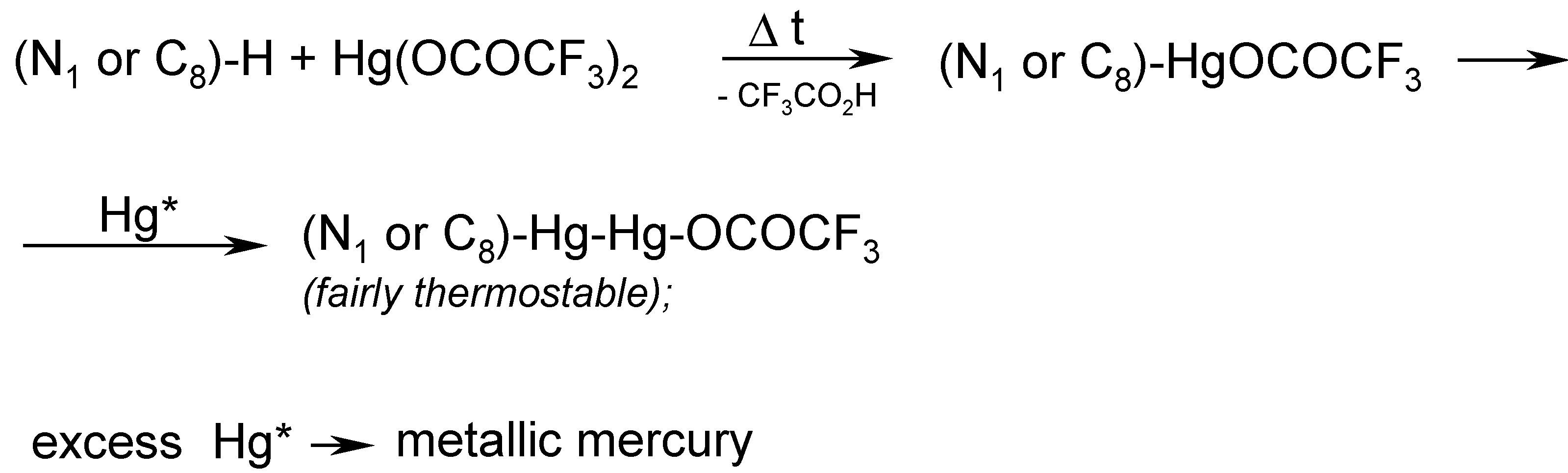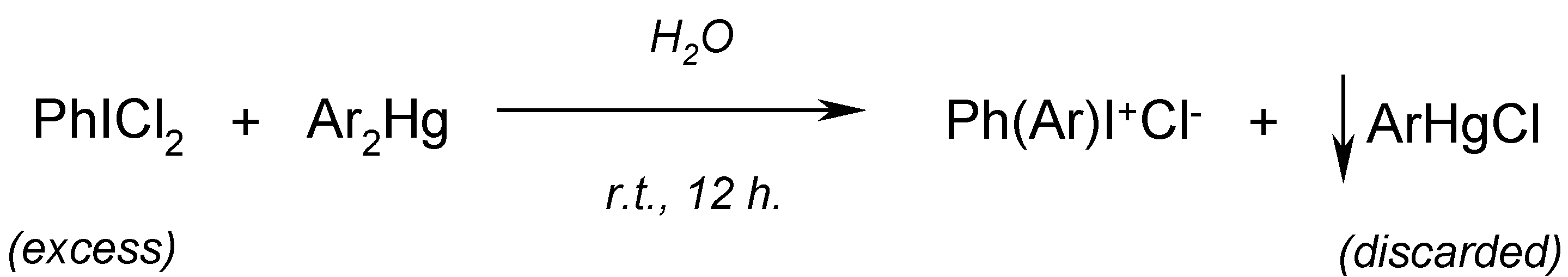1. Introduction
The first organomercury(II) compound (organomercurial) was reported by E. Frankland (1850), who synthesized dimethylmercury, Me2Hg, by the action of methyl iodide on mercury metal under sunlight irradiation. The number of structurally diverse organomercurials that were later synthesized for pharmacological purposes is very large, but their role in chemotherapy has now been completely superseded and their applications as fungicides are also on the decline, owing to their toxicity towards human beings and animals. Nevertheless, numerous organomercurials have found increasing applications as useful reagents for the synthesis of many other organometallics via metal exchange reactions, and they are still attractive as synthetic intermediates which are usually readily available, accommodate essentially all functional groups and possess remarkable thermal and chemical stability towards air, water, dilute acids and bases. These valuable features allow synthetic reactions employing organomercurials to be run under a wide variety of reaction conditions. The major disadvantage of these compounds is the high toxicity of more volatile mercurials. In fact, all heterocyclic (lactamic) organomercurials prepared by us during the course of our research (1980-2000) were nonvolatile and either slightly soluble or practically insoluble in common solvents, hence they were notably less hazardous than e.g. many aliphatic organomercurials. However, due to their very limited solubility in boiling common solvents, they often could not be purified satisfactorily or not be purified at all. This made it difficult (or even impossible) to analyze them reliably for the purpose of making proper structural assignments. Therefore, their possible structures were deduced from subsequent, well known and effective, chemical reactions (usually iodo-demercuration and/or bromo-demercuration reactions) followed by chemical and spectral analysis of the resulting products (i.e. monoiodo and/or monobromo derivatives of the parent aromatics), often also produced in the other known routes, for the sake of a better comparison.
There are a large number of different methods for preparing unsymmetric aromatic organomercurials, ArHgX, but the direct mercuration of aromatic systems is evidently the most simple of them and is thus very often used. Its proper and effective application strongly depends on: (i) the relative reactivity of the reacted aromatic system towards the electrophilic attack by a mercuric salt; (ii) the relative electrophilicity of the mercuric salt applied, e.g. HgCl2 < Hg(OCOCH3)2 < Hg(OCOCF3)2 < Hg(ClO4)2; (iii) the applied reaction conditions: the use of a proper solvent, temperature and reaction time. This is an ordinary electrophilic aromatic substitution and takes place via the arenium ion mechanism to form the corresponding unsymmetric aromatic mercurials, e.g. ArHgCl < ArHgOCOCH3 < ArHgOCOCF3 < ArHgClO4 (their relative reactivities towards the subsequent reactions with various electrophilic reagents being precisely in this order).
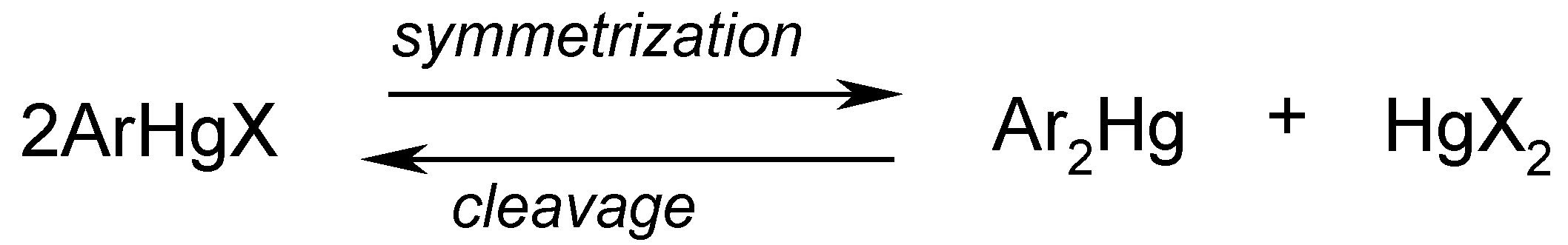
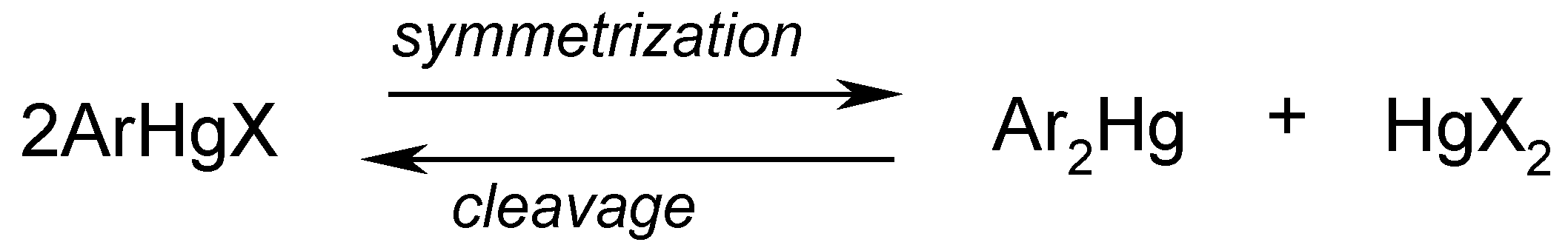
Although more or less effective depending on the particular case considered, several symmetrization methods are known to convert unsymmetric compounds ArHgX to the corresponding symmetric ones, Ar2Hg (usually more reactive than ArHgX). It must be borne in mind that the following equilibrium should be displaced far to the right to cause effective symmetrization:
From a practical point of view, this may be attained either (i) by removal of the mercuric salt HgX2 by strong complexation with e.g. sodium or potassium iodide, sodium thiosulfate, ammonia or EDTA, potassium cyanide, potassium thiocyanate, etc. or (ii) by reduction of HgX2 with hydrazine, sodium stannite, by electrolysis, or otherwise. Consequently, there is no single general procedure applicable to effectively symmetrize the various types of ArHgX, as certain types of ArHgX compounds are symmetrized with great ease and give high yields of Ar2Hg, while others are resistant to particular symmetrization agents, and the desirable Ar2Hg can only be obtained by changes in the applied procedure(s).
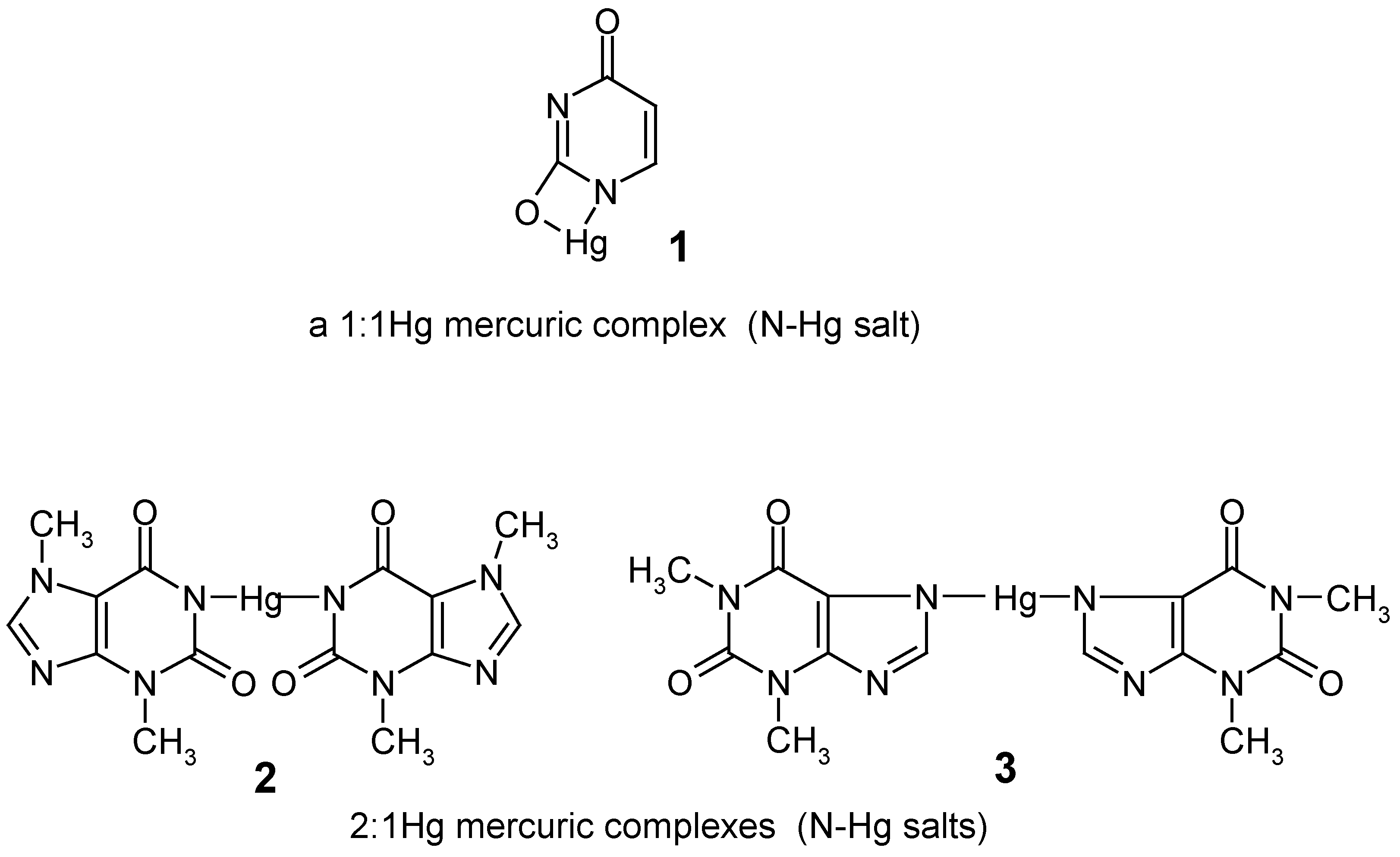
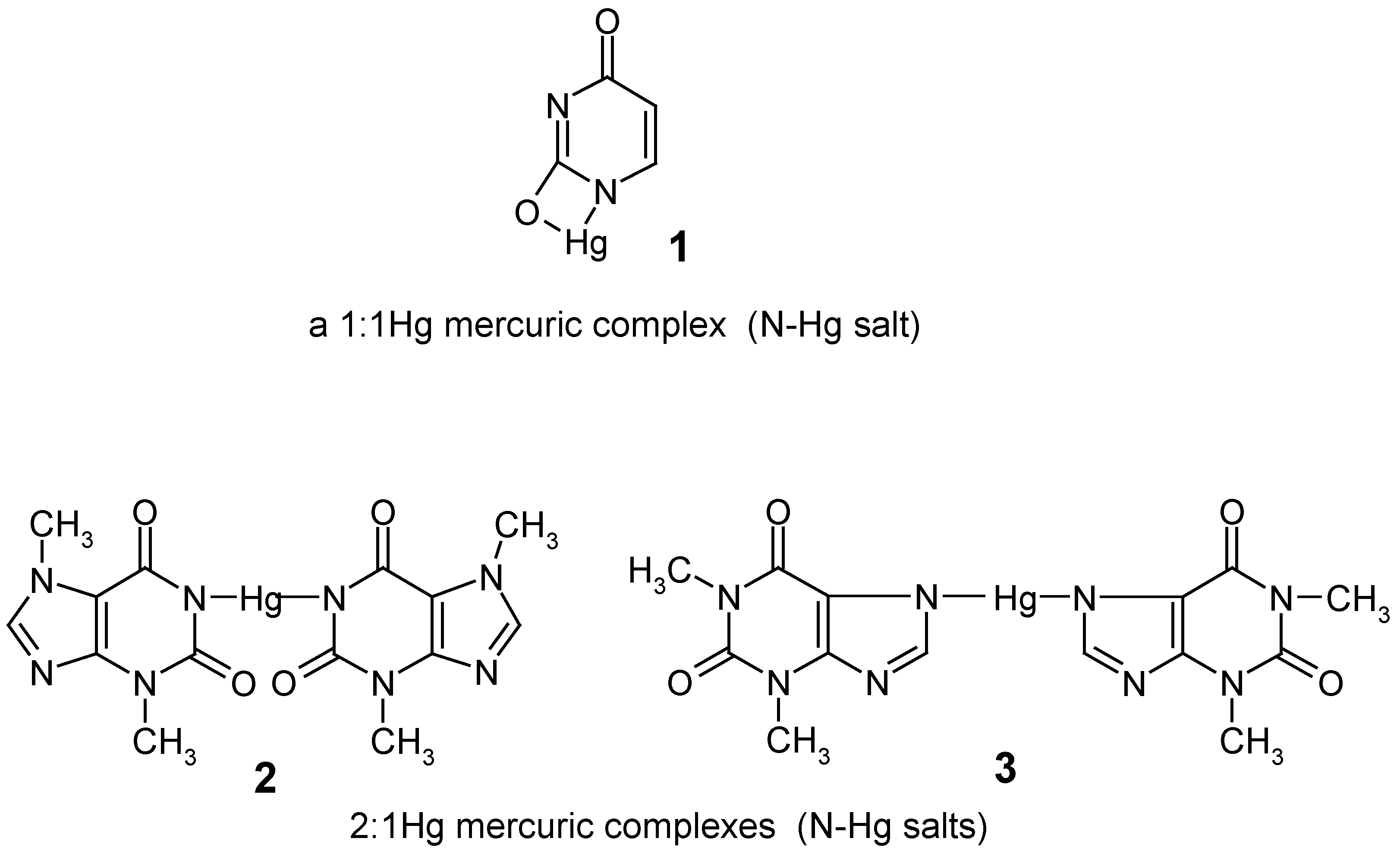
In 1979, when we started our preliminary experiments in this area, the direct mercuration with mercuric salts of some important "model" lactamic heterocycles, e.g. uracil, theobromine and theophylline, had not been previously reported in the literature. The attempted mercuration reactions of these compounds at their C5 or C8 positions, respectively, resulted only in the formation of their insoluble N-Hg salts 1 - 3 (named also 1:1 or 1:2 mercuric ”complexes”), which were precipitated out in full from the mercurating reaction mixtures – this impeded attempts to obtain their true organomercurials, i.e. their mercurated derivatives with the mercury atom joined to the organic residues via C5 or C8 carbon atoms.
In contrast, heterocycles devoid of any
acidic N-H group(s), e.g. caffeine (see
Section 2), 1,3-dimethyluracil (see
Section 6), 2,4-dimethoxypyrimidine (see
Section 7), or 2,3-diphenyl-5-methoxy-benzo[b]furan (see
Section 5) were readily mercurated directly to form the respective
unsymmetric organomercurials, ArHgX, which were often
symmetrized to form Ar
2Hg. Next, the mercurials of the two types were always
iodo-demercurated and/or
bromo-demercurated to form the corresponding iodides or bromides, ArI or ArBr, which after their purification were analyzed and studied by
1H‑NMR and other techniques to confirm the chemical structures of the starting mercurials.
Generally, it is known that mercury(II) ions, consistent with their pronounced electrophilic character, can effectively mercurate a large number of aromatics at their carbon atom(s) to form the corresponding true organomercurials if they are inherently devoid of any acidic S-H, O-H, N-H, Se‑H, and sometimes P-H groupings. Otherwise, Hg(II) ions reveal a greater tendency to combine with those electronegative heteroatoms bearing lone pair(s) of electrons, forming thus some seemingly “mercurated”, sparingly soluble final products or intermediates – which are not true organomercurials, since the mercury atom therein is joined to the organic residue not via carbon atom but via the respective heteroatom. Those containing the O-Hg bond are the least stable. Compounds with the S‑Hg linkage are formed very readily and exhibit particular stability. The strength of the N-Hg bond varies between wide limits, but in most cases it does not exceed that of the S-Hg bond. Organic compounds containing O-Hg, N-Hg, and sometimes even S-Hg bonds are often the first intermediates formed during the direct mercurations of oxygen-, nitrogen-, and some sulfur-containing compounds, and are transformed more or less readily into compounds mercurated at the carbon atom (true organo-mercurials), when this process is carried out under more vigorous conditions, e.g. at a higher temperature or at a lower pH. For example, thioanisole can be C-mercurated in 36.6% yield by means of Hg(OCOCH3)2 on a steam bath, giving thus 4-(acetoxymercurio)thioanisole; for more such examples see Ref. 23, p. 93.
During the course of our systematic, multi-year studies of various
aromatic (carbo- and predominantly heterocyclic) true organomercurials and their reactions, we successfully synthesized
novel organomercurials derived from
theobromine and
theophylline (
Section 9 and
Section 10),
uracil (
Section 8), and also some other mercurials, though mostly in
indirect routes. We also isolated (in ca 28% yield) the supposed-to-be
1,8-bis(acetoxydimercurio)theobromine, which seemingly represents the first
stable organic derivative of
mercury(I), and we subsequently studied its chemical properties (
Section 11). We also discovered several
novel halo- and
cyano-
demercuration reactions; in our opinion, our novel fluoro-, chloro-, and cyano-demercuration reactions are particularly interesting and useful. We also extended, improved and better substantiated Willgerodt`s old method (1897), which enables to synthesize diaryliodonium chlorides by reacting (dichloroiodo)arenes with
symmetric aromatic organomercurials, in stirred hot aqueous suspensions (
Section 14). For more information on our own
published achievements [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16] see the sections that follow.
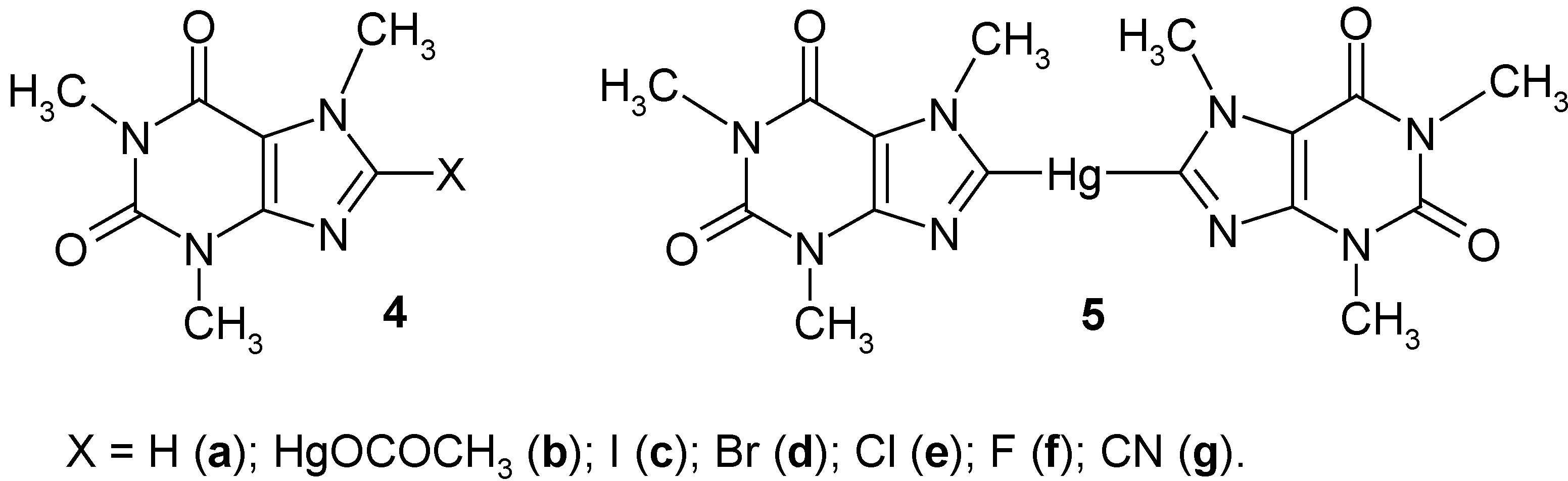
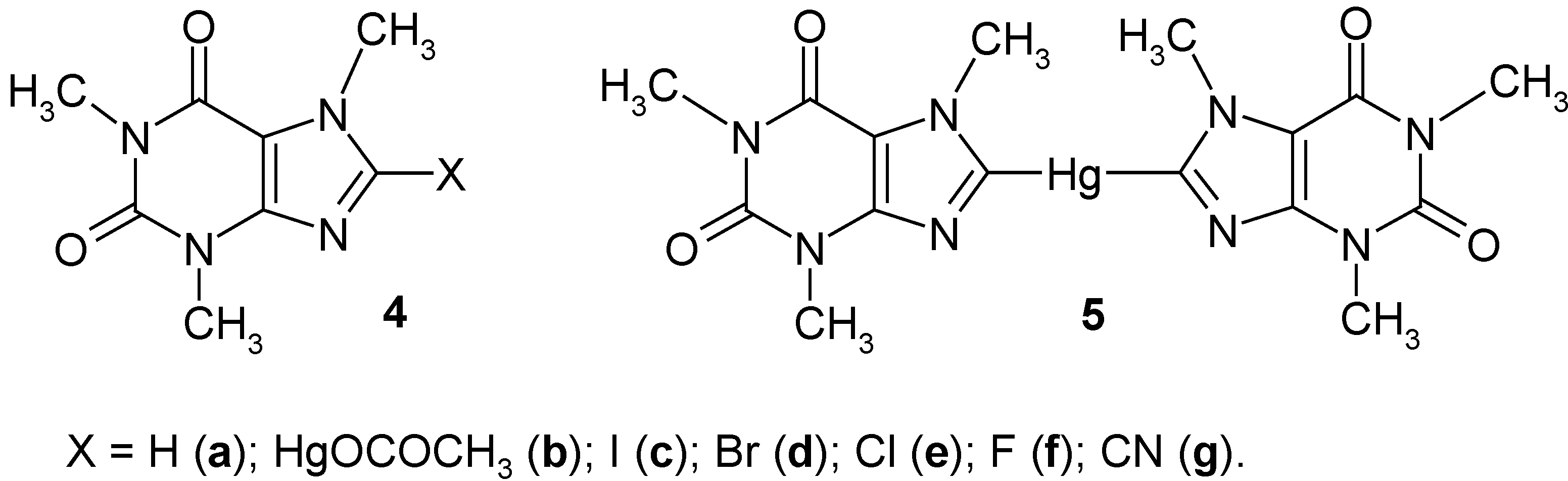
2. Early Results: 8-Substituted Caffeine Mercurials and Their Demercuration Reactions [1,2,11 and 12]
Previously, Rosenthaler [
43] had mercurated
caffeine (
4a) to afford
8-(acetoxymercurio)caffeine (
4b) in 90% crude yield, on boiling
4a for 48 hours with an aq. Hg(OCOCH
3)
2 solution acidified with CH
3CO
2H. Next, he symmetrized
4b with a boiling aq. KI solution for 30 minutes, to give
8,8'-mercuriobis(caffeine) (5) in 50% crude yield; he did not carry out any further reactions with
4b and
5. In our laboratory, we improved the above synthesis of
4b, by adding a few drops of conc. aq. HClO
4, a recommended catalyst [
23,
24,
26,
27,
28], to the mercurating reaction mixture, which shortened considerably the mercuration time to only four hours, while preserving the same 90% crude yield; we also established that the mercurating solutions may be prepared
in situ by dissolving the freshly prepared yellow HgO in hot aq. CH
3CO
2H.
We also improved the crude yield of the symmetrization of
4b from 50% to 83% by using a hot (ca. 90°C) aqueous Na
2S
2O
3 solution, instead of aq. KI. We also
metathesized 4b into sparingly soluble
8‑(chloromercurio)caffeine (83% crude yield) by adding excess aq. NaCl solution to a boiling solution of
4b in water [
1,
11]. On heating with an aq. Na
2S solution, the black precipitate of HgS conclusively proved the presence of mercury in the three aforementioned caffeine mercurials; their chemical structures were confirmed by their
1H-NMR solution spectra as compared with that of caffeine [
1,
11], as well as by subsequent
halo- and
cyano-demercuration reactions discussed below (see also Ref. 11).
Finally, it should be recalled that our many attempts to synthesize either 8-(trichloroacetoxy-mercurio)caffeine or the
more thermostable 8-(chloromercurio)caffeine by a one-pot method suitable for preparing the
thermostable phenylmercury(II) chloride were without success: the latter benzene mercurial was obtained in 70% yield when yellow HgO and CCl
3CO
2H were stirred in thiophene-free benzene for ca one hour at 65-70°C; for the explanation see Ref. 11 as well as
Section 3.
2.1. Halo-demercuration Reactions of Caffeine Mercurials [1, 2, 11 and 12]
Gomberg [
44] had reported the failure to iodinate
caffeine (
4a) with diiodine in chloroform; later on,
8-iodocaffeine (
4c) was obtained in ca 40% yield by heating
caffeine (
4a) with diiodine in a sealed tube at 150°C [
45]. In our laboratory, at first we
iodo-demercurated caffeine mercurials
4b and
5 by applying a widely used procedure [
17,
23,
24,
25,
26,
27,
28]; on heating these mercurials with hot (80°C) aq. KI
3 solutions for 30 minutes, we obtained
4c in the same 95% yield (after purification) from the both substrates
4b and
5 [
1,
2]. Similarly, the sparingly soluble
8-(chloromercurio)caffeine (
4, X = HgCl) was iodo-demercurated to give pure
4c in 65% yield [
11]. The same pure
4c was also obtained by us [
1,
2] in 90% yield from the both substrates
4b and
5, by heating them with a hot (80°C) aq. solution of the freshly sublimated
ICN [
Caution: ICN is highly toxic]; in fact, it is an effective, though less convenient and
unsafe, iodo-demercuration method.
Previously,
caffeine (
4a) was directly brominated to give
8-bromocaffeine (
4d) in high yields [
45,
46]. In our laboratory, the two mercurials
4b and
5 were effectively
bromo-demercurated, by applying the widely-used procedure [
17,
23,
24,
25,
26,
27,
28]; the reactions took place in hot (80°C) aq. KBr
3 solutions
previously adjusted to pH 7, for 30 minutes, to give
4d (purified) in the same 83% yields from the both substrates
4b and
5. We also bromo-demercurated
4b and
5 (at 60°C, for 2 hours) with an aq. slurry of a labile complex (or adduct) of unknown composition,
(KBr-BrCN)x [
Caution: highly toxic], obtained by adding Br
2 to a cooled saturated aq. KCN solution [
47]; this gave compound
4d (purified) in 85% yields from the both substrates
4b and
5. In fact, this represents an effective, albeit less convenient and
unsafe, novel bromo-demercuration method.
The direct chlorination of
caffeine (
4a) to
8-chlorocaffeine (
4e) had been reported as early as 1850; fairly high yields of
4e (ca 80%) were later reported in the literature [
45,
48]. In our laboratory, we obtained the purified compound
4e in 27-90% yields, but only from the
more reactive symmetric mercurial
5 [mercurial
4b did
not react under the same reaction conditions]. At first, we reacted
5 with
neat liquid S2Cl2 or
SCl2 at room temperature for 4 hours to afford
4e in 80% or 70% yields, respectively; though the yield of the latter reaction with SCl
2 was lower, nevertheless the crude
4e thus obtained was easier to purify. When mercurial
5 was similarly suspended in
neat boiling SO2Cl2 for 2 hours, then compound
4e (after its isolation and purification) was obtained in only 27% yield. These
novel chloro-demercuration methods were later on also applied by us to other mercurials (
Section 13.3). Finally, we must admit that we
failed to obtain compound
4e upon passing gaseous Cl
2 through solutions or suspensions of mercurials
4b or
5 in water, aq. KCl solutions, formamide, dimethyl sulfoxide, benzene, etc. The same was true for the action of chlorine
in statu nascendi (generated in reaction: 6HCl + 2KClO
3 → 6Cl + 2KCl + 3H
2O) upon
4b or
5 in aqueous media. For some more reactive organomercurials these chloro-demercuration reactions were successful [
17,
23,
24,
26,
28]; see also
Section 3.
8-Fluorocaffeine (
4f) was not reported in the literature before 1981/1982 [
1,
2]; until then very few organomercurials have been fluoro-demercurated, but only with difluorine diluted with nitrogen gas [
26]. In our laboratory, we suspended the
more reactive mercurial
5 [mercurial
4b did
not react under the same reaction conditions] in liquefied,
neat SF4 (b.p. -40.4°C.
Caution: highly toxic) at -70°C (solid CO
2 + acetone)
for 16 hours. After evaporating SF
4, crude
4f was purified to give 30% yield (after 8 hours only 15%); an increase in the reaction temperature (in an autoclave) considerably diminished the given yield. This
novel and interesting
fluoro-demercuration reaction was later on applied in our laboratory to other
symmetric mercurials; see Ref. 12, where we summarized and commented
all our fluoro-demercuration experiments; see also
Section 13.4.
2.2. Cyano-demercuration Reaction of Caffeine Mercurials [1, 2]
Previously,
8-bromocaffeine (
4d) in the presence of KCN in 80% ethanol yielded
caffeine-8-carboxamide, which by treatment with POCl
3 yielded
8-cyanocaffeine (
4g) [
45,
49]; it is worth mentioning that ICN, when reacted at 110°C with dimethylmercury, yielded mercuric iodide and methyl isocyanide [
50]. In our laboratory, we reacted the
more reactive, symmetric mercurial
5 with freshly purified
BrCN [
Caution: highly toxic]
in water, at 60°C for 2 hours (at higher temperatures some decomposition of
5 was observed, whereas at 40°C the reaction did
not proceed); after purification of the crude product, we obtained pure
4g in 50% yield; the same reaction with unsymmetric mercurial
4b gave pure compound
4g in only 25% yield. It was surprising that by using aq. KBr solutions instead of pure water for dissolving BrCN, the same compound
4g was obtained, instead of the expected
4d (
vide supra). By using aq. KCN solutions instead of water for dissolving BrCN, the yield of this
cyano-demercuration reaction was evidently lowered. We applied this interesting but
unsafe reaction also for
more reactive “model” benzene mercurials (see
Section 3). It should be noted that when we reacted
4b or
5, under widely varied conditions, with (CN)
2 or ClCN solutions, we failed to obtain compound
4g; the latter reagent
decomposed vigorously both
4b and
5 in their solutions or suspensions.
2.3. Unsuccessful Reactions with Caffeine Mercurials and Conclusions [1, 2]
The preparative demercuration methods discussed above show the usefulness of organomercurials in organic synthesis. There are, however,
noticeable differences in the reactivity of the various organomercurials. It has been mentioned several times in the literature [
23,
24,
25,
26,
27,
28,
34] that Ar
2Hg compounds are usually
more reactive as compared with ArHgX. It is seen that
unsymmetric mercurial
4b does
not react with S
2Cl
2, SCl
2, SO
2Cl
2, and SF
4 under the same experimental conditions as does
symmetric 5. The cyano-demercuration reaction of symmetric mercurial
5 furnished twice as much of
4g as compared with unsymmetric mercurial
4b. Only the iodo- and bromo-demercuration reactions furnished
the same yields of
4c and
4d from both
4b and
5. We have also established experimentally that both
4b and
5 did not undergo several well-known demercuration reactions [
23,
24,
25,
26,
27,
28,
34] with HNO
3, (CN)
2, (SCN)
2, Cl
2 or chlorine
in statu nascendi, SOCl
2, and aryldiazonium cations, which were successful with some other, more reactive, organomercurials. Thus, it seems to us that both the caffeine mercurials
4b and
5 are noticeably
less reactive in a number of demercuration reactions as compared with some corresponding organomercurials, ArHgX or Ar
2Hg, in which Ar represent e.g.
o‑nitrophenyl, thienyl or furyl moieties [
23,
24,
25,
26,
27,
28,
34]; cf. particularly our results presented in
Section 3 and
Section 4.
3. “Model” Monosubstituted Benzene Mercurials and Their Demercuration Reactions [7, 11 and 12]
A survey of methods applied so far for the
halo-demercuration of various organomercurials [
17,
24,
25,
26,
27,
28,
34] reveals that dihalogens do decompose them in two stages: (1) R
2Hg + X
2 → RHgX + RX, followed (with an excess of halogens) by (2) RHgX + X
2 → R-X + HgX
2; it has been pointed out [
23] that
symmetric R
2Hg react
more readily than RHgX. In 1870 Dreher and Otto [
51] reacted cold solutions of
diphenylmercury, Ph
2Hg (in ethanol or better in CS
2), with equimolar amounts of I
2 or Br
2, and they obtained the respective halogenomercurio- or halogeno-benzenes, whereas with an excess of the halogens the former were changed into the respective halogenobenzenes and mercuric salts (with necessary heating).
Dry dichlorine gives a vigorous reaction with Ph
2Hg yielding PhHgCl, PhCl and HgCl
2; when Cl
2 is bubbled through a hot aq. suspension of Ph
2Hg or (better) through its hot solution in CS
2, at first PhHgCl, and then more and more PhCl and HgCl
2 are obtained. The German authors [
51] have also remarked that PhHgCl seems to be
less reactive towards the action of Cl
2 than the corresponding PhHgBr and PhHgI towards the action of Br
2 and I
2. A concentrated solution of hypochlorous acid acts like free Cl
2, forming PhHgCl and PhCl from Ph
2Hg. An aqueous solution of PhHgOCOCH
3 heated with an excess of I
2 gives PhI, HgI
2 and CH
3CO
2H [
51].
Later on, the iodo- and the bromo-demercuration reactions have vastly been
improved [
24,
25,
26,
27,
28,
34,
52], e.g. by using KI
3 or KBr
3 solutions, mostly aqueous (cf. Ref. 2), but also with a wide variety of solvents, e.g. alcohols, acetonitrile, DMF, DMSO, pyridine, dioxane or their mixtures with water. Even
permercurated arenes are readily cleaved in this way [
52] by NaI
3 in DMF at room temperature for 3-14 days, by KBr
3 aq. methanolic solutions at room temperature for 1 – 24 hours, or by Cl
2 in DMF at room temperature for 3-8 hours, giving thus the respective periodo-, perbromo-, and perchloro-arenes in good yields. It is also necessary to recall that on a prolonged heating Ph
2Hg with TeCl
2, mercuric telluride and PhCl are formed unexpectedly [
23,
53]. Iodine monochloride reacts according to the reactions: R
2Hg + 2ICl → 2RI + HgCl
2 and RHgX + ICl → RI + HgXCl [
23,
28]; the same diiodocamphor is obtained when mercurated camphor reacts with I
2 or with ICl or with IBr in benzene [
17].
ICN acts preferably as a
iodo-demercuration agent, whereas
BrCN may act
both as a
bromo-demercuration agent as well as a
cyano-demercuration agent depending on the reaction conditions (
vide infra, as well as
Section 2.2). ClCN has been reported to give
no reaction at all with some organomercurials [
17]; cf. however
Section 2.2.
It is of interest to mention that ICN and Me
2Hg in ethereal solution give MeCN at 50°C, and HgI
2 and methyl isonitrile at 110°C [
17,
23,
50]. It has also been reported [
23] that
BrCN does
not cleave the C-Hg bond, but only replaces e.g. the acetoxy group by bromine in α-acetoxymercurio-β-methoxy-β-phenylethane. Pseudohalogens, X
2 = (CNS)
2 and (CN)
2, do react with some
symmetric organomercurials, R
2Hg, giving the respective RX and RHgX compounds; we failed, however, to replace mercury atoms in the
less reactive caffeine mercurials on acting upon them with (CNS)
2 and (CN)
2 (see Ref. 2 as well as
Section 2.3). The reaction with ClN
3 undergoes similarly, viz. R
2Hg + ClN
3 → RHgN
3 + RCl, and it was applied as well with Ph
2Hg (R = C
6H
5) [
17,
24,
25,
26,
27,
28,
34].
In our next paper of the series [
7], we reported the application of several halo- and cyano-demercuration procedures to the
more reactive “model” benzene mercurials
6a,
6b and
7; cf. our results discussed in
Section 2. Thus, d
iphenylmercury, Ph
2Hg (
7), dissolved in ethanol smoothly gave only PhI (
6e) in 72% yield (purified product) on adding pure
ICN and then refluxing the mixture for 3 hours. On refluxing for 3 hours a mixture of
7 with pure
BrCN in
benzene, only PhCN (
6i) was produced in 86% yield – whereas when
7 was refluxed for 3 hours in an ethanolic solution containing
(BrCN-KBr)x, a complex of unknown structure [
47], only PhBr (
6f) was formed in 79% yield. The reaction of
7 with
ClCN, carried out under widely-varied experimental conditions, gave some
composite mixtures, which were not subjected to closer study.
By reacting
7 with large excesses of neat liquid either
S2Cl2 or
SO2Cl2 [
SCl2 was not studied], and leaving overnight at room temperature, we obtained pure PhCl (
6g) in 78% or 47% yields, respectively. The former reaction was also carried out at first at -10°C, and next it was left overnight at room temperature: we obtained PhCl (
6g) in 35% yield together with 30% of isolated PhHgCl (which throws some light on its mechanism). The second reaction was considerably accelerated by a Friedel-Craft catalyst, viz. AlCl
3. Both the symmetric mercurial
7 as well as the two unsymmetric ones,
6a and
6b,
did react with an excess of neat S
2Cl
2 (under the same experimental conditions) to afford PhCl (
6g) in 78%, 50% and 69% yields, respectively; previously (
Section 2), it was supposed that this
new chloro-demercuration procedure was characteristic only of the symmetric organomercurials.
The
novel fluoro-demercuration procedure, applicable only for the
symmetric organomercurial
7, was reported in our other paper [
12]; cf. also
Section 2.1. On reacting
7 with a large excess of neat liquid SF
4, for 8 hours at –(60-70°C), it was possible to obtain PhF (
6h; purified product) in 58% yield; its structure was confirmed by microanalyses and its
1H-NMR spectrum. In this publication we made the following remark (footnote on p. 26): “The calculated yields [for ArF] correspond to ”ideal” reactions: Ar
2Hg + SF
4→ HgF
2 + 2ArF (soluble in organic solvents; often volatile), whereas the “real” reactions probably are
terminated either in full or in part at the
intermediate stage yielding ArF + ArHgF (sparingly soluble in organic solvents; nonvolatile). Since the aim of our investigation [
12] was to obtain the possible highest yields of ArF, therefore we did
not scrutinize the nonextractable residues composed of mercuric salts heavily contaminated by some sulfur-containing side-products and, probably, by the
less-reactive (under the given low-temperature conditions) fluoromercurials, ArHgF.“ In fact, all our fluoro-demercuration procedures, applicable only for Ar
2Hg mercurials, resulted in
moderate yields for the purified ArF, viz. 28-58% [
12].
In our next work [
11] we attempted to mercurate
benzene and caffeine by means of
mercury(II) trichloroacetate; and we submitted therein
a detailed review of the prior attempts to prepare this
thermally unstable mercuric salt. We have come to the conclusion that it is reasonable to prepare this mercuric salt
in situ to obtain appropriate mercurating reaction mixtures. When we suspended yellow HgO in thiophene-free benzene containing Cl
3CCO
2H (a 1:1 molar proportion of Cl
3CCO
2H to HgO seems to be optimal) and the reaction mixture thus obtained was heated for ca one hour at 65-70°C, then we unexpectedly isolated PhHgCl (
6d) in 70% yield (purified product),
instead of the thermolabile mercurial
6c. This method was quite unsuitable for the mercuration of less reactive caffeine. Next, mercurial
6d was readily
iodo-demercurated to give
6e (purified product) in 59% yield, with a hot (80°C) aq. KI
3 solution, for 30 minutes.
4. “Model” 2-Substituted Furan and Thiophene Mercurials and Their Demercuration Reactions [5]
In order to extend the scope of the aforementioned (
Section 2 and
Section 3)
novel chloro-demercuration procedure [the action of neat, liquid
S2Cl2 on mostly
symmetric organomercurials, yielding effectively the corresponding chloro derivatives, ArCl; see however
Section 3], we first synthesized (by the known methods)
2,2'-difurylmercury (
8) and
2,2'-dithienylmercury (
9) (
Scheme 5).
We had expected in advance that they would be
more reactive than those symmetric organomercurials discussed in
Section 2 and
Section 3. However, in our attempted chloro-demercuration reactions we had to use only
freshly-redistilled S2Cl2 to remove the accompanying
SCl2; the latter is known [
54] to equilibrate as follows: 2SCl
2 ![Molecules 06 00927 i001]()
S
2Cl
2 + Cl
2, which is very inconvenient due to the known high sensitivity of furan and thiophene towards the action of free dichlorine; see also our paper [
5] where various chlorinating procedures applicable for furan and thiophene were briefly reviewed, with the relevant references.
At first, we reacted solid finely-powdered mercurials
8 or
9 as well as
2-(chloromercurio)furan or
2-(chloromercurio)thiophene with an excess, as previously (see
Section 2 and
Section 3), of
neat liquid
S2Cl2 with
no solvent, over a wide temperature range from -70°C up to room temperature. Even at -70°C the reactions were
extremely vigorous and could hardly be controlled. Colorless oils, in nearly quantitative yields, were isolated from the reaction mixtures, which were composite mixtures of several highly chlorinated compounds, in part also of open-ring structures, with only small admixtures of 2-monochloro and 2,5-dichloro derivatives of furan or thiophene, respectively. After several attempts we achieved a proper and effective method of
chloro-demercuration mercurials
8 and
9, but only using
carbon disulfide as an
inert diluent – the reactions were completed after two days either at room temperature with
more reactive 8 or, with
less reactive 9, on boiling under a reflux condenser; with still
less reactive 2-(chloromercurio)furan or 2-(chloromercurio)thiophene the reactions did
not proceed under the same experimental conditions. From the reaction mixtures we isolated either pure
2-chlorofuran (
10a) in 60% yield or pure
2-chlorothiophene (
11a) in 70% yield, with
no trace of higher-boiling 2,5-dichlorinated admixtures. Later on, we
considerably simplified the syntheses of compounds
10a and
11a just by mixing together pure furan or thiophene with HgCl
2 previously dissolved in an excess of the freshly-redistilled S
2Cl
2, followed by keeping the mixtures overnight at room temperature. Most likely, the reactions proceeded via some 2-mercurio intermediates, since no 2-chloro derivatives were formed in the
absence of HgCl
2. From the reaction mixtures we isolated pure compounds
10a or
11a in 50% or 60% yields, respectively; see Ref. 5 for experimental details.
The various methods for the
direct iodination of furan and thiophene (and also other heterocycles) were very extensively reviewed in Ref. 55; they were also briefly reviewed in our paper [
5]. We reacted
symmetric mercurials
8 and
9 with the well-known [
23,
26,
28,
34] iodo-demercuration agent, viz. an aq. KI
3 solution, for 30 minutes at 80°C, which gave pure
10b or
11b in 60% or 65% yields, respectively. These yields are considerably
higher than those previously reported, when the same iodo-demercuration method was applied with either 2-(chloromercurio)furan (32% yield was reported [
56]) or 2-(chloromercurio)thiophene (a low yield was reported [
57]). Our
iodo-demercuration reactions confirmed the structures of the starting mercurials
8 and
9 as well as their
higher reactivities as compared with the respective 2-chloromercurio derivatives.
In our paper [
5] we offered the following
general remark (footnote on p. 445): “It is rather a common procedure that after completing several direct mercuration reactions with more electrophilic (than HgCl
2) mercuric acetate or trifluoroacetate, the
less soluble, but least reactive,
chloromercurio derivatives are precipitated out with aq. NaCl or CaCl
2 solutions, in order to increase the
isolated yields of desirable mercurials. In our opinion, it is often more advantageous to collect the respective
symmetric mercurials (usually less soluble as well), which may be obtained by
subsequent addition – if possible – of symmetrizing agents (e.g. KI, Na
2S
2O
3, etc.) directly to the said reaction mixtures, and then to complete the symmetrization reaction in order to obtain the resulting,
more reactive Ar-Hg-Ar compounds”. In fact, we applied this approach
in practice in our works [
6] and [
9]; see
Section 7 and
Section 8 for details.
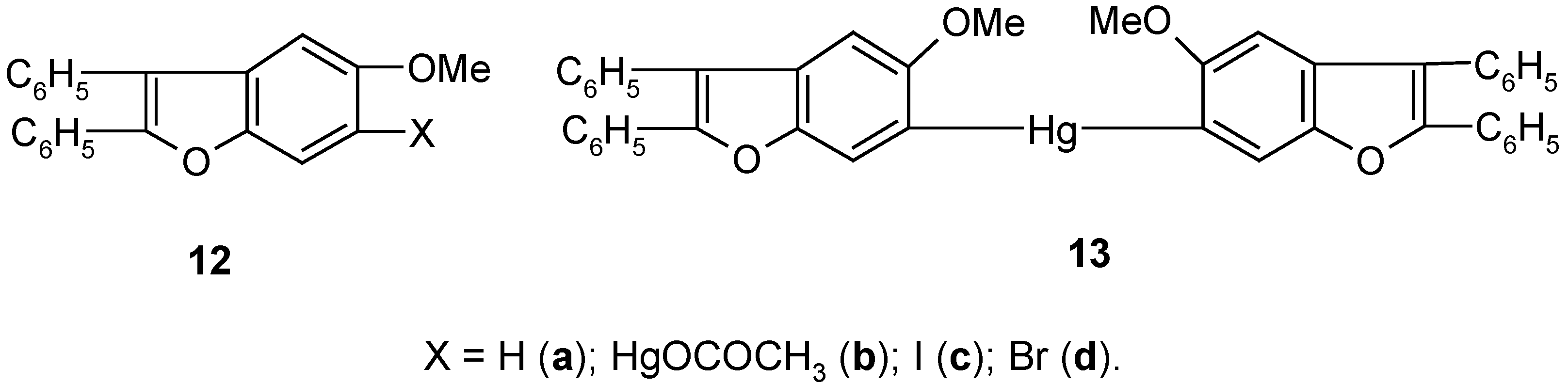
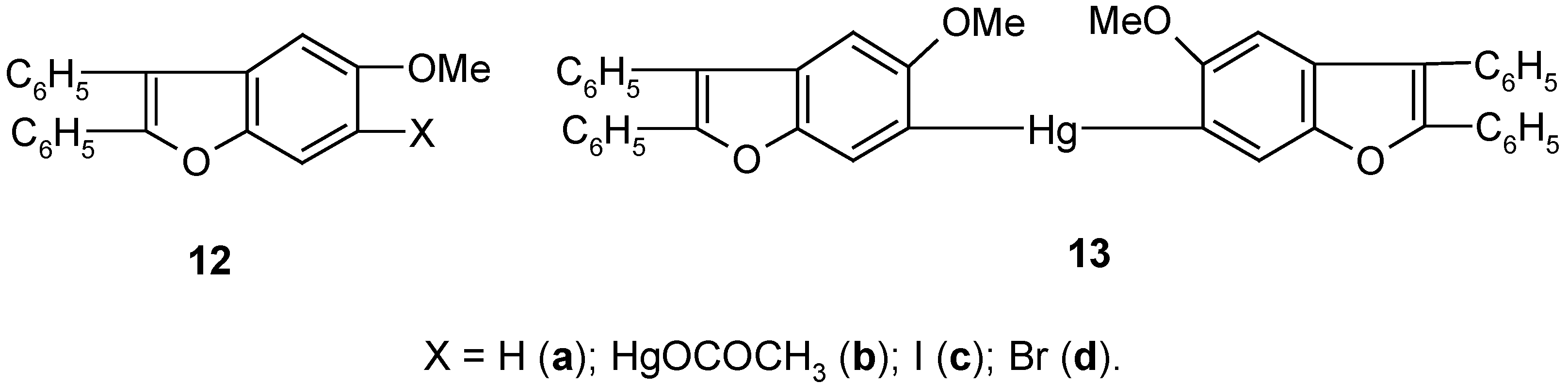
5. 6-Substituted 2,3-Diphenyl-5-methoxybenzo[b]furan Mercurials and Their Demercuration Reactions [10]
Egyptian chemists [
58] had synthesized a number of derivatives of heterocyclic compound
12a, and later they studied their biological activities. They established that in various reactions obeying the S
E-type mechanism, the respective derivatives of
12a were predominantly substituted at its C
6 carbon atom. However, bromination of
12a with Br
2 in CCl
4 gave the 4,6-dibromo derivative of
12a. In co-operation with the Egyptian chemists we decided to synthesize in our laboratory the two mercurials
12b and
13 (
Scheme 6), and then study their bromo- and iodo-demercuration reactions.
The direct mercuration of 12a with Hg(OCOCH3)2 in boiling ethanol, for 2 hours, gave unsymmetric mercurial 12b in ca 70% crude yield, which next was readily symmetrized by a hot (90°C) KI/EtOH/H2O solution, for 2 hours, to afford symmetric mercurial 13 in 60% crude yield. Further proof of the structures of 12b and 13 was given by their routine bromo- and iodo-demercuration reactions, carried out for ca 30 minutes in hot (80°C) aq. KBr3 or KI3 solutions, respectively. These two reactions furnished the respective 6-bromo or 6-iodo derivatives of 12a, i.e. pure compounds 12c and 12d, in 70% yield, the same for the two mercurials 12b and 13, and the same for the two different reactions.
The above syntheses of the mercurials
12b and
13 may open up an easy way for preparing many other 6-substituted derivatives of
12a (e.g.
12, X = F, Cl, CN) by the well-checked by us demercuration procedures explained in
Section 2,
Section 3 and
Section 4, or otherwise. It would also be possible to prepare readily from
12b and/or
13 many metallo- and metalloido-organic derivatives of compound
12a, by using known [
23,
26,
28,
34] methods applicable for ArHgX and/or Ar
2Hg mercurials.
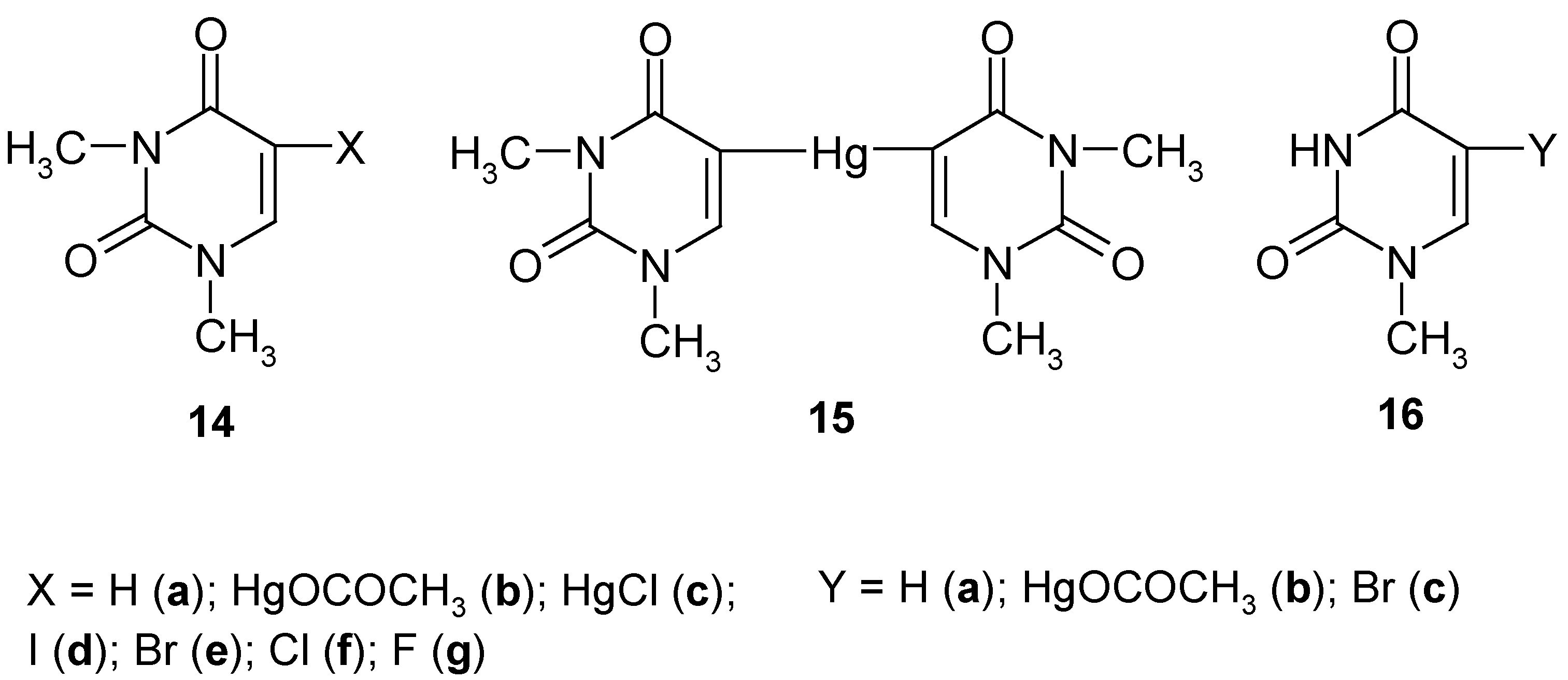
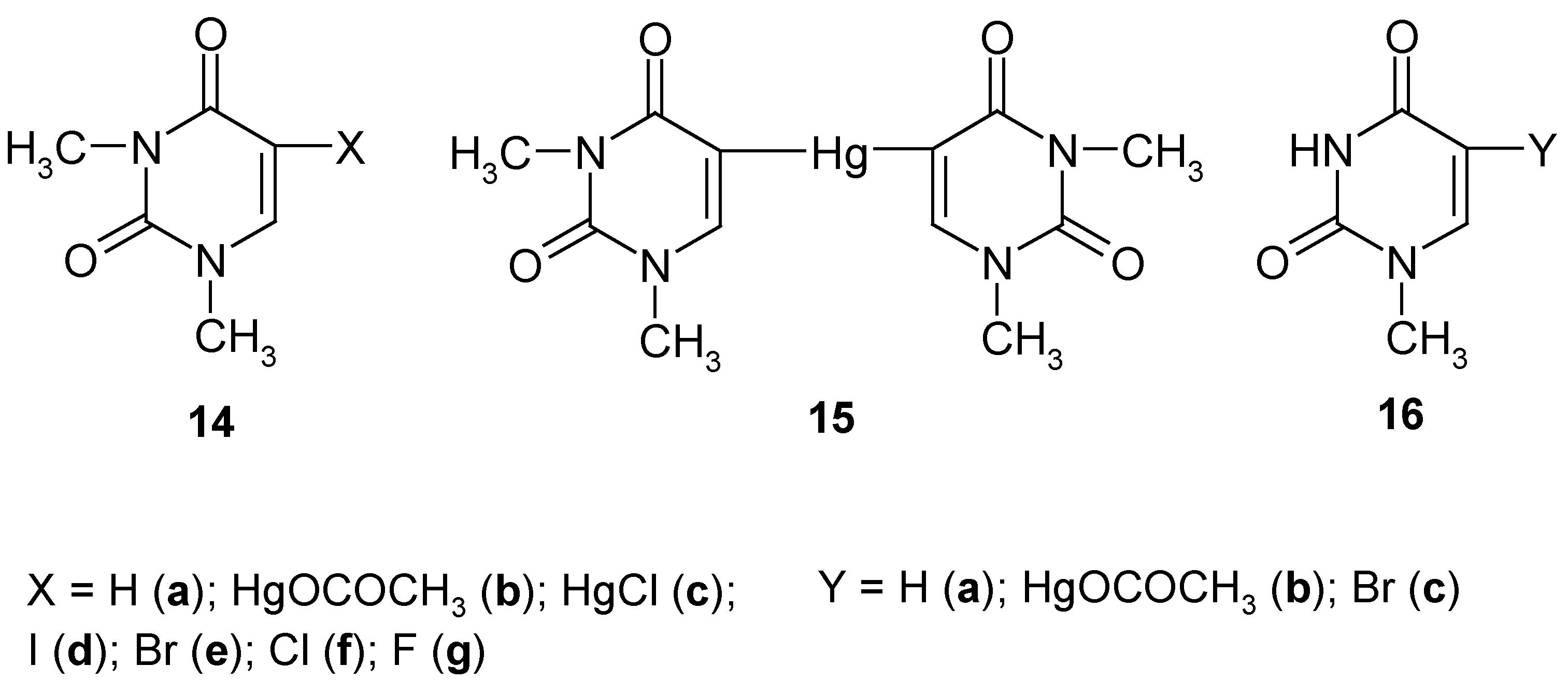
6. 5-Substituted 1,3-Dimethyluracil and 1-Methyluracil Mercurials and Their Demercuration Reactions [6, 8 and 12]
Uracil [pyrimidine-2,4(1
H,3
H)-dione] cannot be directly C-mercurated, since it forms at once a sparingly soluble 1:1 mercury complex (N-Hg salt; formula
1 in
Section 1) [
59] (see however Ref. 60, which will be discussed in
Section 8). Contrariwise,
1,3-dimethyluracil (
14a, Scheme 7) can be readily directly C-mercurated in its 5-position.
We applied routine [
23,
24,
26,
28,
34] mercurating procedures by reacting
14a either with a boiling aq. Hg(OCOCH
3)
2 solution containing CH
3CO
2H, for 4 hours (which gave pure mercurial
14b in 52% yield), or with a boiling buffered C
2H
5OH/H
2O solution of HgCl
2, for 4 hours, which gave pure mercurial
14c in 64% yield. The same compound
14c was also obtained in 45% yield on adding NaCl to a hot aq. solution of
14b. Subsequently, we prepared
symmetric pure mercurial
15 by reacting
14b with a hot boiling aq. KI solution, for 0.5 hour, in 89% yield, after recrystallization from water.
Subsequently, continuing our previous studies related in
Section 2,
Section 3 and
Section 4, we carried out the following chemical experiments on
halo-demercuration of the 1,3- dimethyluracil mercurials
14b and/or
15, viz.
- i)
on reacting 14b or 15 with hot aq. KI3 solutions, we obtained known pure 5-iodo-1,3-dimethyl-uracil (14d) in 88% or 83% yields, respectively;
- ii)
on reacting 14b or 15 with hot (80°C) aq. KBr3 solutions (adjusted in advance to pH = 7), we obtained known pure 5-bromo-1,3-dimethyluracil (14e) in the same 82% yield;
- iii)
on reacting only dry 15 with a large excess of pure liquid S2Cl2, we obtained known pure 5-chloro-1,3-dimethyluracil (14f) in 74% yield;
- iv)
on reacting only dry
15 with a large excess of neat liquefied
SF4 for 48 hours at -(60-70
oC), and following workup explained in Ref. 12, pure compound
14g was produced in 30.3% yield [
12].
So far, there is no easy method of removing N-methyl group(s) from the uracil ring system. Therefore, there is (so far) no easy way of transforming compounds
14d-g, into the respective non-methylated analogues. See however
Section 7, where such the demethylation was possible for 5-substituted 2,4-dimethoxypyrimidines.
Uracil has two N-H groups, which differ chemically and otherwise, e.g. at 25°C [
61]: pK
a (1-N-H) = 9.43; pK'
a (3-N-H) ca. 13.2 (
estimate). In order to check whether or not the 1-N-H group alone must be blocked to accomplish a successful direct mercuration in position 5, we synthesized
1-methyluracil (
16a), and we subsequently carried out its routine mercuration with a boiling, slightly acidified with CH
3CO
2H, aq. Hg(OCOCH
3)
2 solution, for 3.5 hours, which resulted in the formation of mercurial
16b (purified) in 49% yield; by its bromo-demercuration, similarly to that of
14b, we obtained
5-bromo-1-methyluracil (purified) in 78% yield. In fact, also other 1-N-substituted uracils, e.g. uridine [
62] and 2'-deoxyuridine [
63] were readily mercurated in their positions 5 with buffered aq. Hg(OCOCH
3)
2 solutions.
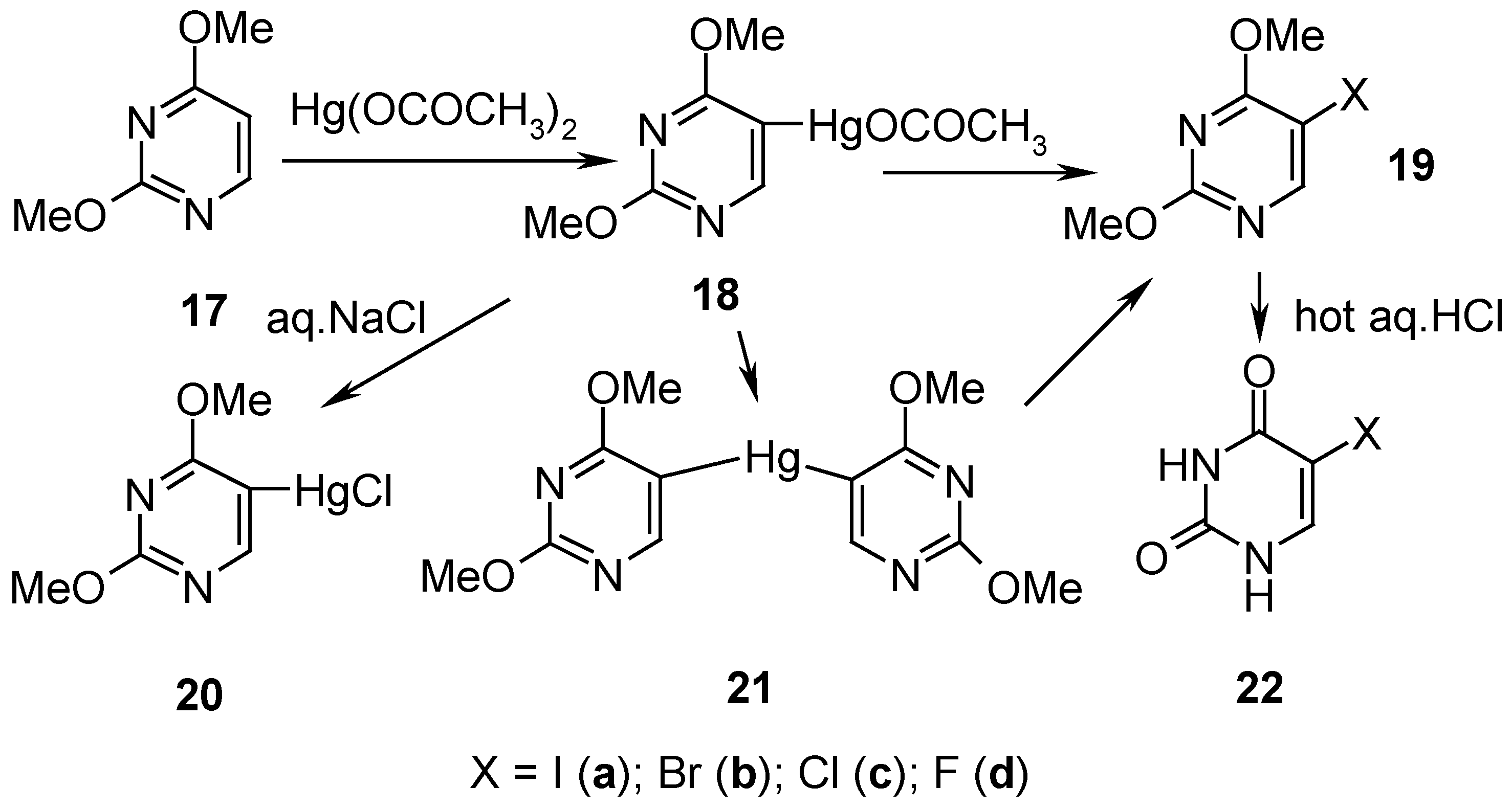
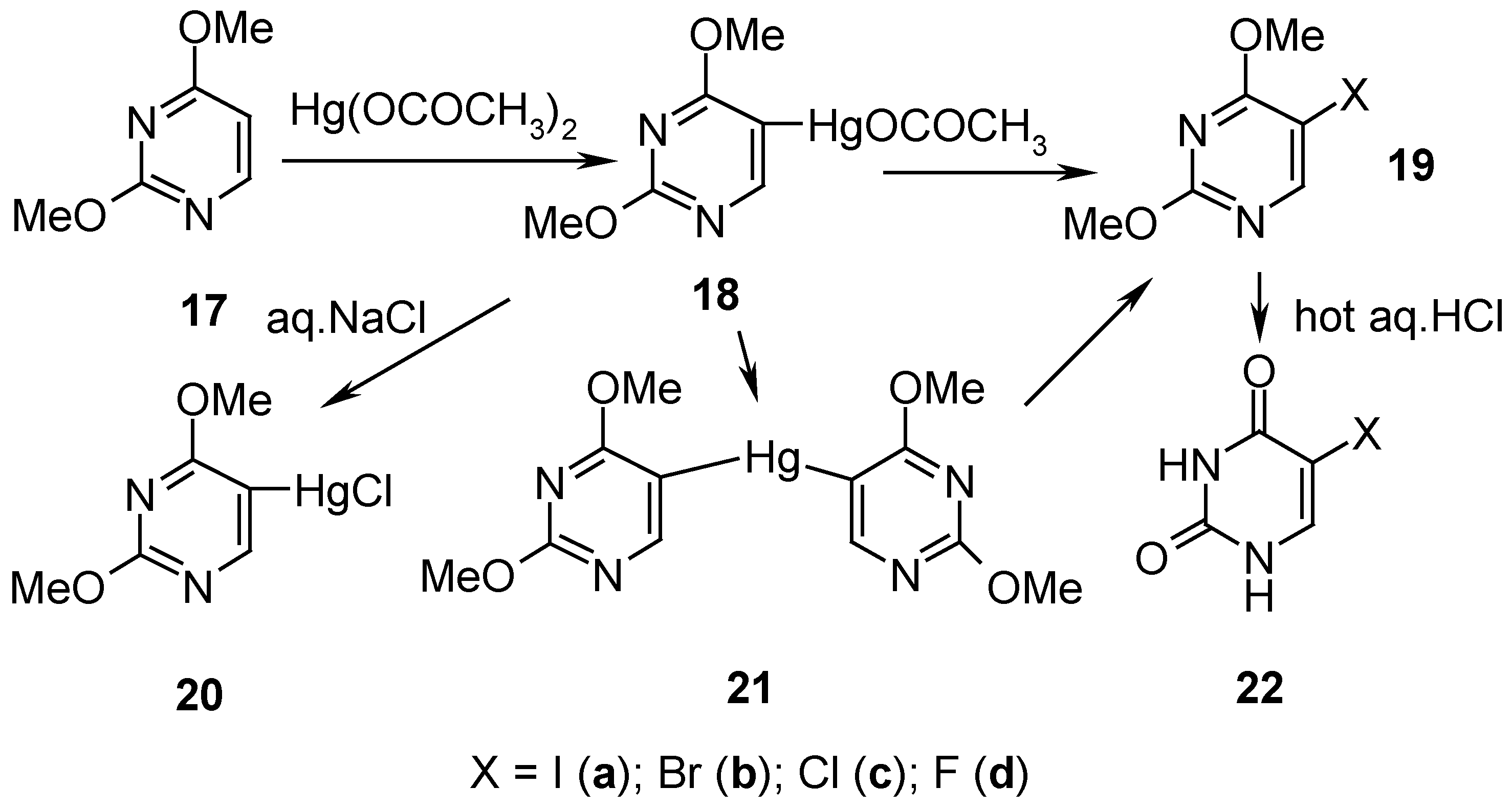
7. 5-Substituted 2,4-Dimethoxypyrimidine Mercurials and Their Demercuration Reactions as well as an Indirect Method for Preparing 5-Halogeno-substituted Uracils [6, 9 and 12]
2,4-Dimethoxypyrimidine (
17, Scheme 8) was readily mercurated with a boiling aq. Hg(OCOCH
3)
2 solution acidified with CH
3CO
2H, for 2 hours, and this
hot solution containing 18 was applied at once in subsequent reactions (
vide infra) [only a small isolated sample of
18 was recrystallized from ethanol, and its
1H-NMR spectrum was run for identification purposes].
By adding an aq. NaCl solution to the aforementioned hot solution containing
18, the
metathesized, insoluble mercurial
20 was precipitated out and collected to give
48% crude yield (
Scheme 8). By adding dropwise a nearly saturated aq. KI solution to the afore-said hot solution containing
18, and refluxing for 0.5 hour, the
symmetrized mercurial
21 (recrystallized from water) was isolated in
79% yield.
Similarly, an appropriate amount of I
2 was added to the boiling aforementioned solution containing
18, which resulted (after cooling) in the isolation of iodo derivative
19a in 58% yield (purified); alternatively, the same compound
19a (purified) was obtained from solid organomercurial
21 by its
iodo-demercuration with a saturated hot (60°C) aq. KI
3 solution, for 45 minutes, which resulted in 52% yield of
19a.
Bromo-demercuration of
18 was attained by adding a
neutralized aq. KBr
3 solution to the afore-said solution containing
18, and next the reaction was carried out at 60-80°C for 45 minutes – isolation of bromo derivative
19b (purified) resulted in 51% yield; also solid organomercurial
21 was similarly
bromo-demercurated to give
19b (purified) in 55% yield. Solid organomercurial
21 was
chloro-demercurated with excess of
freshly redistilled liquid
S2Cl2 (to avoid the undesirable presence of SCl
2; see
Section 4 for the explanation). The reaction transcurred for 8 hours at 20°C, and the reaction mixture was then left overnight – after its two-step workup, pure
5-chloro-2,4-dimethoxypyrimidine (
19c) was produced in 49% yield. On reacting
symmetric mercurial
21 with a large excess of
neat liquefied
SF4 at -(60-70) for 48 hours and the following workup explained in Ref. 12, pure compound
19d was obtained in 32.6 % yield.
5-Halogeno-2,4-dimethoxypyrimidines 19a, 19b, 19c, and
19d, were readily
demethylated by adding them to a 10% aq. hydrochloric acid (used in a large excess), and by evaporating the solutions to dryness on a boiling water bath; the reactions were accompanied by
vigorous foaming. The residues were recrystallized to give pure
5-halogeno-substituted uracils 22a, 22b, 22c, and
22d in 69%, 83%, 69% and 81% [
12] yields, respectively. The title method is called
indirect, because compound
17 is prepared from
uracil, converted first by POCl
3 into 2,4-dichloropyrimidine, which next is reacted upon with CH
3ONa, yielding finally compound
17 [
64]. In reverse, also the direct
demethylation of 2,4-dimethoxypyrimidine (
17) was easily accomplished on heating
17 with hot hydrochloric acid [
65].
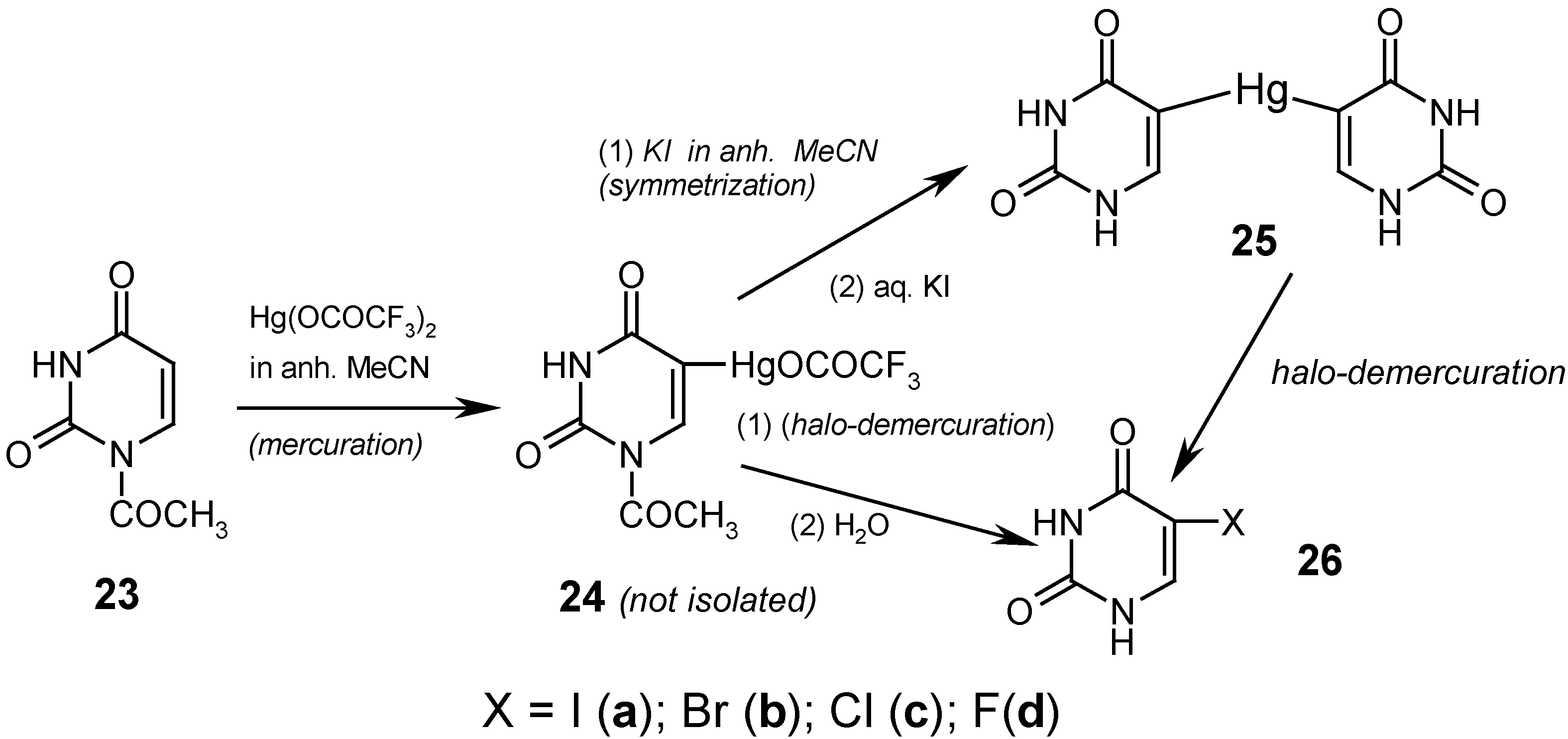
8. 5-Substituted Uracil Mercurials and Their Demercuration Reactions [4, 6, 9, 12 and 14]
In
Section 6 we reported that 1-methyluracil (
16a) may be readily C-mercurated to give 5-(acetoxymercurio)-1-methyluracil (
16b) which next was bromo-demercurated to give 5-bromo-1-methyluracil (
16c). There is (so far) no easy and effective method for removing N-methyl group(s) in the uracil ring system, hence it is not possible to obtain e.g. 5-bromouracil (
26b) from compound
16c. However, it is known [
66] that
1-acetyluracil (
23) is readily
deacetylated even by cold water to form the initial uracil. We made numerous attempts to mercurate
23 in various
anhydrous solvents with the less reactive mercurating agents, viz. HgCl
2 or Hg(OCOCH
3)
2 (
Scheme 9). On prolonged heating only the sparingly soluble N-Hg salt of uracil (formula
1 in
Section 1) was precipitated out in excellent yields, probably with a negligible C-mercuration in position 5.
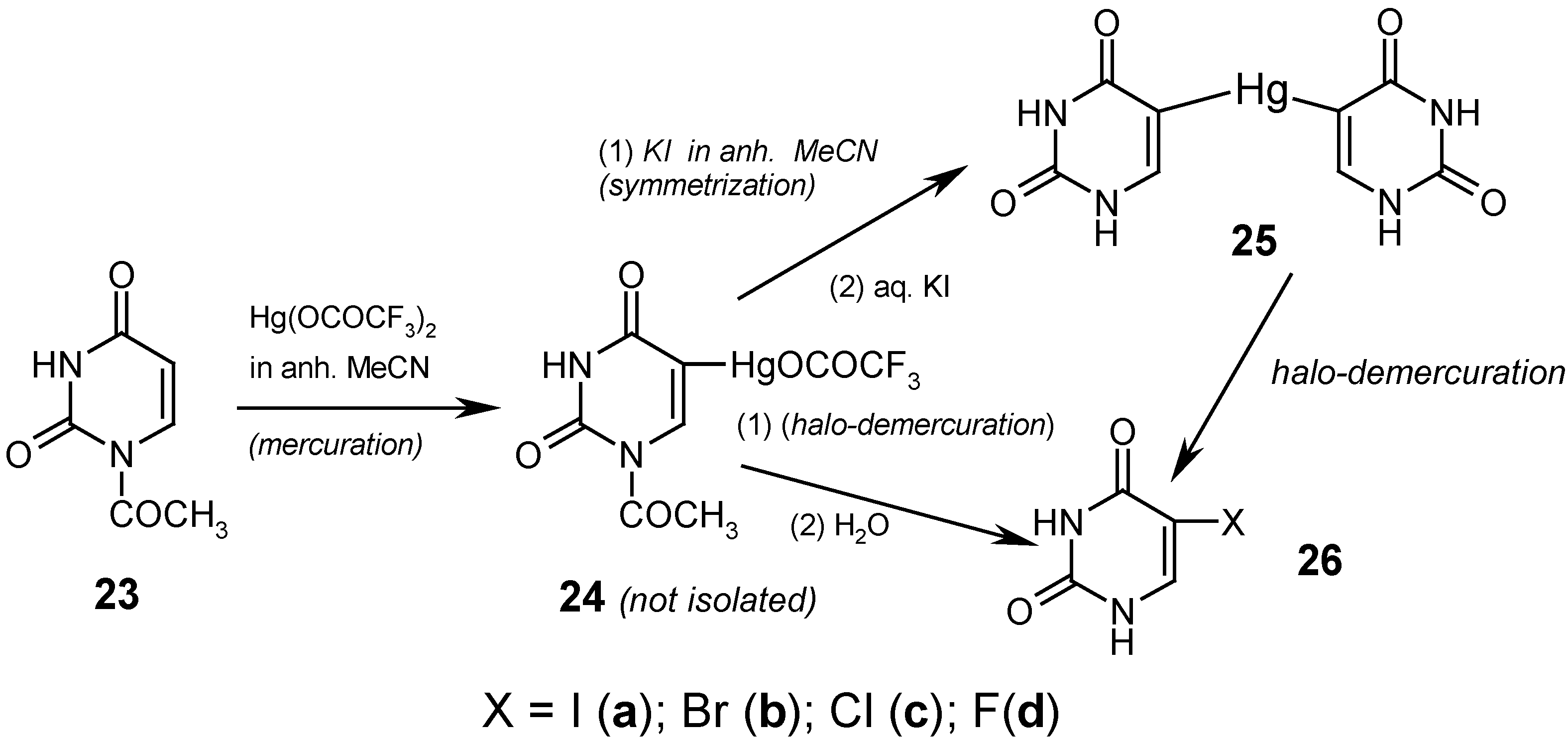
Finally, we effectively C5-mercurated 23 in boiling anhydrous acetonitrile, for 10 hours, but only with the strongly electrophilic Hg(OCOCF3)2. We did not isolate the intermediate mercurial 24, but rather its yellowish, clear hot solutions were applied as such in the following reactions (vide infra); when the same mercuration was carried out, quite similarly, in CD3CN, then the presence of the soluble mercurial 24 was confirmed by 1H-NMR spectroscopy.
If we prolonged for more than ten hours the above mercuration reaction, a white solid was precipitated out in still increasing amounts, and it represented the N-Hg salt 1, but substituted with the HgOCOCF3 group in its position 5 (which was confirmed chemically, spectrally, and in part, analytically). The desirable 5,5'-mercuriobis(uracil) (25) was obtained in 50% crude yield by adding excess KI dissolved in dry acetonitrile to the afore-said yellowish solution of 24. This mixture was refluxed for 4 hours, cooled, and filtered. The collected white precipitate was heated for 30 minutes at 60°C with a saturated aq. KI solution [which also split off the 1-N-acetyl groups] and after cooling, the collected white precipitate was washed well with boiling water, dried, and analyzed. Its structure was confirmed by the routine iodo-demercuration reaction with a hot aq. KI3 solution to give known compound 26a (purified) in 93% yield; see Ref. 4 for details. Alternatively, the same iodo-demercuration reaction was carried out but with using the aforementioned yellowish boiling solution of 24, which was treated at first with diiodine and next the collected white precipitate was heated for 30 minutes, at 600C, with an aq. KI solution to give finally 26a (purified) in 60% yield.
The respective routine
bromo-demercuration reactions with hot aq. KBr
3 solutions (prior adjusted to pH = 7) of either solid
25 or
24 (in its CH
3CN solution) were reported elsewhere [
9], and gave known purified
5-bromouracil (
26b) in 84% and 77% yields, respectively. The above results confirm a somewhat
lesser reactivity of unsymmetric mercurial
24 as compared with symmetric mercurial
25.
The
chloro-demercuration of symmetric mercurial
25 was reported in our paper [
4]. Solid
25 was slowly added to the
freshly–redistilled S2Cl2 (used in excess), and this was left overnight at room temperature. The collected precipitate was washed with dry CH
3CN, recrystallized from ethanol to give known purified
5-chlorouracil (
26c) in 65% yield.
The
novel fluoro-demercuration procedure, presented in Ref. 12, gave
5-fluorouracil (
26d) in 27.1% yield, by reacting
symmetric mercurial
25 with a large excess of
neat liquid
SF4 for 48 hours at ca -60
0C, and the subsequent workup; see
Section 7 for another method of preparing compound
26d.
Note. The
electron-donating groups, viz. the methyl or methoxy groups in caffeine, 2,3-diphenyl-5-methoxybenzo[b]furan, 1,3-dimethyluracil and 1-methyluracil, and 2,4-dimethoxypyrimidine do
increase more or less an electron density in the substituted parent heterocyclic systems, facilitating thus their
direct mercuration even with
less electrophilic mercuric salts, viz. mercuric acetate, sometimes also with HgCl
2 in its buffered aq. solutions. In contrast, any
electron-withdrawing N-acyl groups (e.g. the acetyl, trichloroacetyl or trifluoroacetyl groups), which were introduced by us into the uracil, theophilline or theobromine parent ring systems, render considerably more difficult their direct mercurations with Hg(OCOCH
3)
2, and completely eliminate the use of HgCl
2. It is why we had to use the
strongly electrophilic Hg(OCOCF3)2, usually prior prepared
in situ, to effectively mercurate N-acylated uracil (
Section 8), theophilline (
Section 9), and theobromine (
Section 10) in
anhydrous solvents to obtain possibly highest yields of the desired C-substituted
unsymmetric mercurials.
Visser et al. [
60] have succeded to synthesize
microquantities of radioactive 5-X-uracils (X =
211At or
131I) by reacting 45 μmol of
uracil, dissolved in 1 ml of 0.2 M aq. H
2SO
4, with HgSO
4 (40 μmol), for 3 hours at room temperature, followed with NaCl (90 μmol).
Without isolating the intermediate
5-(chloromercurio)uracil, they added subsequently 0.9 equivalent of
211At/I
2 or
131I/I
2, which resulted in very good
radiochemical yields of final radioactive products. Similar approaches were used, with varying reaction times and temperatures, e.g. for imidazole, thyrosine, phenylalanine, etc. We
scaled up [
14] the aforementioned procedure a
thousandfold (i.e. to the
millimolar scale) as to
directly C
5-mercurate
uracil as well as, for the sake of comparison, 2-thiouracil and theobromine. Only
uracil gave
5-(chloromercurio)uracil in 85% crude yield; its following routine
iodo-demercuration with a hot (80°C) aq. KI
3 solution led to
5-iodouracil in ca 61% crude yield. All our attempts to symmetrize 5-(chloromercurio)uracil by means of hot aq. KI, Na
2S
2O
3 or KSCN solutions as well as by a methanolic solution of hydrazine were not successful. Therefore, the
symmetric mercurial
25 should be prepared in the way explained above. Theobromine reacted as above [
60] but in the millimolar scale, furnished
1-N-(chloromercurio)theobromine in ca 77% crude yield, which being iodo-demercurated gave the initial theobromine in ca 72% yield, with no detectable amount of the expected 8‑iodotheobromine; see also
Section 12. Similarly, 2-thiouracil reacted as above [
60] but on a millimolar scale, forming nearly quantitatively an insoluble crude mercurial, which after its demercuration with an aq. KI solution, yielded solely the initial 2-thiouracil with no detectable amount of any C-mercurated product. We
concluded [
14] the above experiments as follows: “the direct mercuration procedure offered in
Ref. 60 should always be tried in the future, since it is relatively simple and less hazardous than the other ones (cf.
Section 11), though it is
less general than it has been expected and wanted”.
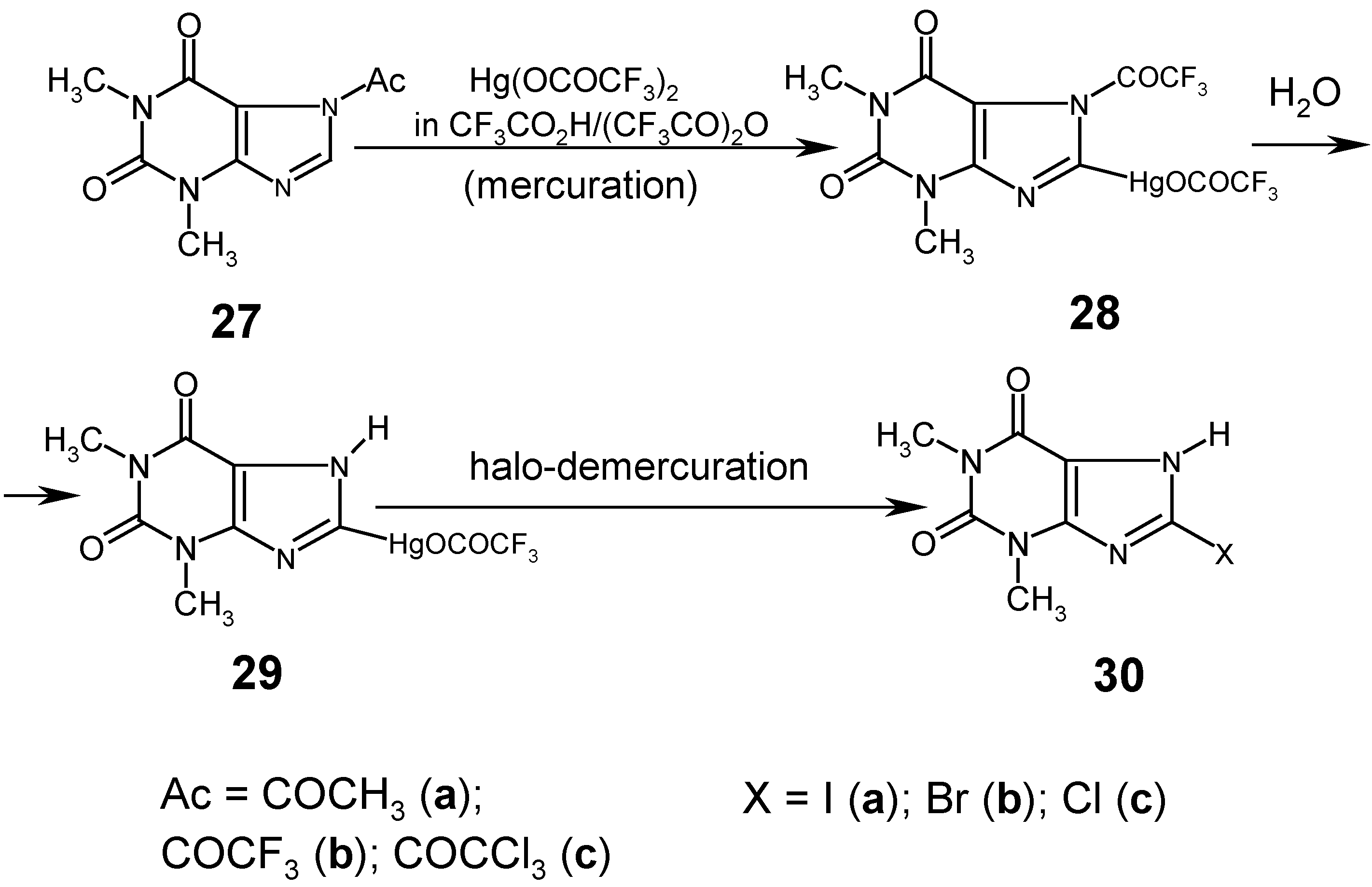
9. 8-Substituted Theophylline Mercurials and Their Demercuration Reactions [3, 4]
When
theophylline as well as theobromine and uracil, all having
acidic N-H groups, are reacted with Hg(II) salts, then their sparingly soluble N-Hg salts (see formulae
1 – 3 in
Section 1) are immediately precipitated out from the mercurating solutions [
63,
66], and their effective C-mercuration cannot be performed; see
Section 8, where this topic is discussed. N-Acetyl derivatives of theophylline and theobromine [
67], are
easily hydrolyzable by the action of water, likewise as does
1‑acetyluracil (
Section 8).
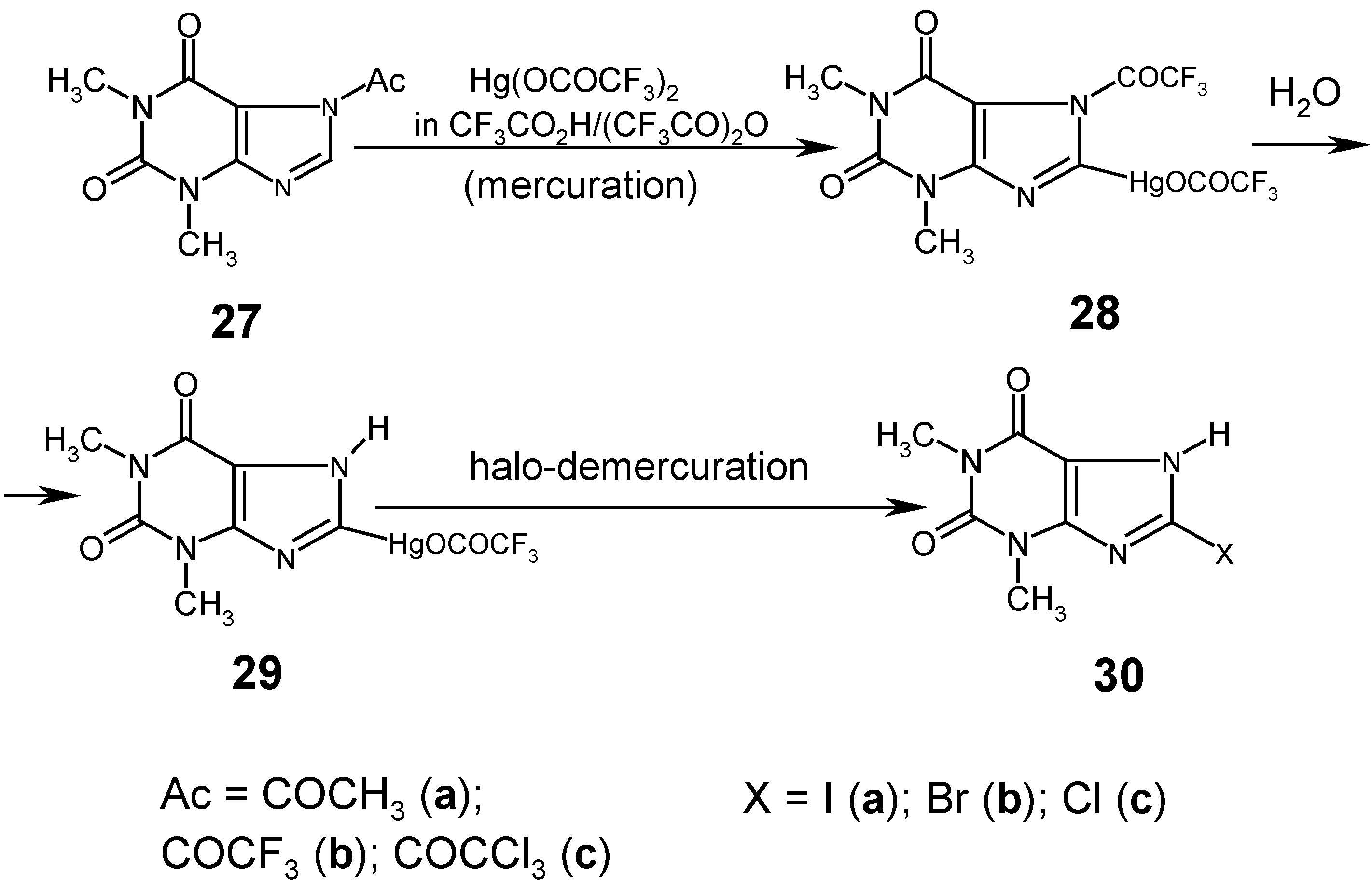
We made many attempts to mercurate
7-acetyltheophylline (
27a) in various
anhydrous media, e.g. in CH
3CO
2H/(CH
3CO)
2O mixtures, with
less reactive mercurating agents, viz. HgCl
2 or Hg(OCOCH
3)
2, but all our experiments were unsuccessful for the same reasons as those explained for 1-acetyluracil (
Section 8). We succeeded in C-mercurating the theophylline system in its 8-position (which is somewhat more reactive than the same position in theobromine and caffeine), but only by using the
strongly electrophilic Hg(OCOCF3)2 dissolved in boiling
anhydrous mixtures made of CF
3CO
2H and (CF
3CO)
2O. The reaction was complete after 10 hours, the resulting reaction mixture was concentrated under dimished pressure,
water was added to the viscous residue, and a white precipitate collected was recrystallized from water to give
8-(trifluoracetoxymercurio)theophylline (
29) in 53% yield. We also
simplified the above procedure as follows. Theophylline was refluxed with (CF
3CO)
2O for 2 hours [thus,
7-(trifluoroacetyl)theophylline (
27b) was produced there
in situ], then a solution of Hg(OCOCF
3)
2 in CF
3CO
2H/(CF
3CO)
2O was added, and the same mercuration reaction was carried out under a reflux condenser for 10 hours; the subsequent workup was the same as previously described to afford mercurial
29 in 64% yield. This yield was later increased to 70%, when
7‑(trichloroacetyl)theophylline (
27c) was used as the starting substrate (Ref. 4
; see footnote on p. 386).
Next, we carried out the routine iodo- and bromo-demercuration reactions with mercurial 29 in hot aq. KI3 or KBr3 solutions (previously adjusted to pH = 7), at 80°C for 30 minutes, which gave the purified compounds 30a and 30b in 95% and 96% yields, respectively. However, our attempts to prepare 8-chlorotheophilline (30c) from dry unsymmetric mercurial 29, using pure liquid S2Cl2 or SCl2 as the chloro-demercuration agents, were unsuccessful.
11. Preparation of 1,8-bis(Acetoxydimercurio)theobromine and Its Reactions [13]
Australian chemists [
52] have prepared numerous fully mercurated (permercurated) arenes by the reaction of an excess of
molten Hg(OCOCF3)2 with suitable arenes at ca 180-245°C; the reaction temperature used depended on the reactivity of a given starting arene. The
crude (i.e.
not analyzed)
permercurated arenes were subsequently halo-demercurated to give the corresponding
perhalogenated arenes. Therefore, we expected that the same
melting chemical procedure would be suitable for the direct permercuration of several fairly stable, N-H acidic,
lactamic heterocycles, e.g. theobromine, theophylline, xanthine, hypoxanthine, uracil, etc. For our preliminary study we chose
theobromine, whose mercurials are discussed in
Section 10.
In order to accomplish the said melting chemical procedure, we intimately mixed pure theobromine with an excess of molten Hg(OCOCF3)2 (prepared in situ). A vigorous reaction was observed with the evolution of gaseous, strongly toxic products as well as tiny droplets of metallic mercury. This is why, in our opinion, the organic derivatives of mercury(I) would readily be formed in the hot reaction mixture.
Note. We submitted in our paper [
13] the following
assumption (see footnote on p. 30): Mercury
in statu nascendi (Hg*), probably formed during the thermal decomposition of Hg(OCOCF
3)
2, would, at a guess, react readily with intermediates of mercury(II), forming the resulting compounds of mercury(I), viz.
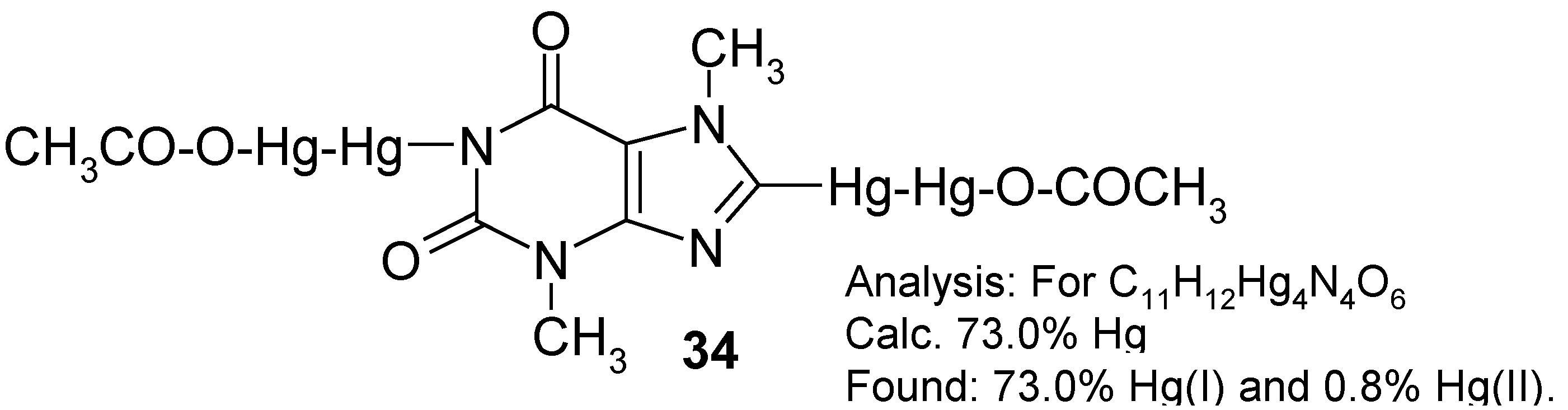
The main problem was how to properly control the reaction temperature, since the aforementioned reaction mixture thickened continuosly, until it had solidified. In hindsight, it would probably be desirable in the future to identify some suitable
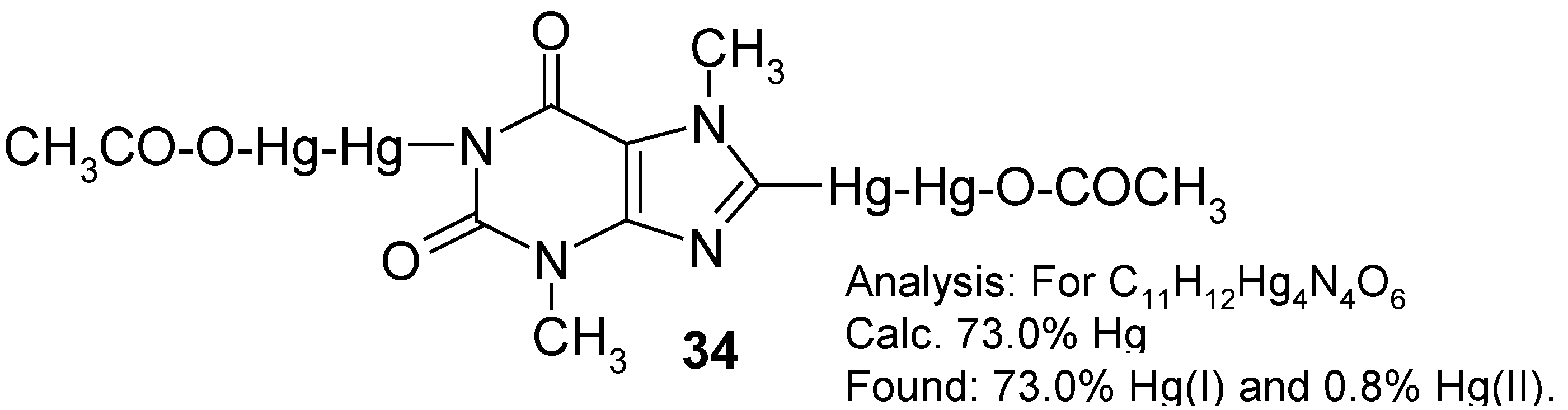
inert solvents, e.g. some highly boiling hydrocarbons or perfluorinated hydrocarbons, in order to better control the reaction. For the time being, we obtained some
crude melt, evidently containing tiny droplets of metallic mercury as well as some highly mercurated products, most likely
1,8-bis(trifluoroacetoxydimercurio)theobromine. The latter tentative opinion was further indirectly supported by the subsequent reaction.
On boiling the powdered
crude melt with glacial acetic acid – where the following
metathesis would take place: -Hg-Hg-OCOCF
3 + excess CH
3COOH → -Hg-Hg-OCOCH
3 + CF
3CO
2H – a considerably purer
yellow product was obtained (after the concentration of the CH
3CO
2H extract under reduced pressure) in
28% yield calculated with respect to the starting theobromine. Its analysis is given in the Scheme above. Its
1H‑NMR spectrum (in CF
3COOD) shows the absence of any 8-
H low-field proton signal at ca. 8.5 ppm, and the presence of two N-C
H3 three-proton signals (at 3.38 and 4.08 ppm) characteristic of theobromine [
3]. At ca 1.8 ppm there is, however, an additional and intense six-proton
singlet derived from two, apparently spectrally equivalent, C
H3COO groups. Thus, it may be guessed that the two assumed –Hg-Hg-OCOCH
3 groups are substituted in mercurial
34 in its 1 and 8 positions. The lack of any 1-N
H proton signal is insignificant, since it might have been due to a quick isotopic exchange with the deuterated solvent. Hence, the said substitution in position 1 is better explained by the full absence of any characteristic N-H absorption band over the 3070-3150 cm
-1 IR range which, in contrast, is found at 3120 cm
-1 in the comparative IR spectrum of theobromine taken also in Nujol.
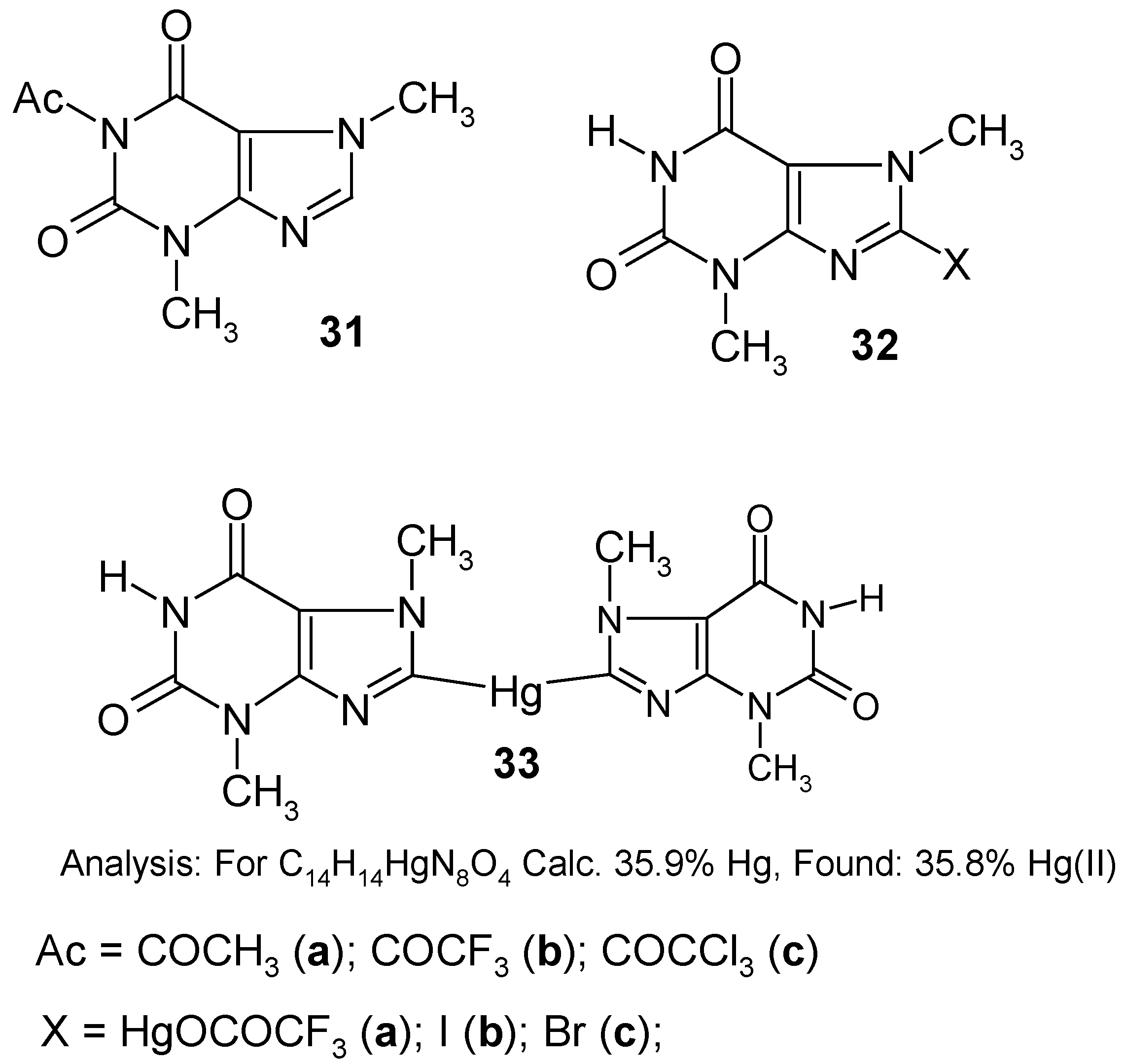
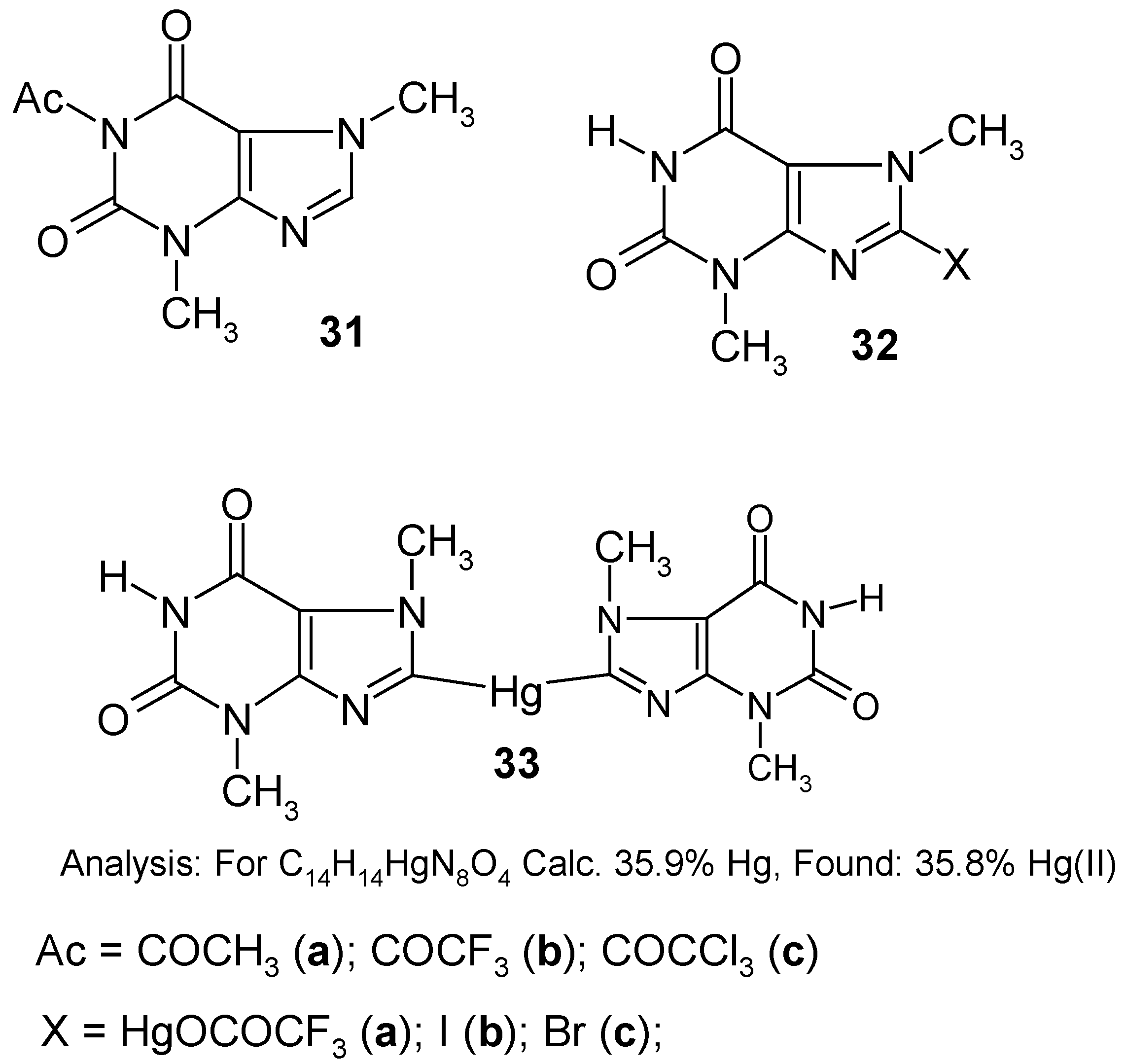
This considerably purified product
34 was used
as such in the following reactions without further purification. Compound
34 was readily
iodo- and
bromo-demercurated with either a boiling I
2 solution in dry CH
3CN, for 1.5 hours, or by an aq. KBr
3 solution, at 80°C for one hour, to give finally either
8‑iodotheobromine (
32b) (purified) in 97% yield, or
8-bromotheobromine (
32c) (purified) in 92% yield; in the course of both demercuration procedures, the (N
1)-Hg-Hg-OCOCH
3 groups are exchanged in full by the hydrogen atoms (proto-demercuration), whereas those attached to the C
8 atom of
34 are replaced by the respective halogen atoms (halo-demercuration). The same difference was observed in the course of our attempted symmetrization of compound
34 by a hot ethanolic solution of
hydrazine (the other symmetrizing agents tried were either ineffective or gave worse results). Only the C
8-substituted –Hg-Hg-OCOCH
3 groups were all engaged in the formation of
-Hg- bridging present in the formed
8,8'-mercuriobis(theobromine) (
33), which was obtained, after its purification, in 88% yield [its structure was confirmed analytically, spectrally as well as by its subsequent iodo- and bromo-demercuration reactions; see
Section 10], whereas those groups substituted in 1-N position were readily split off in favor of the hydrogen atom. All these reactions are new in the field of organic derivatives of
mercury(I) – to the best of our knowledge (
Note: Organic C-derivatives of
mercury(I) so far are rare and generally regarded as
unstable. For example, controlled electrolysis of 1,6-dibromohexane with a mercury cathode gives the dimer [Me(CH
2)
5Hg]
2 [
68]).
It is impossible not to mention that the permercurated arenes prepared by the Australian chemists [
52] had been neither purified nor analyzed, but they were immediately used as such in the subsequent halo-demercuration reactions, resulting in numerous, purified and analyzed, perhalogenated arenes. Nobody knows whether the said intermediate permercurated arenes were, in fact, the derivatives of mercury(II), mercury(I) or, possibly, were composite mixtures of both; this should be elucidated in future.
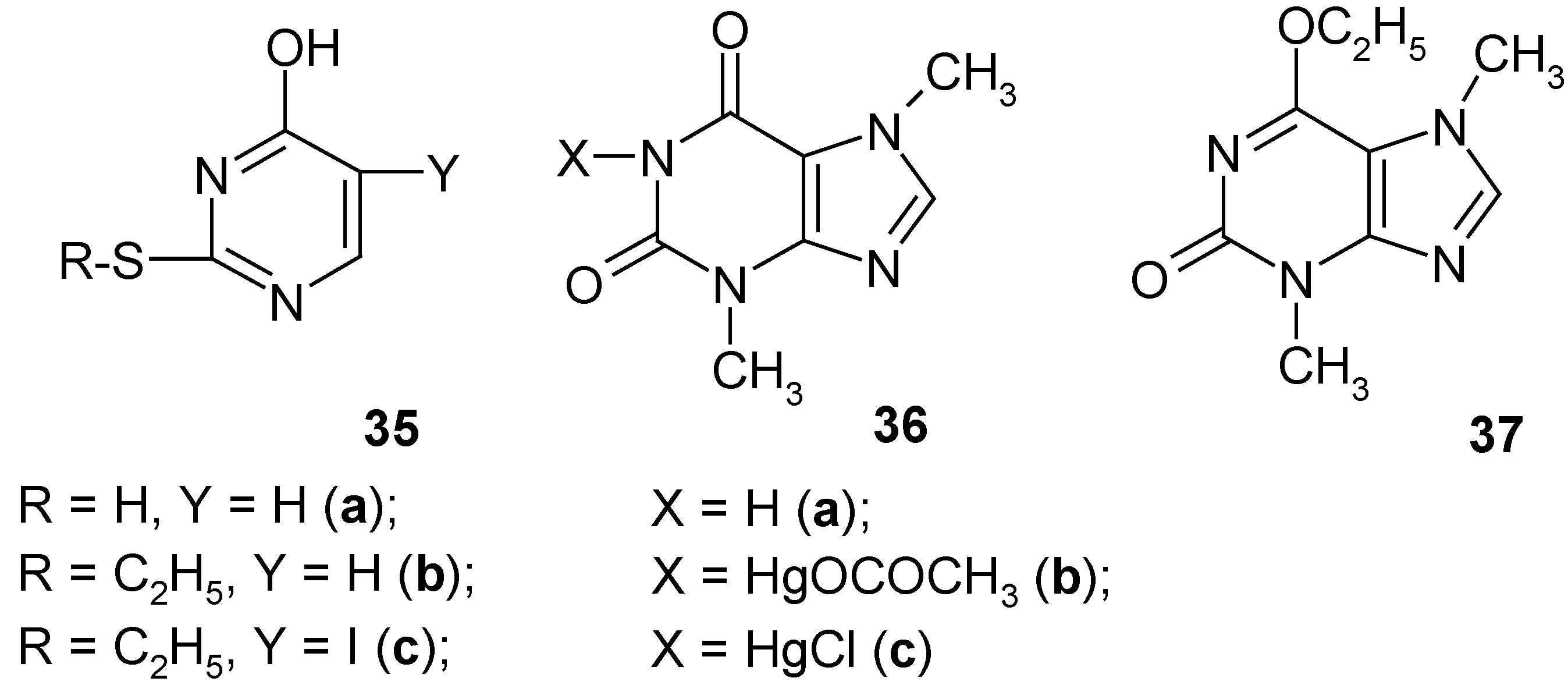
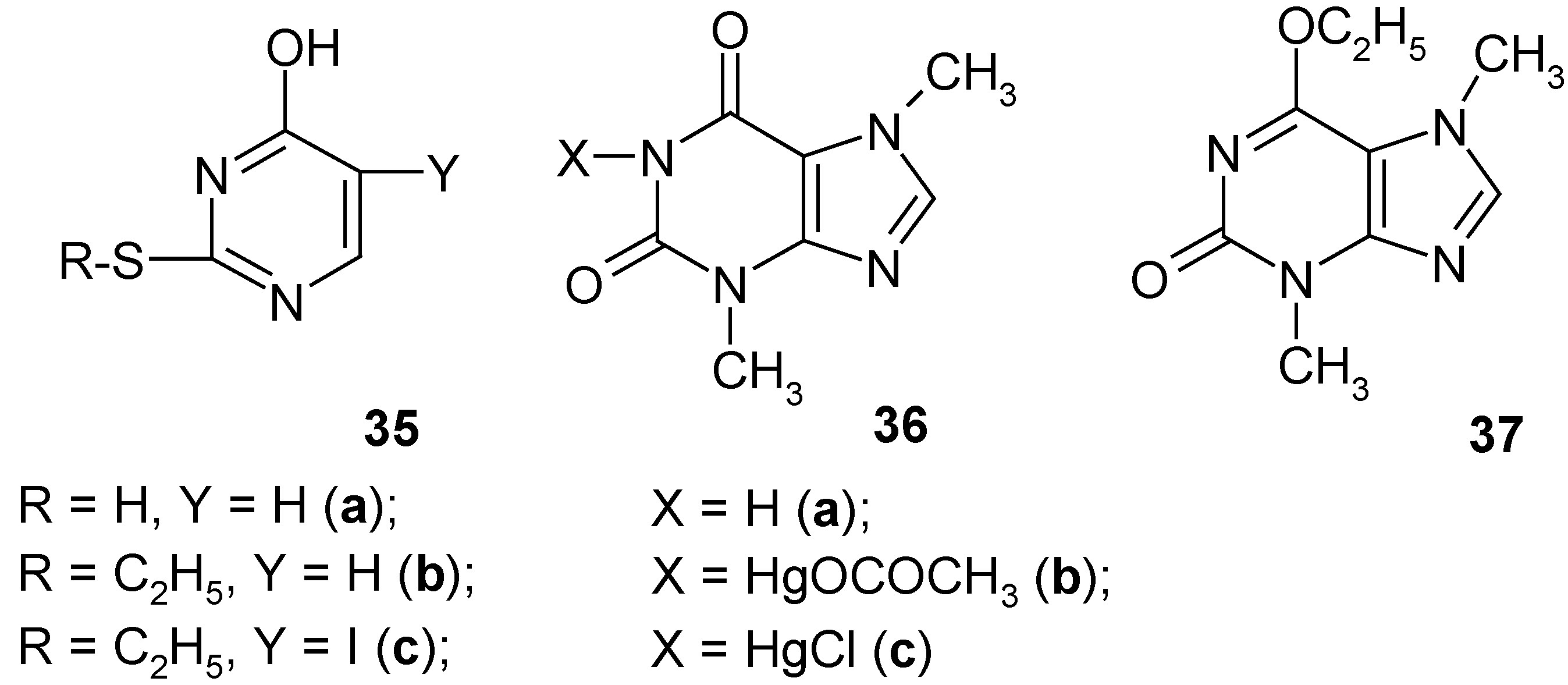
12. Further Studies on Some Heteroaromatic Mercurials [14]
Using various known methods of
direct C-mercuration we attempted to mercurate
uracil,
thiouracil (
35a),
S-ethyl-2-thiouracil (
35b),
theobromine (
36a) and
6-O-ethyltheobromine (
37). Only
uracil was effectively C
5-mercurated giving the crude
5-(chloromercurio)uracil (
Section 8) in ca 85% crude yield, whereas the rest formed (in good yields) either S-Hg bonds (compounds
35a and
35b) or N-Hg bonds (compounds
36a and
37); in the latter its 6-O-ethyl groups were
completely split off under the reaction conditions. All the newly obtained
crude mercurials were next
iodo-demercurated by the routine methods [
23,
24,
25,
34,
42]. This furnished from S-ethyl-2-thiouracil mercurial the corresponding C-iodinated product, i.e
. S-ethyl-5-iodo-2-thiouracil (
35c) in ca 64% crude yield.
The other mercurials were mostly
proto-demercurated, though with some noticeable exceptions which are discussed below. The main purpose of our work [
14] was to detect whether the aforementioned
crude mercurials do contain (or not) any detectable amounts of C-mercurated admixtures, which would then have been transformed into recognizable
C-iodinated derivatives formed in the subsequent iodo-demercuration reactions.
It has been shown in
Section 7 that
2,4-dimethoxypyrimidine (
17) was effectively C
5-mercurated on boiling with an aq. Hg(OCOCH
3)
2 solution acidified with CH
3CO
2H, for 2 hours. Hence, we
did expect that compound
37 would behave similarly to give a
true mercurial C-substituted in position 8. Thus, compound
37 was refluxed for 55 hours with a Hg(OCOCH
3)
2 solution in boiling glacial acetic acid (the reaction was monitored with TLC). Quite unexpectedly, we obtained a
new mercurial 36b, i.e.
1-(acetoxymercurio)theobromine, in 83% yield, whose structure was confirmed by chemical tests and its IR and
1H-NMR spectra; no
1H-NMR spectral evidence was found for any C
8-mercuration. The 6-O-ethyl groups in compound
37 were
completely split off under the reaction conditions, momentarily forming the parent
tautomeric theobromine (
36a), which
immediately reacted with Hg(OCOCH
3)
2 to give the new mercurial
36b. Our assumption was supported as follows: when compound
37 was refluxed for 48 hours with
neat CH
3CO
2H; after cooling, we isolated
theobromine (
36a) in 83% yield, which was proven chemically and spectroscopically. We also obtained another
new mercurial
36c, i.e.
1-(chloromercurio)-theobromine in ca 77% crude yield (
Section 8). The two crude mercurials
36b and
36c were refluxed for 2 hours with excess diiodine dissolved in
dry CH
3CN; after workup this furnished
theobromine (
36a) in 76% and 72% yields, respectively. No detectable amounts of known 8‑iodotheobromine – which would have supported the sought C
8-mercuration – were found in the both reaction mixtures which furnished only theobromine.
It should be added that, so far, only one
theobromine mercurial with a 2:1 Hg ratio has been reported [
69]; its structure is shown in
Section 1. In the same paper [
69] other N-Hg mercurials prepared from
theophylline,
hypoxanthine,
xanthine,
guanine and
uracil (the latter is a 1:1 Hg complex as shown in
Section 1) were also reported.
The new S-ethylthiouracil mercurial (supposedly a 1:1 Hg complex) was synthesized as follows: compound 35b was dissolved in CH3OH acidified with two drops of added conc. aq. HClO4. Then a solution of Hg(OCOCH3)2 in methanol was added to the former solution, the mixture was refluxed for 8 hours, and then left overnight. The collected white precipitate was practically insoluble in common solvents, and was obtained in ca 42% crude yield. When this novel crude mercurial was boiled with a conc. aq. KI solution until the combined solution was clear and slightly yellowish, then after cooling we isolated the recovered compound 35b in 99% yield. But the same crude mercurial upon refluxing with an aq. KI3 solution for 30 – 40 minutes, unexpectedly gave (after cooling) the iodo derivative 35c in ca 64% crude yield; it was recrystallized from ethanol and 2-propanol yielding pure 35c.
14. Improved Syntheses of Some Diaryliodonium Salts from Symmetric Diarylmercurials and (Dichloroiodo)arenes (Willgerodt`s Method) [16]
Willgerodt [
70,
71,
72] had reacted cold (or hot [
71])
aqueous suspensions of
equal masses [in practical terms this means that the mercurials were used
in a deficit] of powdered PhICl
2 with powdered Ar
2Hg (where Ar = phenyl, 2- and 4-tolyl, and 2-naphthyl) to afford the respective
diaryliodonium chlorides (yields were not reported); sparingly soluble ArHgCl and other admixtures, e.g. PhIO [
73], were hot-filtered off and discarded, viz.
Beringer and Lillien [
74] applied the Willgerodt method to obtain three unsymmetric diaryliodonium chlorides. They obtained only
4-acetamidophenyl(phenyl)iodonium chloride (which was precipitated out as its sparingly soluble
iodide, isolated in
10% crude yield) by reacting
equimolar amounts of Ph
2Hg with 4-AcNHC
6H
4ICl
2 in hot water (40-50
oC) for 12 hours. We obtained [
16] the same
iodide, but in
80% crude yield, by reacting
equal masses of PhICl
2 with symmetric 4,4
'-mercuriobis(acetanilide) suspended in stirred hot water (40-50
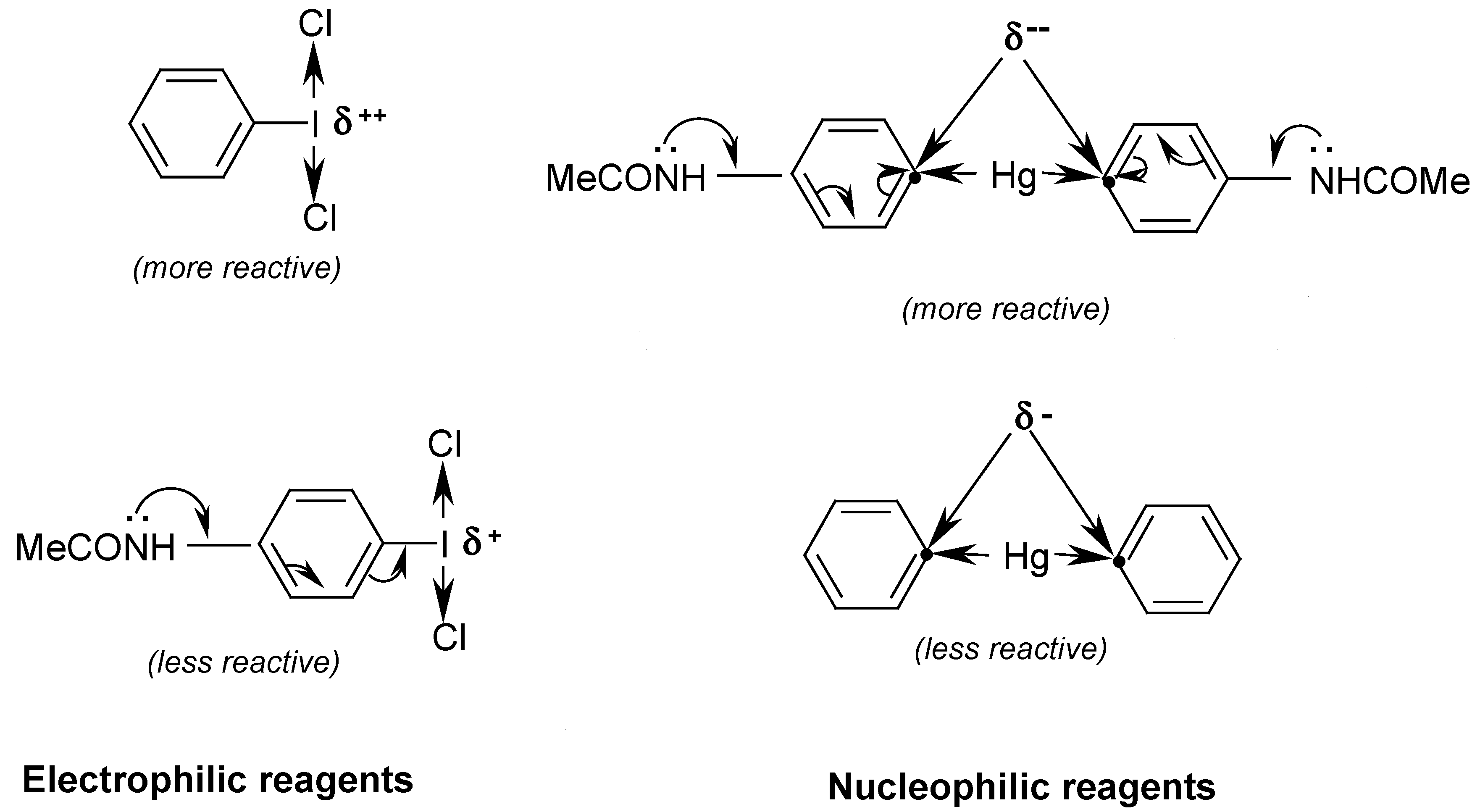
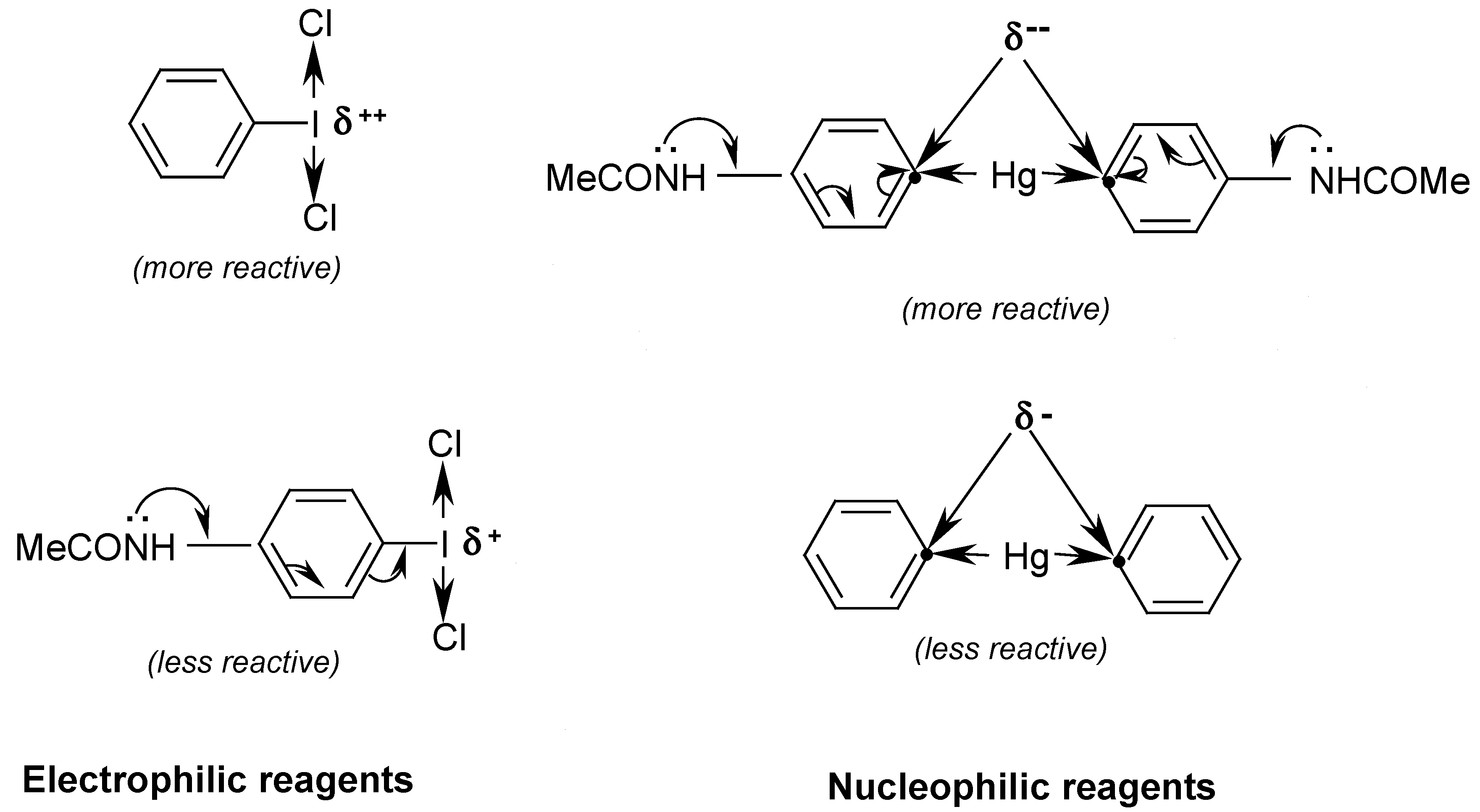
oC) for 12 hours. We explained this evident yield increase as follows:
Consequently, by reacting
equal masses of PhICl
2 with symmetric 4,4
'-mercuriobis(
N,N-dimethylaniline), suspended in stirred hot water (40-50
oC) for 12 hours, we isolated from the hot filtrate, after its cooling,
4-dimethylaminophenyl(phenyl)iodonium chloride in
60% crude yield. Previously, Beringer and Lillien [
74] failed to obtain this iodonium salts,
para-substituted with only one NMe
2 group; the same failure was also reported by Neiland [
75]. A similar iodonium salt bearing the two
p,p’-substituted NMe
2 groups was synthesized by quite a different route [
76]; this synthesis is shown (
Scheme 7) in our paper [
16].
We also attempted,
without effect, to synthesize various 8-(aryliodonio)caffeine halides with using the Willgerodt method. Hence, we used our short-cut, oxidative method [
77] to obtain
8-(4-methoxy-phenyliodonio)caffeine bromide (49% crude yield) by acidic coupling of the previously oxidized 8‑iodocaffeine with anisole. This is, in fact, the first
iodine(III) derivative of caffeine, which may open up novel routes for preparing 8-substituted caffeines by its reactions with various nucleophiles [
78].
16. Conclusions
This review shows our small research group`s main interests in developing
novel (or considerably improved) preparative procedures, mainly in the class of aromatic heterocyclic mercurials, which afforded a number of both unsymmetric, ArHgX, and symmetric, Ar
2Hg, C-mercurated compounds, mostly
not reported in the former literature. In our opinion, most interesting are those
indirect preparative C-mercuration methods, which made possible the syntheses of organomercurials derived from
uracil, theophilline, and
theobromine; they also open up new ways for preparing other similar organomercurials from many aromatic activated systems having N-H
acidic groups, which when reacted with mercuric salts, usually form at once the
insoluble N-Hg salts that precipitate out from the reaction mixtures,
instead of forming the expected
true organomercurials (with the mercury atoms joined to the organic residues via carbon atoms). In order to better confirm the chemical structures of the new organomercurials synthesized by us, they were next
iodo- and/or
bromo-demercurated (by known halo-demercuration procedures) to form in high yields a considerable number of the respective (purified) iodo and bromo derivatives (
Section 13), whose structures were well established chemically, spectrally, and by comparison with the available literature data. We also discovered same
novel halo- and
cyano-
demercuration procedures, which enabled us to obtain a number of the corresponding (purified) aromatic halides and nitriles in high yields (
Section 13). Also very interesting is our synthesis of
1,8-bis(acetoxydimercurio)theobromine, seemingly the first
stable organic derivative of
mercury(I), as well as its novel reactions (
Section 11). Finally, we considerably
improved the old Willgerodt method (1897), which enables to synthesize in high yields diaryliodonium chlorides from appropriate (dichloroiodo)arenes and symmetric aromatic organomercurials (
Section 14). We hope that our preparative procedures, disscussed and explained in the present review, will be applied either
as such, or they would be further improved and extended in other organic chemical laboratories.

{kind=link}
{kind=link}
{kind=link}
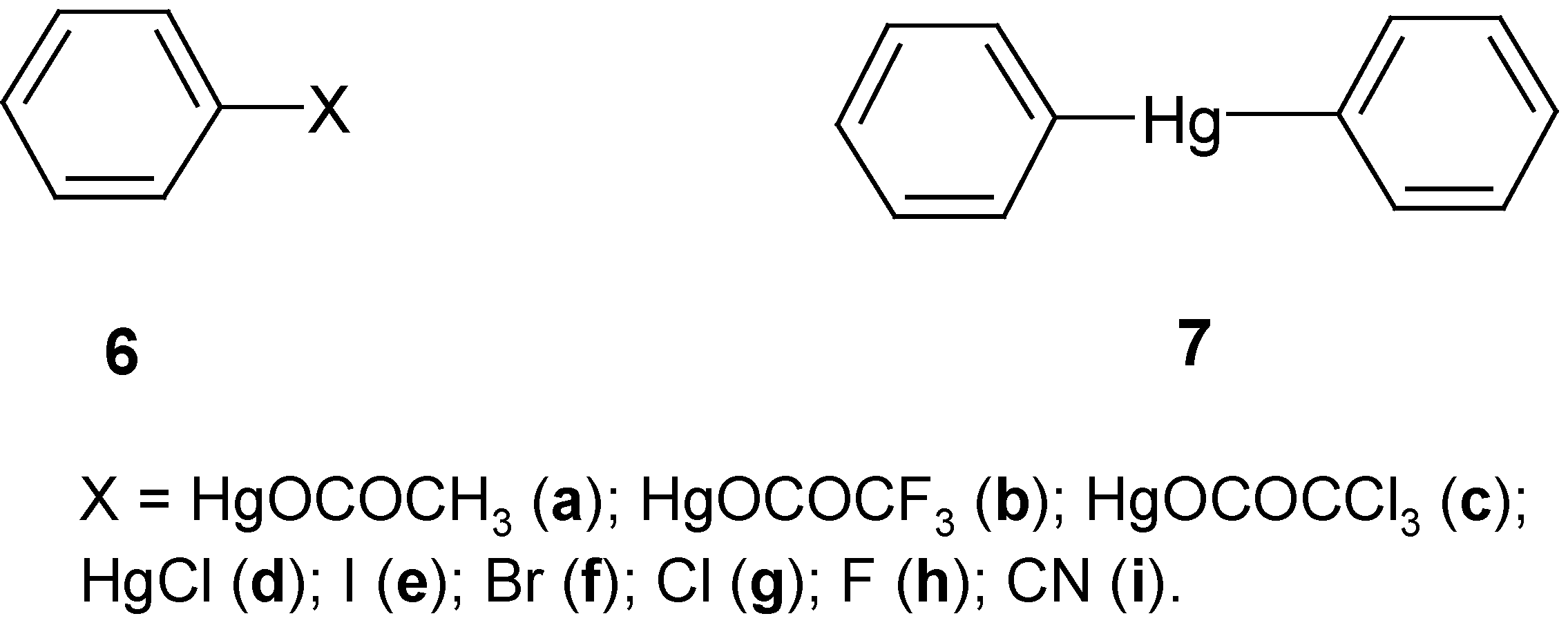{kind=link}
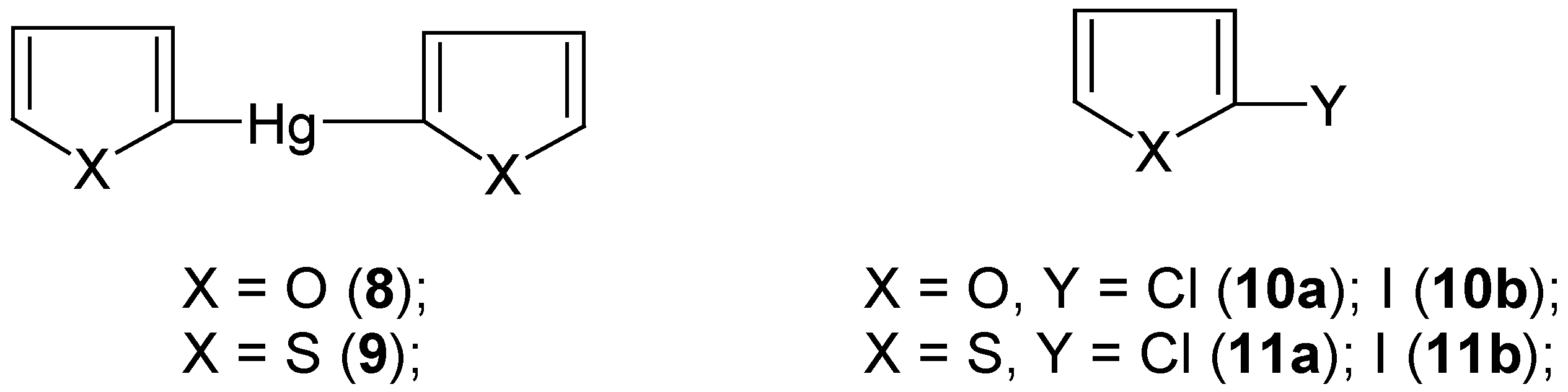{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
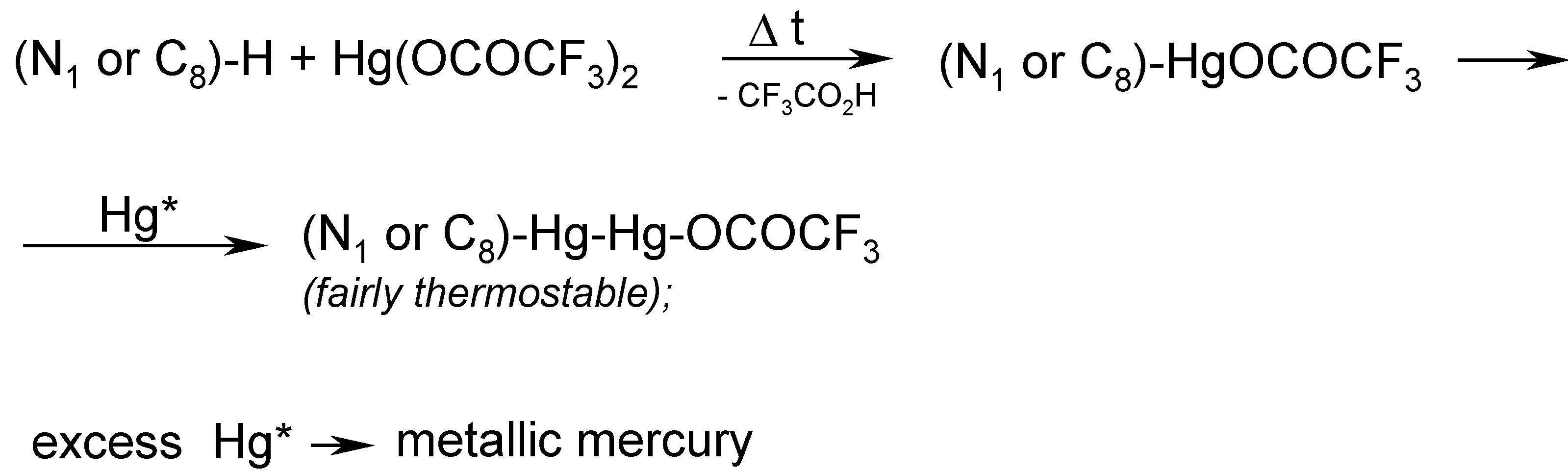{kind=link}
{kind=link}
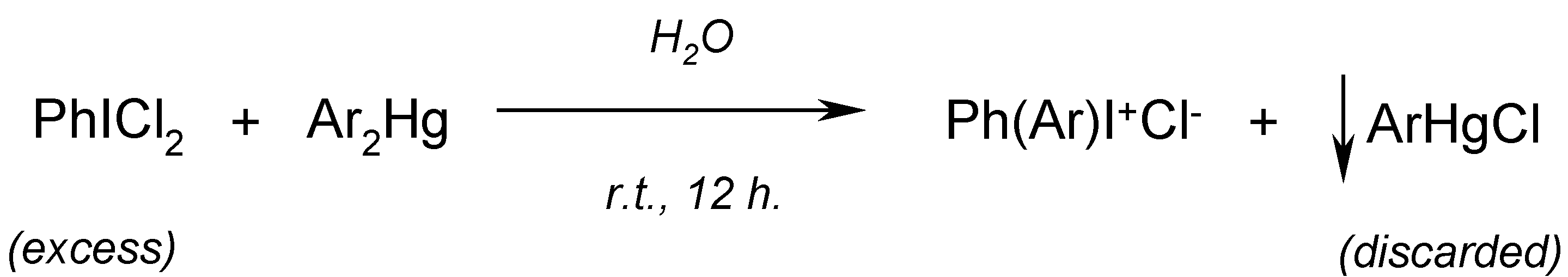{kind=link}
{kind=link}
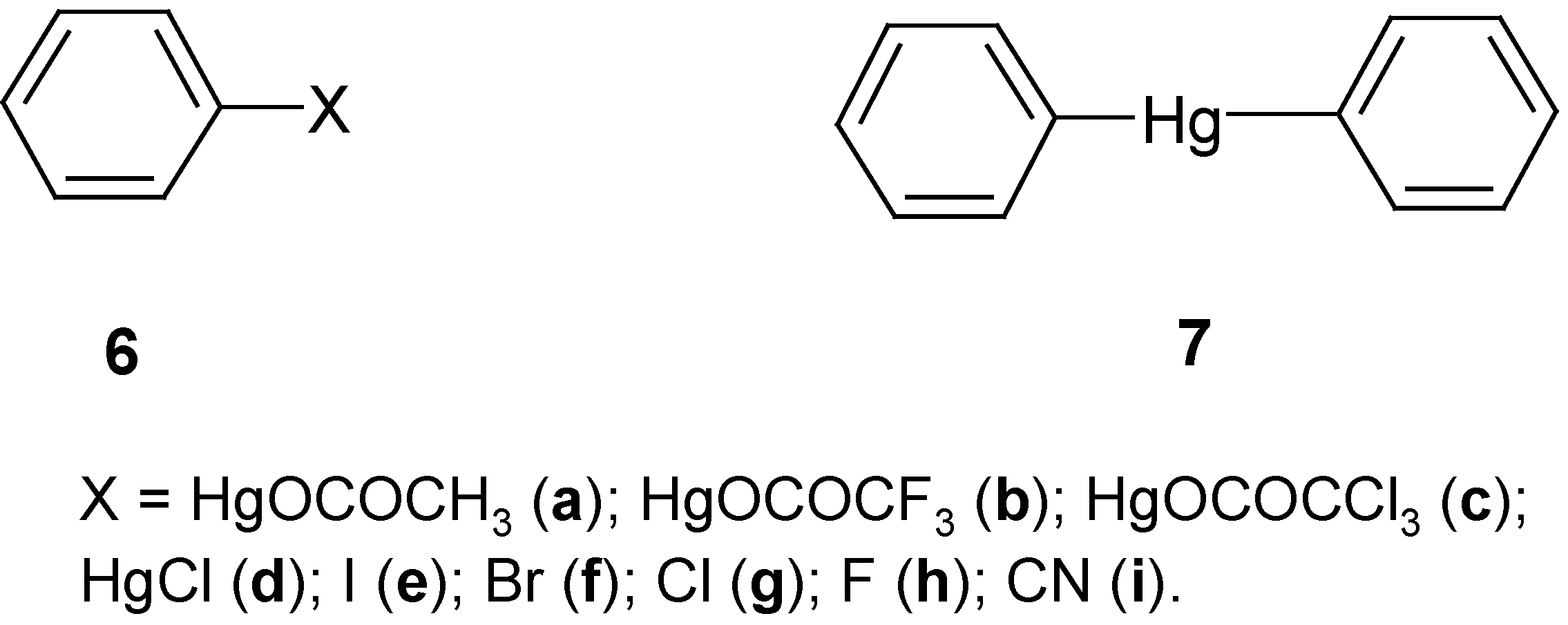
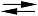 S2Cl2 + Cl2, which is very inconvenient due to the known high sensitivity of furan and thiophene towards the action of free dichlorine; see also our paper [5] where various chlorinating procedures applicable for furan and thiophene were briefly reviewed, with the relevant references.
S2Cl2 + Cl2, which is very inconvenient due to the known high sensitivity of furan and thiophene towards the action of free dichlorine; see also our paper [5] where various chlorinating procedures applicable for furan and thiophene were briefly reviewed, with the relevant references.