
Synthesis and Biological Evaluation of Isomeric Artemisinin Trimers as Novel Antitumor Agents

 , , and
, , and
Abstract
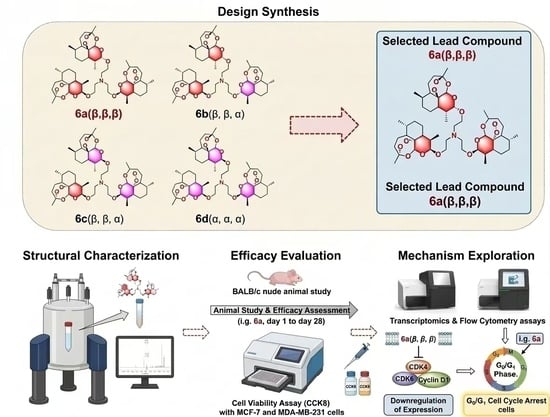
1. Introduction
2. Results and Discussion
2.1. Synthesis of Artemisinin Trimers 6a (β, β, β), 6b (β, β, α), 6c (β, α, α) and 6d (α, α, α)
2.2. H NMR and 13C NMR of Compounds 6a (β, β, β), 6b (β, β, α), 6c (β, α, α) and 6d (α, α, α)
2.3. Structural Diagrams of Compounds 6a (β, β, β), 6b (β, β, α), 6c (β, α, α) and 6d (α, α, α)
2.4. Antitumor Effects of 6a (β, β, β), 6b (β, β, α), 6c (β, α, α) and 6d (α, α, α) in the MCF-7 Model
2.5. The Effect of 6a (β, β, β) on Cell Cycle Arrest
3. Materials and Method
3.1. General Chemical Procedures
3.2. Synthesis of Compounds
3.2.1. Compound 4b (β, α)
3.2.2. Compound 4c (α, α)
3.2.3. Compound 5b
3.2.4. Compound 6b (β, β, α)
3.2.5. Compound 6c (β, α, α)
3.2.6. Compound 6d (α, α, α)
3.3. Biological Methods
3.3.1. Cell Viability Assay
3.3.2. Colony Formation Assay
3.3.3. Animal Experiments
3.3.4. Transcriptome Sequencing and Analysis
3.3.5. Cell Cycle Analysis
3.3.6. Western Blot
3.3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zheng, L.; Ding, Y.; Xu, X.; Wang, H.; Shi, G.; Li, Y.; He, Y.; Gong, Y.; Zhang, X.; Wei, J.; et al. CDH17-targeting CAR-NK cells synergize with CD47 blockade for potent suppression of gastrointestinal cancers. Acta Pharmacol. Sin. B 2025, 15, 2559–2574. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xu, R.; Shi, J.; Yang, Z.; Zheng, J.; Wei, D. Artesunate induces ferroptosis in osteosarcoma through NCOA4-mediated ferritinophagy. FASEB J. 2025, 39, e70488. [Google Scholar] [CrossRef]
- Vakhrusheva, O.; Zhao, F.; Markowitsch, S.D.; Slade, K.S.; Brandt, M.P.; Tsaur, I.; Cinatl, J., Jr.; Michaelis, M.; Efferth, T.; Blaheta, R.A.; et al. Artesunate Inhibits Metastatic Potential in Cisplatin-Resistant Bladder Cancer Cells by Altering Integrins. Cells 2025, 14, 570. [Google Scholar] [CrossRef] [PubMed]
- Yuan-Ce, L.; Qi, Z.; Hong-Yang, Z.; Yan-Wen, W.; Yu-Mei, S.; Bi-Juan, Y.; Jun-Lin, Y. Artesunate, as an Hsp90 inhibitor, inhibits the proliferation of Burkitt’s lymphoma cells by inhibiting AKT and ERK. Front. Pharmacol. 2023, 14, 1218467. [Google Scholar] [CrossRef]
- Ling, H.; Tang, J.; Zhu, Y.; Niu, K.; Qiu, P.; Chen, R.; Chen, L. Dihydroartemisinin inhibits NSCLC by targeting MDM2/c-Myc axis independent of p53. Biochem. Pharmacol. 2025, 241, 117191. [Google Scholar] [CrossRef]
- Dai, X.; Chen, W.; Qiao, Y.; Chen, X.; Chen, Y.; Zhang, K.; Zhang, Q.; Duan, X.; Li, X.; Zhao, J.; et al. Dihydroartemisinin inhibits the development of colorectal cancer by GSK-3β/TCF7/MMP9 pathway and synergies with capecitabine. Cancer Lett. 2024, 582, 216596. [Google Scholar] [CrossRef]
- Augustin, Y.; Staines, H.M.; Krishna, S. Artemisinins as a novel anti-cancer therapy: Targeting a global cancer pandemic through drug repurposing. Pharmacol. Ther. 2020, 216, 107706. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Xiong, L.; Yan, G.; Du, S.; Liu, J.; Shi, Y. Susceptibility of cervical cancer to dihydroartemisinin-induced ferritinophagy-dependent ferroptosis. Front. Mol. Biosci. 2023, 10, 1156062. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, J.; Ren, W.; Liu, Z.L.; Cheng, Y.F.; Zhang, X.M. Combination treatment with artemisinin and oxaliplatin inhibits tumorigenesis in esophageal cancer EC109 cell through Wnt/β-catenin signaling pathway. Thorac. Cancer 2020, 11, 2316–2324. [Google Scholar] [CrossRef]
- Farhan, M.; Silva, M.; Xingan, X.; Zhou, Z.; Zheng, W. Artemisinin Inhibits the Migration and Invasion in Uveal Melanoma via Inhibition of the PI3K/AKT/mTOR Signaling Pathway. Oxid. Med. Cell Longev. 2021, 2021, 9911537. [Google Scholar] [CrossRef]
- Li, J.; Feng, L.; Yuan, Y.; He, T.; Zou, X.; Su, B.; Liu, K.; Yang, X. Inhibition of HOXC11 by artesunate induces ferroptosis and suppresses ovarian cancer progression through transcriptional regulation of the PROM2/PI3K/AKT pathway. World J. Surg. Oncol. 2024, 22, 268. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Dai, H.Q.; Huang, X.W.; Feng, J.; Deng, J.H.; Wang, Z.X.; Yang, X.M.; Liu, Y.J.; Wu, Y.; Chen, P.H.; et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol. Sin. 2021, 42, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhu, X.; Zhao, Q.; Shen, Y.; Tao, L.; Zhang, J.; Lin, H.; Zhuge, W.; Cho, Y.C.; Cui, R.; et al. Dihydroartemisinin, a potential PTGS1 inhibitor, potentiated cisplatin-induced cell death in non-small cell lung cancer through activating ROS-mediated multiple signaling pathways. Neoplasia 2024, 51, 100991. [Google Scholar] [CrossRef]
- Ismail, M.; Yang, W.; Li, Y.; Chai, T.; Zhang, D.; Du, Q.; Muhammad, P.; Hanif, S.; Zheng, M.; Shi, B. Targeted liposomes for combined delivery of artesunate and temozolomide to resistant glioblastoma. Biomaterials 2022, 287, 121608. [Google Scholar] [CrossRef]
- Okamoto, H.; Yoshikawa, K.; Shimada, A.; Sano, R.; Inukai, D.; Yamanaka, S.; Suzuki, S.; Ueda, R.; Ueda, H.; Fujimoto, Y.; et al. Artesunate and cisplatin synergistically inhibit HNSCC cell growth and promote apoptosis with artesunate-induced decreases in Rb and phosphorylated Rb levels. Oncol. Rep. 2023, 50, 154. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Zhang, S.; Zhang, D.; Feng, R.; Li, N.; Chen, W.; Chen, X.; Yang, S. Effects and Mechanisms of Anlotinib and Dihydroartemisinin Combination Therapy in Ameliorating Malignant Biological Behavior of Gastric Cancer Cells. Curr. Pharm. Biotechnol. 2021, 22, 523–533. [Google Scholar] [CrossRef]
- Raphela-Choma, P.P.; Lukhwareni, R.; Simelane, M.B.C.; Motadi, L.R.; Choene, M.S. Antitumor effect of Iso-mukaadial acetate on MCF-7 breast cancer mice xenograft model. Sci. Rep. 2024, 14, 13744. [Google Scholar] [CrossRef]
- Yao, Y.; Guo, Q.; Cao, Y.; Qiu, Y.; Tan, R.; Yu, Z.; Zhou, Y.; Lu, N. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-β signaling in breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 282. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Y.; Gai, L.; Zhang, Z.; Zhang, Y.; Zhang, G.; Du, K.; Gao, C. Artesunate regulates malignant progression of breast cancer cells via lncRNA TUG1/miR-145-5p/HOXA5 axis. PLoS ONE 2025, 20, e0329490. [Google Scholar] [CrossRef]
- Chatterjee, R.; Shukla, A.; Chakrabarti, K.; Chatterji, U. CLEC12A sensitizes differentially responsive breast cancer cells to the anti-cancer effects of artemisinin by repressing autophagy and inflammation. Front. Oncol. 2023, 13, 1242432. [Google Scholar] [CrossRef]
- Zhang, R.; Tang, C.; Liu, H.C.; Ren, Y.; Xu, C.H.; Ke, C.Q.; Yao, S.; Huang, X.; Ye, Y. Ainsliatriolides A and B, Two Guaianolide Trimers from Ainsliaea fragrans and Their Cytotoxic Activities. J. Org. Chem. 2018, 83, 14175–14180. [Google Scholar] [CrossRef]
- Maslivetc, V.; Laguera, B.; Chandra, S.; Dasari, R.; Olivier, W.J.; Smith, J.A.; Bissember, A.C.; Masi, M.; Evidente, A.; Mathieu, V.; et al. Polygodial and Ophiobolin A Analogues for Covalent Crosslinking of Anticancer Targets. Int. J. Mol. Sci. 2021, 22, 11256. [Google Scholar] [CrossRef]
- Raposo Moreira Dias, A.; Pina, A.; Dal Corso, A.; Arosio, D.; Belvisi, L.; Pignataro, L.; Caruso, M.; Gennari, C. Multivalency Increases the Binding Strength of RGD Peptidomimetic-Paclitaxel Conjugates to Integrin α(V) β(3). Chemistry 2017, 23, 14410–14415. [Google Scholar] [CrossRef]
- Zhang, D.; Xiao, Z.; Wang, N.; Huang, A.; Wen, J.; Kong, L.; Luo, J. Trisarcglaboids A and B, two cytotoxic lindenane sesquiterpenoid trimers with a unique polymerization mode isolated from Sarcandra glabra. Bioorg. Chem. 2024, 146, 107259. [Google Scholar] [CrossRef]
- Khongsti, K.; Pasupuleti, B.G.; Das, B.; Bez, G. 1,2,3-Triazole tethered 1,2,4-trioxane trimer induces apoptosis in metastatic cancer cells and inhibits their proliferation, migration and invasion. Bioorg. Chem. 2021, 112, 104952. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.-W.; Cheung, M.-K.; Lam, W.-L.; Soo, M.-K.; Tsang, H.-W.; Voerste, A.; Williams, I.D. C-10 Ester and Ether Derivatives of Dihydroartemisinin − 10-α Artesunate, Preparation of Authentic 10-β Artesunate, and of Other Ester and Ether Derivatives Bearing Potential Aromatic Intercalating Groups at C-10. Eur. J. Org. Chem. 2002, 2002, 113–132. [Google Scholar] [CrossRef]
- Beekman, A.C.; Barentsen, A.R.W.; Woerdenbag, H.J.; Van Uden, W.; Pras, N.; Konings, A.W.T.; El-Feraly, F.S.; Galal, A.M.; Wikström, H.V. Stereochemistry-Dependent Cytotoxicity of Some Artemisinin Derivatives. J. Nat. Prod. 1997, 60, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Pan, Y.; Wang, J.; Yue, L.; Luo, Y.; Lv, F.; Lv, J.; Chen, J.; Zhao, Q.; Lin, H. Design, Synthesis, and Antitumor Activities of Isomers of Artemisinin Dimer Derivatives. Chem. Biodivers. 2023, 20, e202300615. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, J.; Yue, L.; Zhang, Z.; Lv, J.; Chen, J.; Cao, J.; Guo, F.; Zhao, Q. Anti-tumor effects of artemisinin-based oligomers: From monomer to trimer as a novel drug-enhancing strategy. Eur. J. Med. Chem. 2025, 287, 117313. [Google Scholar] [CrossRef]
- Zuo, J.; Jiang, Y.; Zhang, E.; Chen, Y.; Liang, Z.; Zhu, J.; Zhao, Y.; Xu, H.; Liu, G.; Liu, J.; et al. Synergistic effects of 7-O-geranylquercetin and siRNAs on the treatment of human breast cancer. Life Sci. 2019, 227, 145–152. [Google Scholar] [CrossRef]
- Gangangari, K.K.; Váradi, A.; Majumdar, S.; Larson, S.M.; Pasternak, G.W.; Pillarsetty, N.K. Imaging Sigma-1 Receptor (S1R) Expression Using Iodine-124-Labeled 1-(4-Iodophenyl)-3-(2-adamantyl)guanidine ([(124)I]IPAG). Mol. Imaging Biol. 2020, 22, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, Z.; He, Y.; He, Z.; Ban, Z.; Zhu, Y.; Ding, L.; Yang, C.; Jeong, J.H.; Yuan, W.; et al. EphA2 promotes tumorigenicity of cervical cancer by up-regulating CDK6. J. Cell Mol. Med. 2021, 25, 2967–2975. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Thakar, T.; Nicolae, C.M.; Moldovan, G.L. PARP14 regulates cyclin D1 expression to promote cell-cycle progression. Oncogene 2021, 40, 4872–4883. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position |  |  |  |  | ||||
|---|---|---|---|---|---|---|---|---|
| δH Mult (J in Hz) | δC | δH Mult (J in Hz) | δC | δH Mult (J in Hz) | δC | δH Mult (J in Hz) | δC | |
| 1/1′/1″ | / | 104.5 104.3 | / | 104.2 104.0 | / | 104.3 104.1 | / | 104.3 |
| 2/2′/2″ | 2.36 td, (14.1, 3.9) 2.04–2.00, m | 36.6 36.4 | 2.38–2.30, m 2.02–1.99, m | 36.5 36.3 | 2.38–2.30, m 2.08–1.95, m | 36.6 36.4 | 2.40–2.29, m 2.01 dt, (14.5, 3.7) | 36.7 36.5 |
| 3/3′/3″ | 1.89–1.85, m 1.52–1.47, m | 24.8 24.8 24.5 | 1.87–1.84, m 1.53–1.47, m | 24.7 24.5 | 1.90–1.80, m 1.55–1.46, m | 24.8 24.8 | 1.92–1.84, m 1.56–1.46, m | 24.9 |
| 4/4′/4″ | 1.25–1.22, m | 52.7 52.4 | 1.33–1.18, m | 52.6 51.7 | 1.32–1.19, m | 52.7 51.7 | 1.33–1.24, m | 52.1 51.8 |
| 5/5′/5″ | 1.35–1.30, m | 37.5 37.4 | 1.33–1.18, m | 37.4 37.3 | 1.32–1.19, m | 37.5 37.4 | 1.33–1.24, m | 37.5 37.4 |
| 6/6′/6″ | 1.65–1.61, m | 34.7 34.3 | 1.77–1.60, m | 34.7 34.3 | 1.75–1.60, m | 34.7 34.3 | 1.77–1.66, m | 34.6 34.4 |
| 7/7′/7″ | 1.80–1.70, m | 24.8 24.8 24.5 | 1.87–1.84, m | 22.2 | 1.90–1.80, m | 22.3 | 1.92–1.84, m | 22.3 |
| 8/8′/8″ | 1,45–1.43, m | 44.5 | 1.53–1.47, m | 45.4 44.5 | 1.55–1.46, m | 45.4 44.6 | 1.56–1.46 m | 46.6 45.5 |
| 9/9′/9″ | 2.62–2.59, m | 30.9 | 2.61–2.57, m | 32.6 30.9 | 2.61–2.54, m | 32.7 31.0 | 2.40–2.29, m | 32.8 31.7 |
| 10/10′/10″ | 4.76, d, (3.3) | 102.6 100.6 | 4.75, d, (3.1) 4.41, d, (9.2) | 102.2 100.3 | 4.74, d, (3.2) 4.41, d, (9.2) | 102.3 100.3 | 4.94, d, (4.2) 4.44, d, (8.9) | 103.2 100.4 |
| 11/11′/11″ | 5.39, s | 88.1 | 5.38, s 5.30, s | 87.9 91.2 | 5.37, s 5.30, s | 88.0 91.2 | 5.44, s 5.32, s | 91.3 89.1 |
| 12/12′/12″ | / | 81.2 80.5 | / | 81.1 80.3 | / | 81.2 80.4 | / | 81.7 80.5 |
| 13/13′/13″ | 1.42, s | 26.3 26.1 | 1.41, s | 26.2 26.0 | 1.40, s | 26.3 26.1 | 1.42, d, (6.4) | 26.2 26.1 |
| 14/14′/14″ | 0.95, d, (6.3) | 20.5 20.4 | 0.93, d, (6.2) | 20.4 20.3 | 0.93, d, (6.0) | 20.5 20.4 | 0.96–0.93, m | 20.4 20.2 |
| 15/15′/15″ | 0.89, d, (7.3) | 12.7 13.2 | 0.89–0.83, m | 13.1 12.7 | 0.88–0.83, m | 13.2 12.8 | 0.87, d, (7.1) 1.18, d, (7.3) | 12.9 19.7 |
| 16/16′/16″ | 3.92–3.86, m 3.45–3.42, m | 66.6 65.8 | 3.89–3.83, m 3.49–3.41, m | 67.8 67.4 | 3.98–3.93, m 3.88–3.82, m 3.53–3.42, m | 67.8 67.4 | 4.04–3.88, m 3.61–3.49, m | 67.8 67.5 |
| 17/17′/17″ | 2.82–2.69, m | 51.7 | 2.79–2.72, m | 54.7 54.3 | 2.84–2.72, m | 54.9 54.4 | 2.88–2.77, m | 54.7 54.6 |
| Compound | IC50/(μM) | |
|---|---|---|
| MCF-7 | MDA-MB-231 | |
| 4a (β, β) | 3.62 ± 0.05 | 6.24 ± 1.06 |
| 4b (β, α) | 0.33 ± 0.04 | 5.25 ± 2.79 |
| 4c (α, α) | 2.70 ± 0.06 | >10 |
| 6b (β, β, α) | 0.03 ± 0.005 | >10 |
| 6c (β, α, α) | 0.05 ± 0.002 | 3.89 ± 0.02 |
| 6d (α, α, α) | 0.06 ± 0.01 | 2.85 ± 0.24 |
| 6a (β, β, β) | 0.09 ± 0.03 | 0.11 ± 0.06 |
| Cisplatin | 2.41 ± 0.27 | 8.57 ± 1.83 |
| Gemcitabine | 4.33 ± 0.36 | 0.73 ± 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Zhang, Z.; Li, A.; Jiang, B.; Bejoma, T.; Zhao, Y.; Guo, F.; Li, Y.; Li, H.; Zhao, Q. Synthesis and Biological Evaluation of Isomeric Artemisinin Trimers as Novel Antitumor Agents. Molecules 2026, 31, 1228. https://doi.org/10.3390/molecules31081228
Zhang Z, Li A, Jiang B, Bejoma T, Zhao Y, Guo F, Li Y, Li H, Zhao Q. Synthesis and Biological Evaluation of Isomeric Artemisinin Trimers as Novel Antitumor Agents. Molecules. 2026; 31(8):1228. https://doi.org/10.3390/molecules31081228
Chicago/Turabian StyleZhang, Zejin, Along Li, Bingying Jiang, Typhaine Bejoma, Yongxi Zhao, Fujiang Guo, Yajuan Li, Huiyu Li, and Qingjie Zhao. 2026. "Synthesis and Biological Evaluation of Isomeric Artemisinin Trimers as Novel Antitumor Agents" Molecules 31, no. 8: 1228. https://doi.org/10.3390/molecules31081228
APA StyleZhang, Z., Li, A., Jiang, B., Bejoma, T., Zhao, Y., Guo, F., Li, Y., Li, H., & Zhao, Q. (2026). Synthesis and Biological Evaluation of Isomeric Artemisinin Trimers as Novel Antitumor Agents. Molecules, 31(8), 1228. https://doi.org/10.3390/molecules31081228