
Eco-Friendly Synthesis of 2-Styryl-benzo[d][1,3]oxazin-4-ones from N-Cinnamoyl-Anthranilic Acids



Abstract
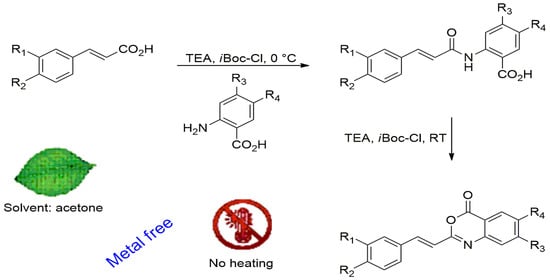
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Preparation of N-Cinnamoyl-Anthranilic Acids
3.2.1. For Cinnamic Acids Lacking Free Phenolic Groups, Such as 1a, 1c, 1d, and Piperonylic Acid
3.2.2. For Cinnamic Acids with a Single Phenolic Group, Such as 1b and 1e
3.2.3. For Cinnamic Acids with Two Phenolic Groups, Such as Caffeic Acid, 1f
3.2.4. Formation of N-Cinnamoyl-Anthranilic Acids
3.2.5. Formation of 2-Styryl-benzo[d][1,3]oxazin-4-ones
3.2.6. Carbonate Deprotection and Ring Opening of Benzo[d][1,3]oxazin-4-ones
Reaction in MeOH
Reaction in EtOAc
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DMF | Dimethylformamide |
| HCAs | Hydroxycinnamic acids |
| iBoc-Cl | Isobutyl chloroformate |
| Etoc-Cl | Ethyl Chloroformate |
References
- Moussa, Z.; Ramanathan, M.; Al-Masri, H.T.; Ahmed, S.A. Recent progress in the synthesis of benzoxazin-4-ones, applications in N-directed ortho-functionalizations, and biological significance. Molecules 2024, 29, 5710. [Google Scholar] [CrossRef]
- Von Nussbaum, F.; Li, V.M. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Bioorg. Med. Chem. Lett. 2015, 25, 4370−4381. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L.; Moorman, A.R.; Dobbs, J.; Abeles, R.H. Suicide inactivation of chymotrypsin by benzoxazinones. Biochemistry 1984, 23, 1753. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, P.; Bryson, A.; Hickling, R.; Rissanen, A.; Rossner, S.; Toubro, S.; Valensi, P. Cetilistat (ATL-962), a novel lipase inhibitor: A 12-week randomized, placebo-controlled study of weight reduction in obese patients. Int. J. Obes. 2007, 31, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Mohamed, A.A.; Ibrahim, R.A. Design, synthesis, molecular docking and biological studies of 4H-Benzo[d][1,3]oxazin-4-one derivatives and their uses in heterocyclic synthesis. ChemistrySelect 2022, 7, e202200325. [Google Scholar] [CrossRef]
- Coppola, G. The chemistry of 4H-3,1-benzoxazin-4-ones. J. Heterocycl. Chem. 1999, 36, 563–588. [Google Scholar] [CrossRef]
- Khan, S.A.; Yang, H.; Ying, F.; Chu, C.N.; Lee, T.K.W. Polypharmacology-driven discovery of ZAK-I-57: A potent multi-targeted benzoxazinone small molecule for hepatocellular carcinoma therapy. MedComm 2025, 6, e70291. [Google Scholar] [CrossRef]
- Kumar, P.; Dutta, S.; Batra, D.; Mishra, D.; Kumar, D.; Bhalla, G.; Singh, B.K. Selective functionalization of 2-phenyl-4H-benzo[d][1,3]oxazin-4-ones via C–H activation. Tetrahedron 2025, 173, 134466. [Google Scholar] [CrossRef]
- Kumar, P.; Gupta, M.; Bahadur, V.; Parmar, V.S.; Singh, B.K. Radical-induced, palladium-catalyzed C–H activation: An approach to functionalize 4H-benzo[d][1,3]oxazin-4-one derivatives by using toluenes, aldehydes, and benzyl alcohols. Eur. J. Org. Chem. 2018, 2018, 1552–1558. [Google Scholar] [CrossRef]
- Marasini, B.P.; Rahim, F.; Perveen, S.; Karim, A.; Mohammed Khan, K.; Atta-Ur-Rahman, C.M.I. Synthesis, structure-activity relationships studies of benzoxazinone derivatives as α-chymotrypsin inhibitors. Bioorg. Chem. 2017, 70, 210–221. [Google Scholar] [CrossRef]
- Goel, P.; Jumpertz, T.; Tichá, A.; Ogorek, I.; Mikles, D.C.; Hubalek, M.; Pietrzik, C.U.; Strisovsky, K.; Schmidt, B.; Weggen, S. Discovery and validation of 2-styryl substituted benzoxazin-4-ones as a novel scaffold for rhomboid protease inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1417–1422. [Google Scholar] [CrossRef]
- Gataullin, R.R. Advances in the synthesis of aryl-fused six- to nine-membered N,O-heterocycles. Russ. Chem. Bull. 2025, 74, 585–622. [Google Scholar] [CrossRef]
- Yan, J.; Yang, Z.; Guo, J.; Xue, Q.; Chen, P. Efficient synthesis of 2-aryl-4H-benzo[d][1,3]oxazin-4-ones by copper-catalyzed decarboxylation of α-keto acids with anthranilic acids. Asian J. Org. Chem. 2024, 13, e202400157. [Google Scholar] [CrossRef]
- Annor-Gyamfi, J.K.; Bunce, R.A. 4H-Benzo[d][1,3]oxazin-4-ones and dihydro analogs from substituted anthranilic acids and ortho-esters. Molecules 2019, 24, 3555. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-X.; Wei, T.-Q.; Xu, P.; Wang, S.-Y.; Ji, S.-J. I2/TBHP-mediated oxidative coupling of amino-based bisnucleophiles and isocyanides: Access to 2-aminobenzoxazinones, 2-aminobenzoxazines, and 2-aminoquinazolies under metal-free conditions. J. Org. Chem. 2018, 83, 13491–13497. [Google Scholar] [CrossRef]
- Zheng, Y.; Dong, M.; Qu, E.; Bai, J.; Wu, X.F.; Li, W. Pd-catalyzed carbonylative synthesis of 4H-Benzo[d][1,3]oxazin-4-ones using benzene-1,3,5-triyl triformate as the CO source. Chem. Eur. J. 2021, 27, 16219. [Google Scholar] [CrossRef]
- Dhawa, U.; Kaplaneries, N.; Ackermann, L. Green strategies for transition metal-catalyzed C–H activation in molecular syntheses. Org. Chem. Front. 2021, 8, 4886–4913. [Google Scholar] [CrossRef]
- Zarrelli, A.; Longobardo, L. Improved chemical synthesis of avenanthramides family and its analogs by mixed anhydride method. Eur. J. Org. Chem. 2025, 28, e202500169. [Google Scholar] [CrossRef]
- Petrillo, F.; Maione, A.; Spampinato, M.; Massa, L.D.; Guida, M.; Zarrelli, A.; Galdiero, E.; Longobardo, L. Antifungal and antibiofilm activities of 2-aminobenzoic acid derivatives against a clinical ocular Candida albicans isolate for biomedical applications. Antibiotics 2025, 14, 432. [Google Scholar] [CrossRef]
- Longobardo, L.; Di Fabio, G.; Zarrelli, A. Sustainable chemical derivatization of hydroxycinnamic acids. ChemistrySelect 2024, 9, e202305046. [Google Scholar] [CrossRef]
- DellaGreca, M.; Longobardo, L. Protection and Activation of Hydroxycinnamic Acids in Water. ChemistrySelect 2020, 5, 4588. [Google Scholar] [CrossRef]
- Collins, F.W. Oat phenolics: Avenanthramides, novel substituted N-cinnamoylanthranilate alkaloids from oat groats and hulls. J. Agric. Food Chem. 1989, 37, 60–66. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Zarrelli, A.; Longobardo, L. Eco-Friendly Synthesis of 2-Styryl-benzo[d][1,3]oxazin-4-ones from N-Cinnamoyl-Anthranilic Acids. Molecules 2026, 31, 709. https://doi.org/10.3390/molecules31040709
Zarrelli A, Longobardo L. Eco-Friendly Synthesis of 2-Styryl-benzo[d][1,3]oxazin-4-ones from N-Cinnamoyl-Anthranilic Acids. Molecules. 2026; 31(4):709. https://doi.org/10.3390/molecules31040709
Chicago/Turabian StyleZarrelli, Armando, and Luigi Longobardo. 2026. "Eco-Friendly Synthesis of 2-Styryl-benzo[d][1,3]oxazin-4-ones from N-Cinnamoyl-Anthranilic Acids" Molecules 31, no. 4: 709. https://doi.org/10.3390/molecules31040709
APA StyleZarrelli, A., & Longobardo, L. (2026). Eco-Friendly Synthesis of 2-Styryl-benzo[d][1,3]oxazin-4-ones from N-Cinnamoyl-Anthranilic Acids. Molecules, 31(4), 709. https://doi.org/10.3390/molecules31040709