
Recent Advances in the Enantioselective Organocatalytic [4+2] Cycloadditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Organocatalytic Diels–Alder Reactions
2.1. Secondary Amines as Catalysts
2.2. Amine Catalyst Other than Hayashi’s and Jørgensen’s Catalysts
2.3. Hydrogen Bonding in Organocatalyzed Diels–Alder Reactions
2.3.1. Urea and Thiourea Catalysis
2.3.2. Squaramide-Based Catalysis
2.3.3. Brønsted Acid Catalysis
3. Conclusions
Funding
Conflicts of Interest
References
- Jurczak, J.; Bauer, T.; Chapuis, C. Intermolecular [4+2] Cycloadditions. In Stereoselective Synthesis of Organic Compounds; Helmchen, G., Mulzer, R.W.H.J., Schaumann, E.G., Eds.; Thieme Verlag: Stuttgart, Germany; New York, NY, USA, 1995; Volume 5, pp. 2735–2871. [Google Scholar]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Bhowmick, K.C.; Bihani, M.; Zhao, J.C.G. Organocatalyzed Asymmetric Diels-Alder Reactions in Aqueous or Semi-Aqueous Media. Mini-Rev. Org. Chem. 2018, 15, 3–19. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric organocatalytic cycloadditions. Tetrahedron 2012, 68, 2197–2232. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinol Silyl Ethers as Efficient Organocatalysts for the Asymmetric Michael Reaction of Aldehydes and Nitroalkenes. Angew. Chem. Int. Ed. Engl. 2005, 44, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Marigo, M.; Wabnitz, T.C.; Fielenbach, D.; Jørgensen, K.A. Enantioselective Organocatalyzed α Sulfenylation of Aldehydes. Angew. Chem. Int. Ed. 2005, 44, 794–797. [Google Scholar] [CrossRef]
- Klier, L.; Tur, F.; Poulsen, P.H.; Jørgensen, K.A. Asymmetric cycloaddition reactions catalysed by diarylprolinol silyl ethers. Chem. Soc. Rev. 2017, 46, 1080–1102. [Google Scholar] [CrossRef]
- Gotoh, H.; Uchimaru, T.; Hayashi, Y. Two Reaction Mechanisms via Iminium Ion Intermediates: The Different Reactivities of Diphenylprolinol Silyl Ether and Trifluoromethyl-Substituted Diarylprolinol Silyl Ether. Chemistry 2015, 21, 12337–12346. [Google Scholar] [CrossRef]
- Łukasik, B.; Ossowski, J.; Frankowski, S.; Albrecht, Ł. Asymmetric [4+2]-Cycloaddition of Anthracene Derivatives via Hydrazone Activation. Adv. Synth. Catal. 2023, 366, 704–709. [Google Scholar] [CrossRef]
- Hayashi, Y.; Bondzic, B.P.; Yamazaki, T.; Gupta, Y.; Ogasawara, S.; Taniguchi, T.; Monde, K. Asymmetric Diels-Alder Reaction of α-Substituted and β,β-Disubstituted α,β-Enals via Diarylprolinol Silyl Ether for the Construction of All-Carbon Quaternary Stereocenters. Chem. Eur. J. 2016, 22, 15874–15880. [Google Scholar] [CrossRef]
- Li, J.L.; Yang, K.C.; Li, Y.; Li, Q.; Zhu, H.P.; Han, B.; Peng, C.; Zhi, Y.G.; Gou, X.J. Asymmetric synthesis of bicyclic dihydropyrans via organocatalytic inverse-electron-demand oxo-Diels-Alder reactions of enolizable aliphatic aldehydes. Chem. Commun. 2016, 52, 10617–10620. [Google Scholar] [CrossRef]
- Yang, L.; Huang, W.; He, X.H.; Yang, M.C.; Li, X.; He, G.; Peng, C.; Han, B. Stereoselective Synthesis of Hydropyrano [3,2-b]indoles via Organocatalytic Asymmetric Inverse-Electron-Demand Oxa-Diels–Alder Reaction. Adv. Synth. Catal. 2016, 358, 2970–2975. [Google Scholar] [CrossRef]
- Džambaski, Z.; Tzaras, D.I.; Lee, S.; Kokotos, C.G.; Bondzic, B.P. Enantioselective Organocatalytic Enamine C−H Oxidation/Diels- Alder Reaction. Adv. Synth. Catal. 2019, 361, 1792–1797. [Google Scholar] [CrossRef]
- Bondžić, B.P.; Daskalakis, K.; Taniguchi, T.; Monde, K.; Hayashi, Y. Stereoselective Construction of Fluorinated Quaternary Stereogenic Centers via an Organocatalytic Asymmetric exo-Selective Diels-Alder Reaction in the Presence of Water. Org. Lett. 2022, 24, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Shibatomi, K.; Kawasaki, Y.; Iwasa, S. Organocatalytic enantioselective Diels–Alder reaction of 4,4,4-trifluorocrotonaldehyde. J. Fluor. Chem. 2015, 179, 77–82. [Google Scholar] [CrossRef]
- Barlose, C.L.; Ostergaard, N.L.; Bitsch, R.S.; Iversen, M.V.; Jørgensen, K.A. A Direct Organocatalytic Enantioselective Route to Functionalized trans-Diels-Alder Products Having the Norcarane Scaffold. Angew. Chem. Int. Ed. Engl. 2021, 60, 18318–18327. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Barlose, C.; Jørgensen, J.; Carlsen, B.D.; Jorgensen, K.A. Asymmetric Catalytic Aza-Diels-Alder/Ring-Closing Cascade Reaction Forming Bicyclic Azaheterocycles by Trienamine Catalysis. Chem. Eur. J. 2017, 23, 38–41. [Google Scholar] [CrossRef]
- Pawar, T.J.; Maqueda-Cabrera, E.E.; Alonso-Castro, A.J.; Olivares-Romero, J.L.; Cruz Cruz, D.; Villegas Gomez, C. Enantioselective synthesis of tetrahydrocarbazoles via trienamine catalysis and their anxiolytic-like activity. Bioorg. Med. Chem. Lett. 2020, 30, 127063–127066. [Google Scholar] [CrossRef]
- Gu, B.-Q.; Yang, W.-L.; Wu, S.-X.; Wang, Y.-B.; Deng, W.-P. Organocatalytic asymmetric synthesis of tetrahydrocarbazoles via an inverse-electron-demand Diels–Alder reaction of 2,3-indole-dienes with enals. Org. Chem. Front. 2018, 5, 3430–3434. [Google Scholar] [CrossRef]
- Wu, S.X.; Gu, B.Q.; Xu, H.; Zheng, X.; Luo, X.; Deng, W.P. Organocatalytic Asymmetric Inverse-Electron-Demand Diels-Alder Reaction of Pyrrolidone-Dienes with Enals. Adv. Synth. Catal. 2019, 361, 4302–4313. [Google Scholar] [CrossRef]
- Kim, B.; Lee, S.; Yunmi Lee, S. Organocatalytic Enantio- and Diastereoselective Diels-Alder Reaction between 2,4-Dienals and α,β-Unsaturated Esters. Adv. Synth. Catal. 2023, 365, 3887–3896. [Google Scholar] [CrossRef]
- Fulton, M.G.; Bertron, J.L.; Reed, C.W.; Lindsley, C.W. Formal Total Synthesis of Pericoannosin A. J. Org. Chem. 2019, 84, 12187–12191. [Google Scholar] [CrossRef]
- Pauvert, Y.; Charette, A.B. Asymmetric Synthesis of (-)-Cannabidiol (CBD), (-)-Delta(9)-Tetrahydrocannabinol (Delta(9)-THC) and Their cis Analogs Using an Enantioselective Organocatalyzed Diels-Alder Reaction. Org. Lett. 2024, 26, 6081–6085. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.S.; Bharani, M.S.; Jha, M.A.K.; Naik, M.S.C.; Sivaprakash, M.; Chowhan, L.R. Enantioselective Synthesis of Octahydrofuranoindole Core of Aspidosperma Alkaloids via a Diels-Alder/Reduction/Fluoroetherification Reaction Sequence. Chem. Asian J. 2023, 18, e202300419. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Inoshita, T.; Kitajima, M.; Ishikawa, H. Asymmetric Total Synthesis of Senepodine F. Org. Lett. 2023, 25, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Ushida, N.; Nagai, N.; Adachi, M.; Nishikawa, T. Concise Stereocontrolled Synthesis of an α-Carbagalactose Segment of RCAI-56, a Candidate Anticancer Agent. Synlett 2019, 30, 977–981. [Google Scholar] [CrossRef]
- Ren, J.W.; Zhou, Z.F.; Xiao, J.A.; Chen, X.Q.; Yang, H. Acid-Relayed Organocatalytic exo-Diels–Alder Cycloaddition of Cyclic Enones with 2-Vinyl-1H-indoles. Eur. J. Org. Chem. 2016, 2016, 1264–1268. [Google Scholar] [CrossRef]
- Pasha, M.; Tanaka, F. Organocatalytic diastereo- and enantioselective oxa-hetero-Diels-Alder reactions of enones with aryl trifluoromethyl ketones for the synthesis of trifluoromethyl-substituted tetrahydropyrans. Org. Biomol. Chem. 2021, 19, 9242–9250. [Google Scholar] [CrossRef]
- Mose, R.; Jensen, M.E.; Preegel, G.; Jørgensen, K.A. Direct Access to Multifunctionalized Norcamphor Scaffolds by Asymmetric Organocatalytic Diels-Alder Reactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 13630–13634. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Krishna, P.M. Asymmetric Synthesis of Nature-Inspired Bioactive Spiro Compounds through Organocatalytic Diels–Alder Reactions. Asian J. Org. Chem. 2016, 5, 729–734. [Google Scholar] [CrossRef]
- Maity, R.; Pan, S.C. Dienamine-Mediated Asymmetric Inverse-Electron-Demand Hetero-Diels–Alder Reaction of Linear Deconjugated Enones: Diversity-Oriented Synthesis of 3,4-Dihydropyrans. Eur. J. Org. Chem. 2017, 2017, 871–874. [Google Scholar] [CrossRef]
- Vishwanath, M.; Vinayagam, P.; Gajulapalli, V.P.R.; Kesavan, V. Asymmetric Organocatalytic Assembly of Oxindoles Fused with Spiro-3,4-dihydropyrans with Three Contiguous Stereocenters Consisting of Vicinal Quaternary Centers. Asian J. Org. Chem. 2016, 5, 613–616. [Google Scholar] [CrossRef]
- Min, C.; Lin, C.T.; Seidel, D. Catalytic enantioselective intramolecular aza-diels-alder reactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 6608–6612. [Google Scholar] [CrossRef] [PubMed]
- Frankowski, S.; Skrzyńska, A.; Sieroń, L.; Albrecht, Ł. Deconjugated-Ketone-Derived Dienolates in Remote, Stereocontrolled, Aromative aza-Diels-Alder Cycloaddition. Adv. Synth. Catal. 2020, 362, 2658–2665. [Google Scholar] [CrossRef]
- Bania, N.; Barman, D.; Pan, S.C. Organocatalytic Asymmetric Inverse-Electron-Demand Diels-Alder Reaction between Alkylidene Pyrazolones and Allyl Ketones: Access to Tetrahydropyrano[2,3-c]pyrazoles. J. Org. Chem. 2023, 88, 9584–9593. [Google Scholar] [CrossRef]
- Gao, T.-P.; Liu, D.; Lin, J.-B.; Hu, X.-Q.; Wang, Z.-Y.; Xu, P.-F. Direct construction of chiral quaternary dihydropyranones through highly enantioselective organocatalytic hetero-Diels–Alder reactions of olefinic azlactones. Org. Chem. Front. 2016, 3, 598–602. [Google Scholar] [CrossRef]
- Joshi, H.; Yadav, A.; Das, A.; Singh, V.K. Organocatalytic Asymmetric Hetero-Diels-Alder Reaction of in Situ Generated Dienes: Access to α,β-Unsaturated delta-Lactones Featuring CF(3)-Substituted Quaternary Stereocenter. J. Org. Chem. 2020, 85, 3202–3212. [Google Scholar] [CrossRef]
- Khan, Z.A.; Singh, V.K. Asymmetric Organocatalytic Diels-Alder Reaction of Olefinic Azlactones with Unsaturated Thiazolones: Access to Spirocyclohexenone Thiazolone Skeletons. J. Org. Chem. 2024, 89, 15271–15281. [Google Scholar] [CrossRef]
- Yang, K.X.; Ji, D.S.; Zheng, H.; Gu, Y.; Xu, P.F. Organocatalytic inverse-electron-demand Diels-Alder reaction between 5-alkenyl thiazolones and β,gamma-unsaturated carbonyl compounds. Chem. Commun. 2022, 58, 4227–4230. [Google Scholar] [CrossRef]
- Bauer, T.; Hakim, Y.Z.; Morawska, P. Recent Advances in the Enantioselective Radical Reactions. Molecules 2023, 28, 6252. [Google Scholar] [CrossRef]
- Dell’Amico, L.; Vega-Penaloza, A.; Cuadros, S.; Melchiorre, P. Enantioselective Organocatalytic Diels-Alder Trapping of Photochemically Generated Hydroxy-o-Quinodimethanes. Angew. Chem. Int. Ed. Engl. 2016, 55, 3313–3317. [Google Scholar] [CrossRef]
- Xiang, M.; Li, C.-Y.; Zhang, J.; Zou, Y.; Li, W.-S.; Tian, F.; Wan, W.-J.; Wang, L.-X. Organocatalytic enantioselective Diels–Alder reaction between hydroxymaleimides and in situ generated nitrosoalkenes for direct preparation of chiral hemiketals with 1,2-oxazine skeleton. Org. Chem. Front. 2021, 8, 6215–6219. [Google Scholar] [CrossRef]
- Huang, L.J.; Weng, J.; Wang, S.; Lu, G. Organocatalytic Diels–Alder Reaction of 2-Vinylindoles with Methyleneindolinones: An Efficient Approach to Functionalized Carbazolespirooxindoles. Adv. Synth. Catal. 2015, 357, 993–1003. [Google Scholar] [CrossRef]
- Lin, Y.; Hou, X.Q.; Li, B.Y.; Du, D.M. Organocatalytic Remote Asymmetric Inverse-Electron-Demand Oxa-Diels-Alder Reaction of Allyl Ketones with Isatin-Derived Unsaturated Keto Esters. Adv. Synth. Catal. 2020, 362, 5728–5735. [Google Scholar] [CrossRef]
- Weilbeer, C.; Sickert, M.; Naumov, S.; Schneider, C. The Brønsted Acid-Catalyzed, Enantioselective Aza-Diels-Alder Reaction for the Direct Synthesis of Chiral Piperidones. Chem. Eur. J. 2017, 23, 513–518. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Hofmann, F.; Moock, D.; Schneider, C. Intramolecular Aza-Diels-Alder Reactions of ortho-Quinone Methide Imines: Rapid, Catalytic, and Enantioselective Assembly of Benzannulated Quinolizidines. Angew. Chem. Int. Ed. Engl. 2018, 57, 4774–4778. [Google Scholar] [CrossRef]
- Zhao, J.J.; Sun, S.B.; He, S.H.; Wu, Q.; Shi, F. Catalytic asymmetric inverse-electron-demand oxa-Diels-Alder reaction of in situ generated ortho-quinone methides with 3-methyl-2-vinylindoles. Angew. Chem. Int. Ed. Engl. 2015, 54, 5460–5464. [Google Scholar] [CrossRef]
- Xie, Y.; List, B. Catalytic Asymmetric Intramolecular [4+2] Cycloaddition of In Situ Generated ortho-Quinone Methides. Angew. Chem. Int. Ed. Engl. 2017, 56, 4936–4940. [Google Scholar] [CrossRef]
- Ghosh, S.; Das, S.; De, C.K.; Yepes, D.; Neese, F.; Bistoni, G.; Leutzsch, M.; List, B. Strong and Confined Acids Control Five Stereogenic Centers in Catalytic Asymmetric Diels-Alder Reactions of Cyclohexadienones with Cyclopentadiene. Angew. Chem. Int. Ed. Engl. 2020, 59, 12347–12351. [Google Scholar] [CrossRef]
- Gatzenmeier, T.; van Gemmeren, M.; Xie, Y.; Höfler, D.; Leutzsch, M.; List, B. Asymmetric Lewis acid organocatalysis of the Diels–Alder reaction by a silylated C–H acid. Science 2016, 351, 949–952. [Google Scholar] [CrossRef]
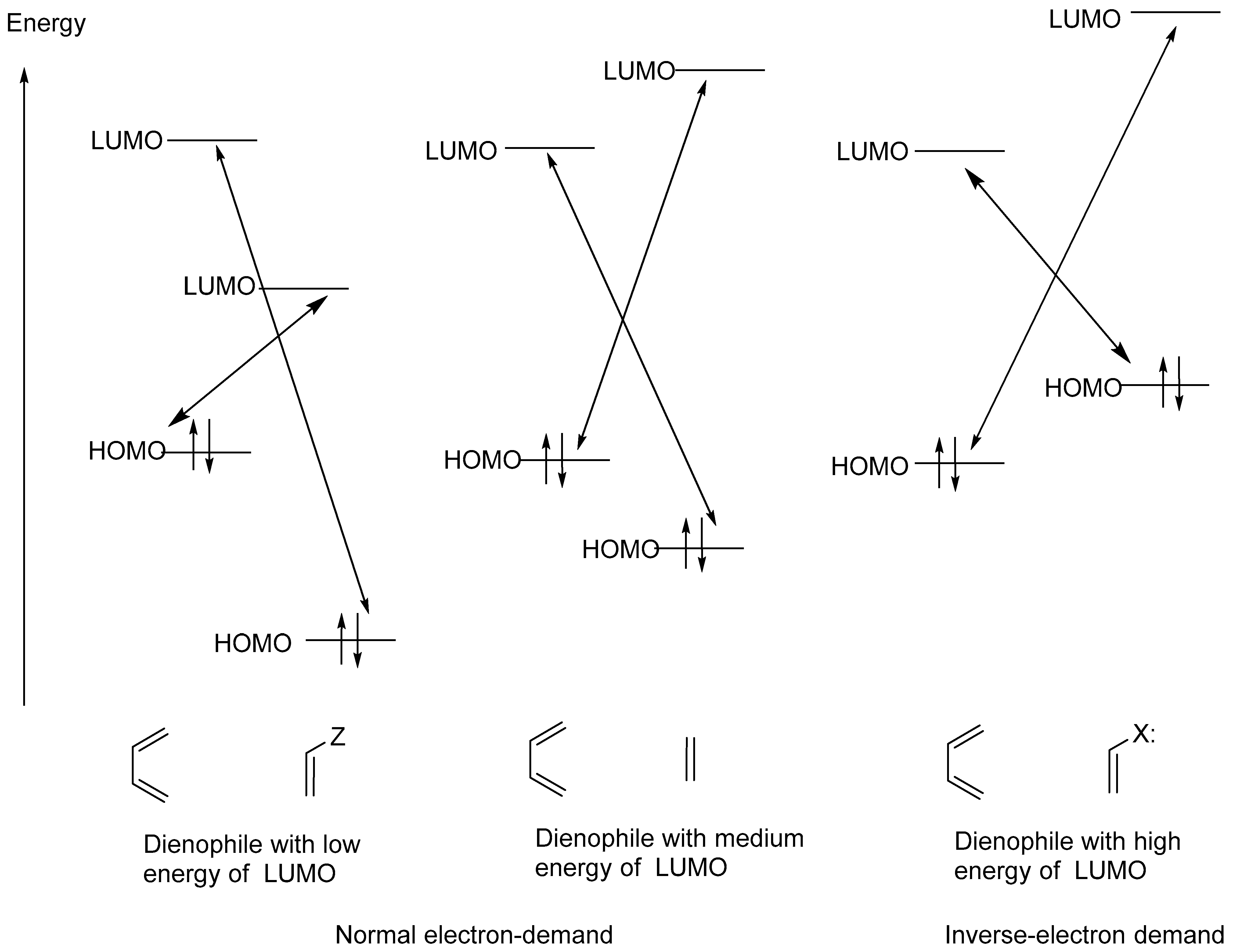
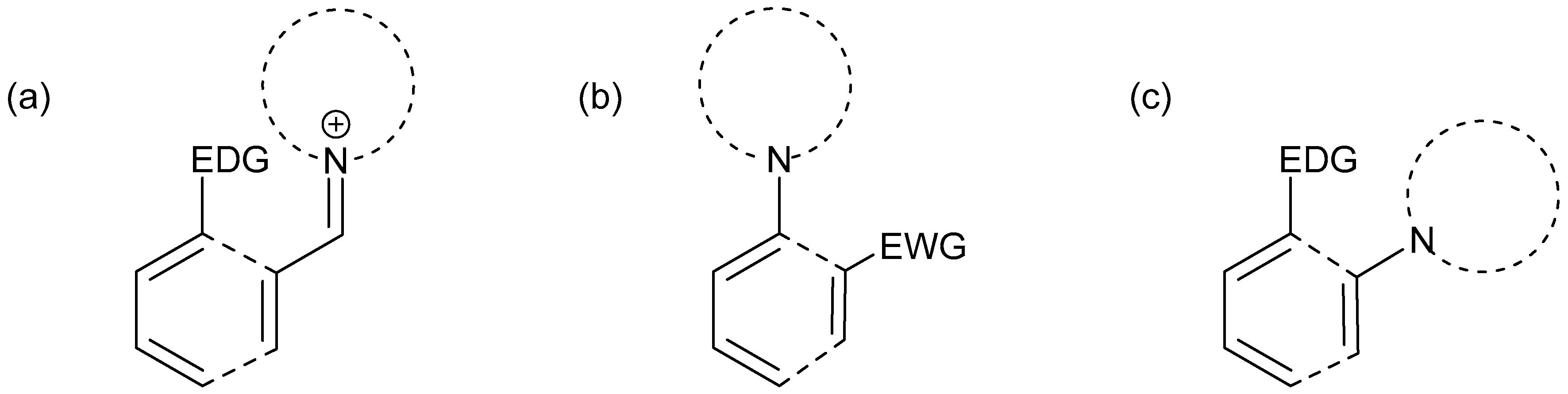
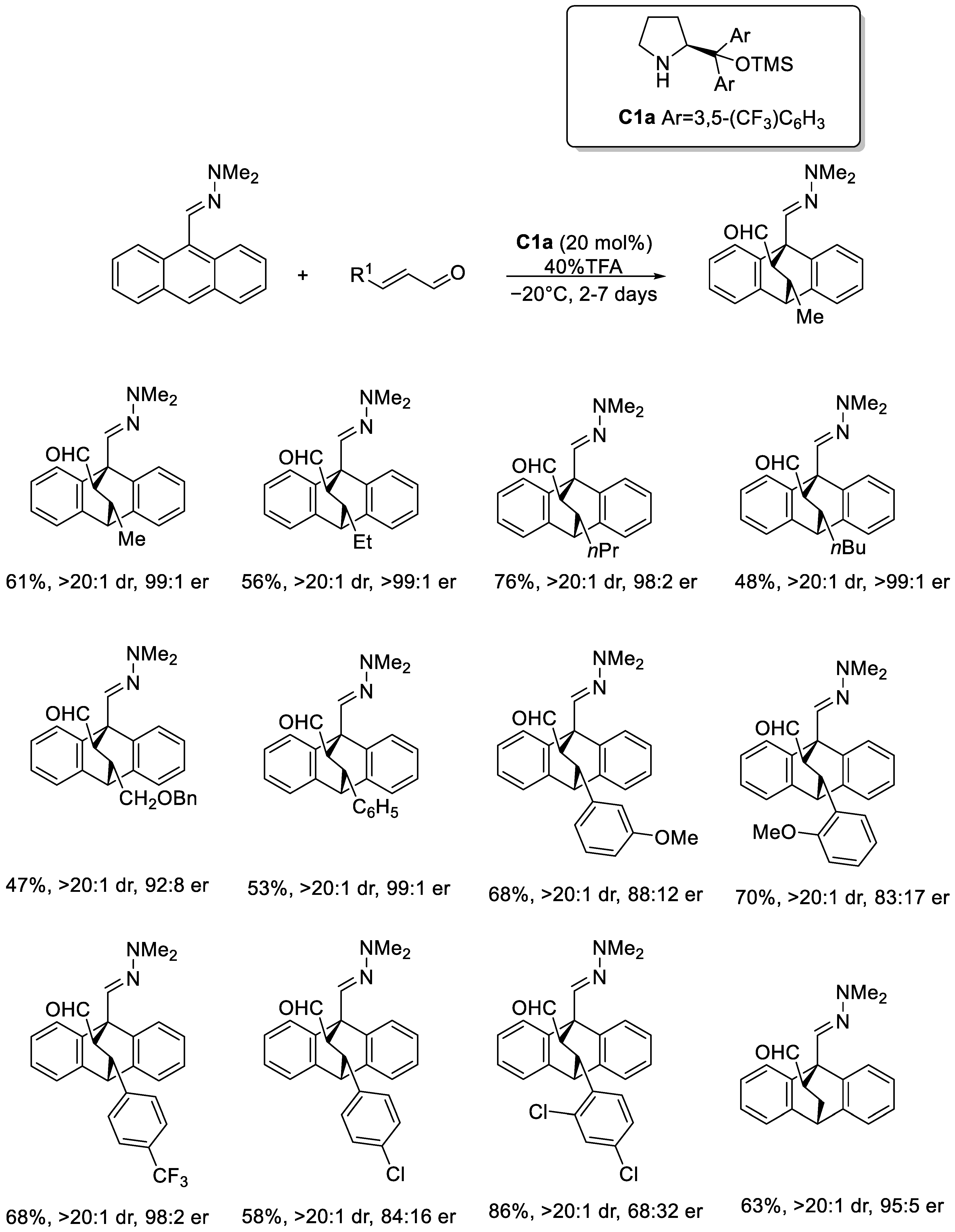
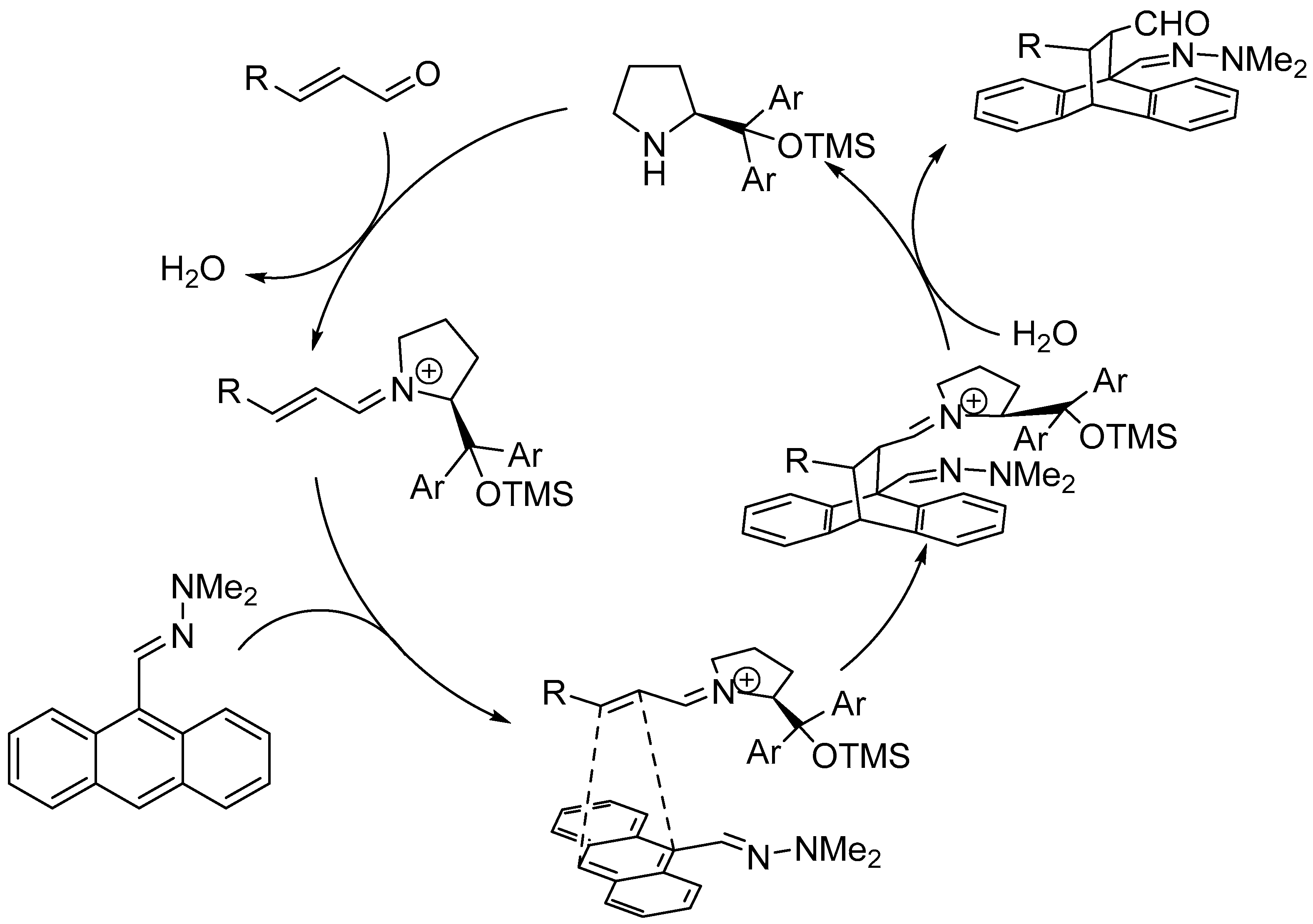
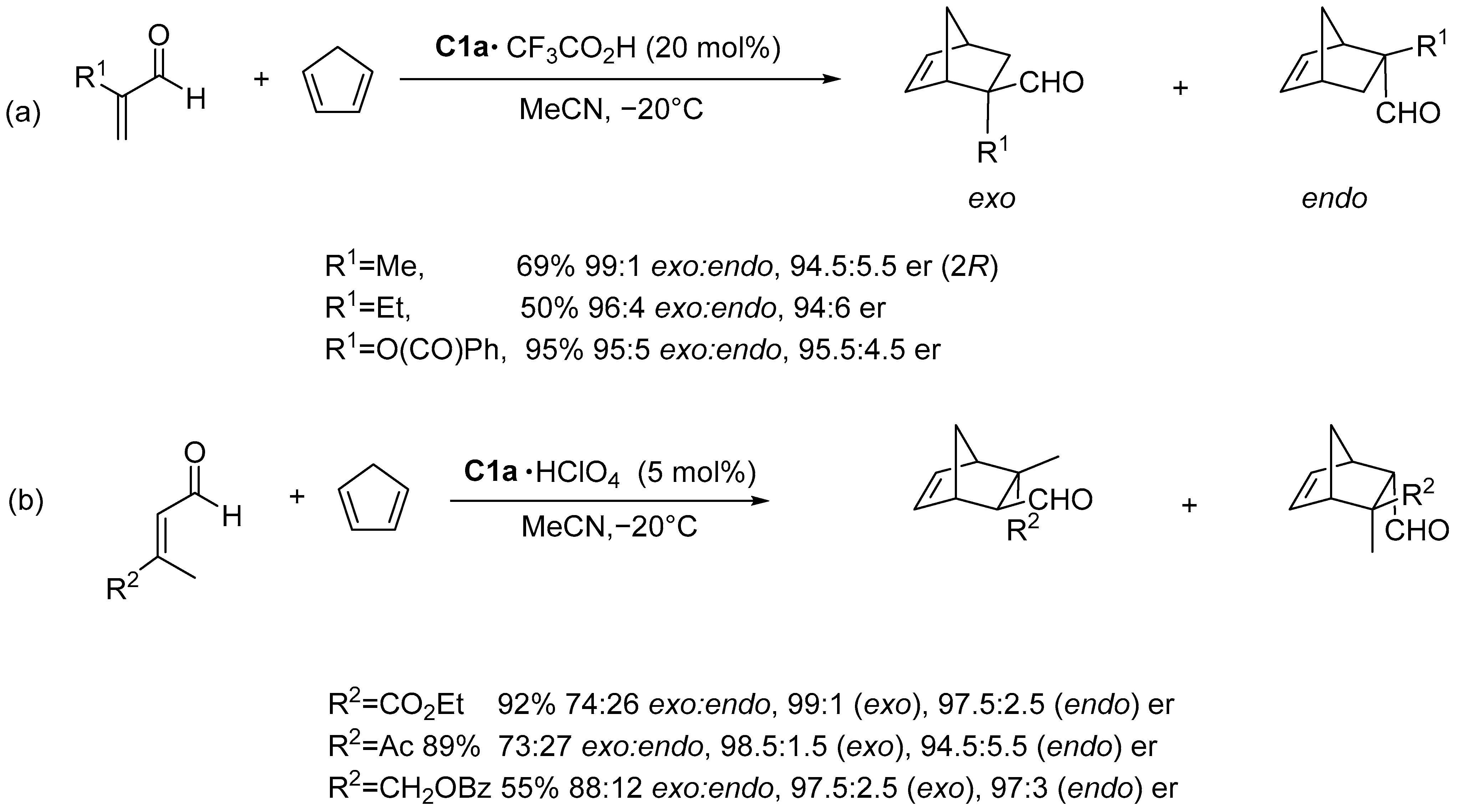
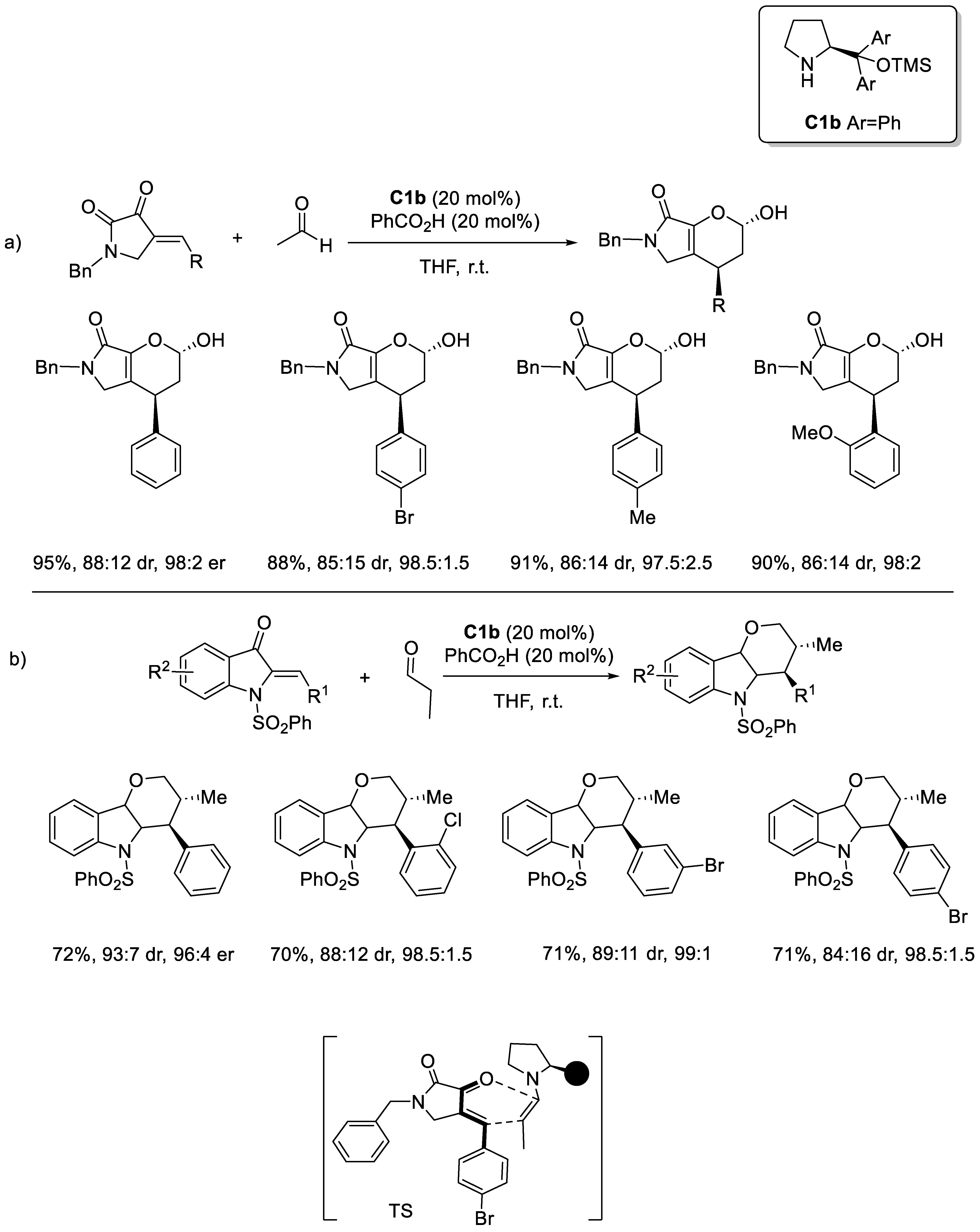
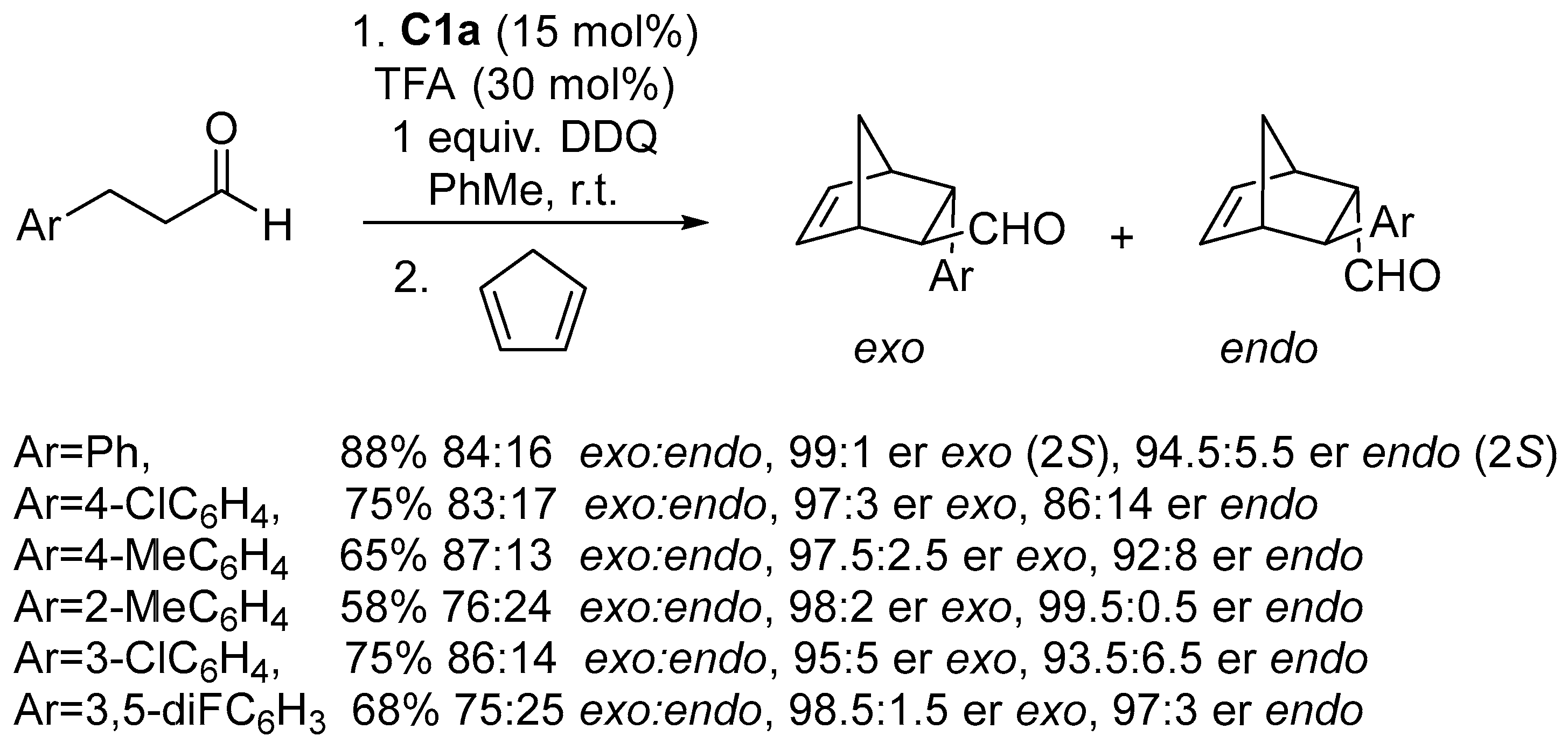
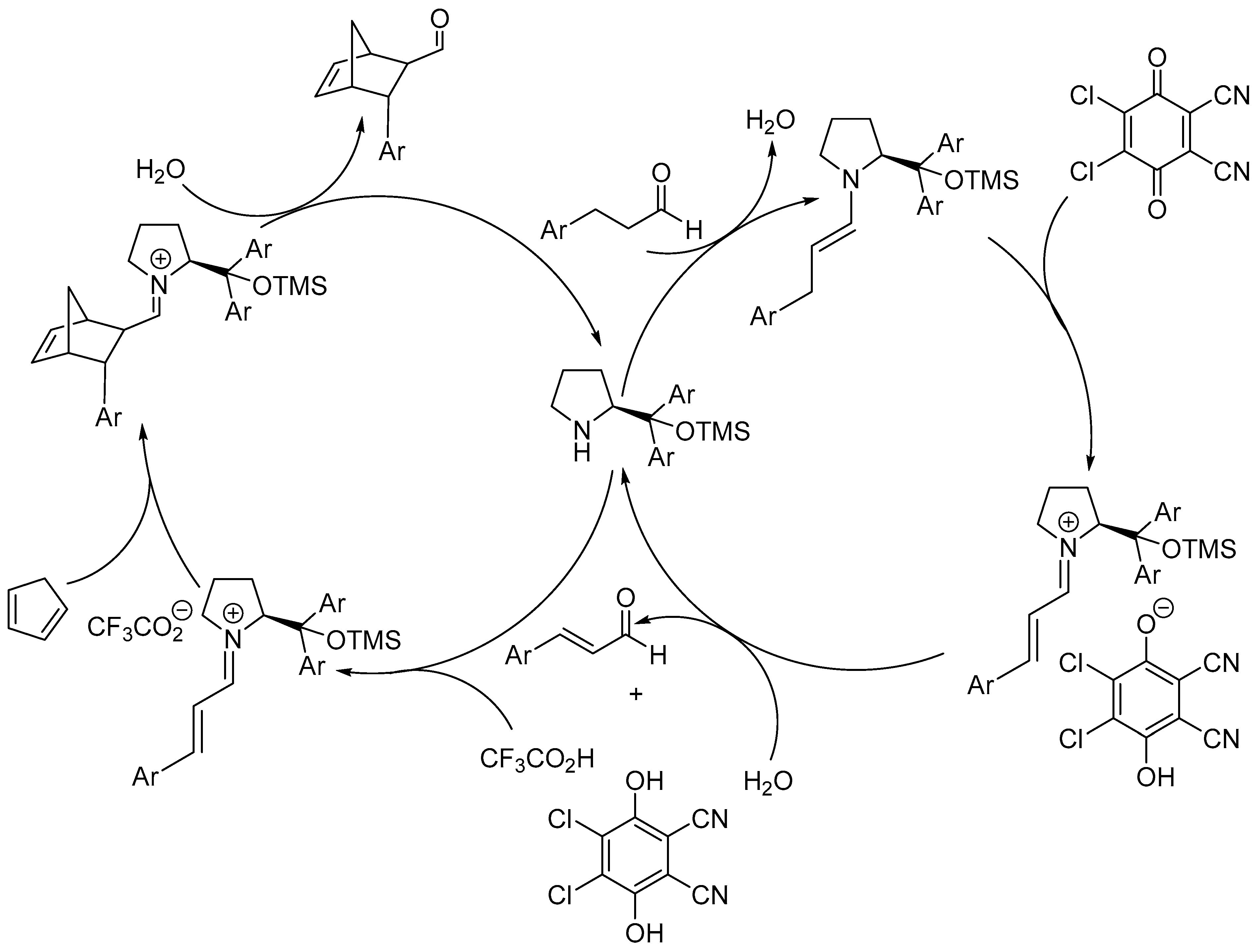
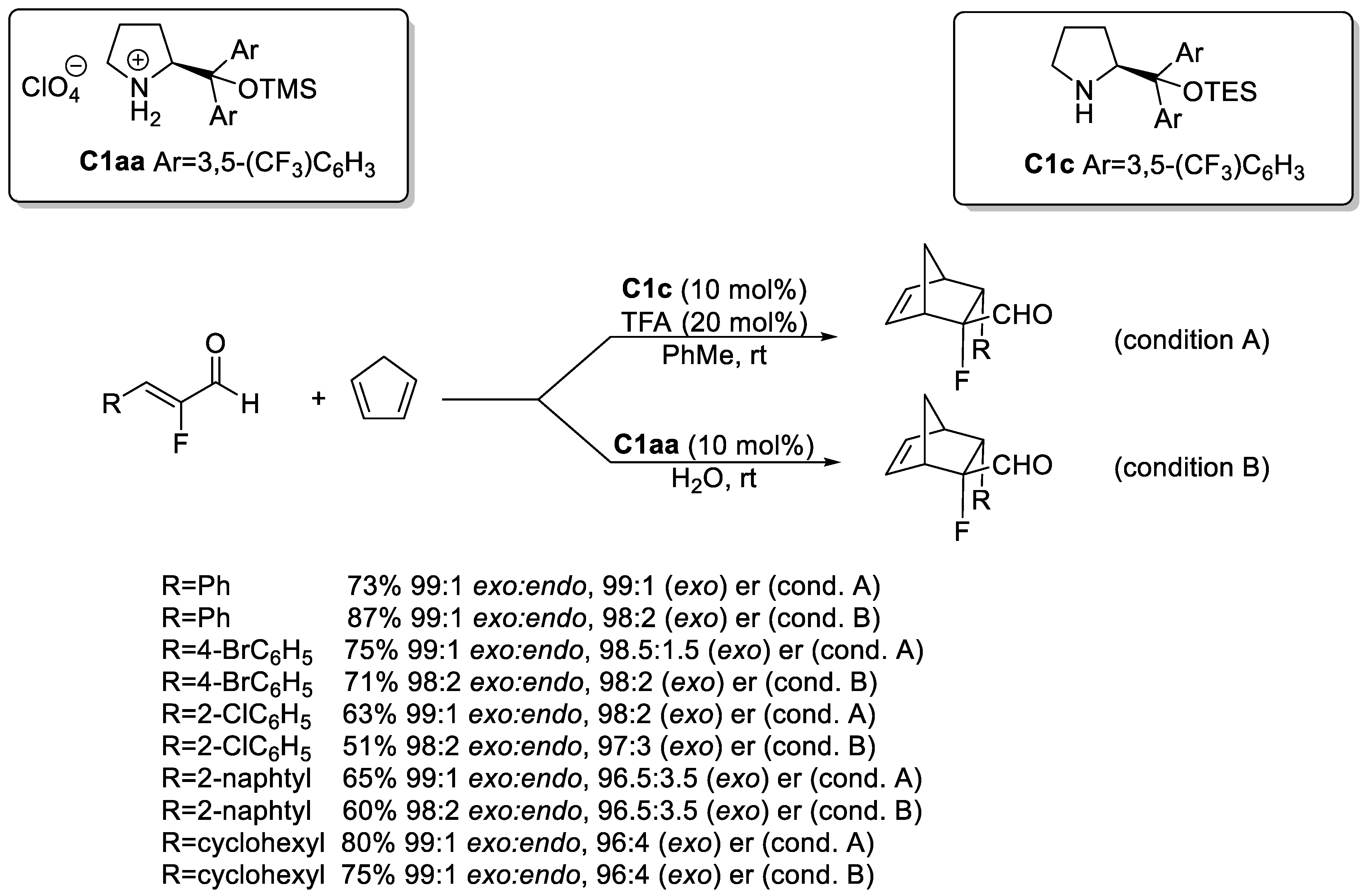
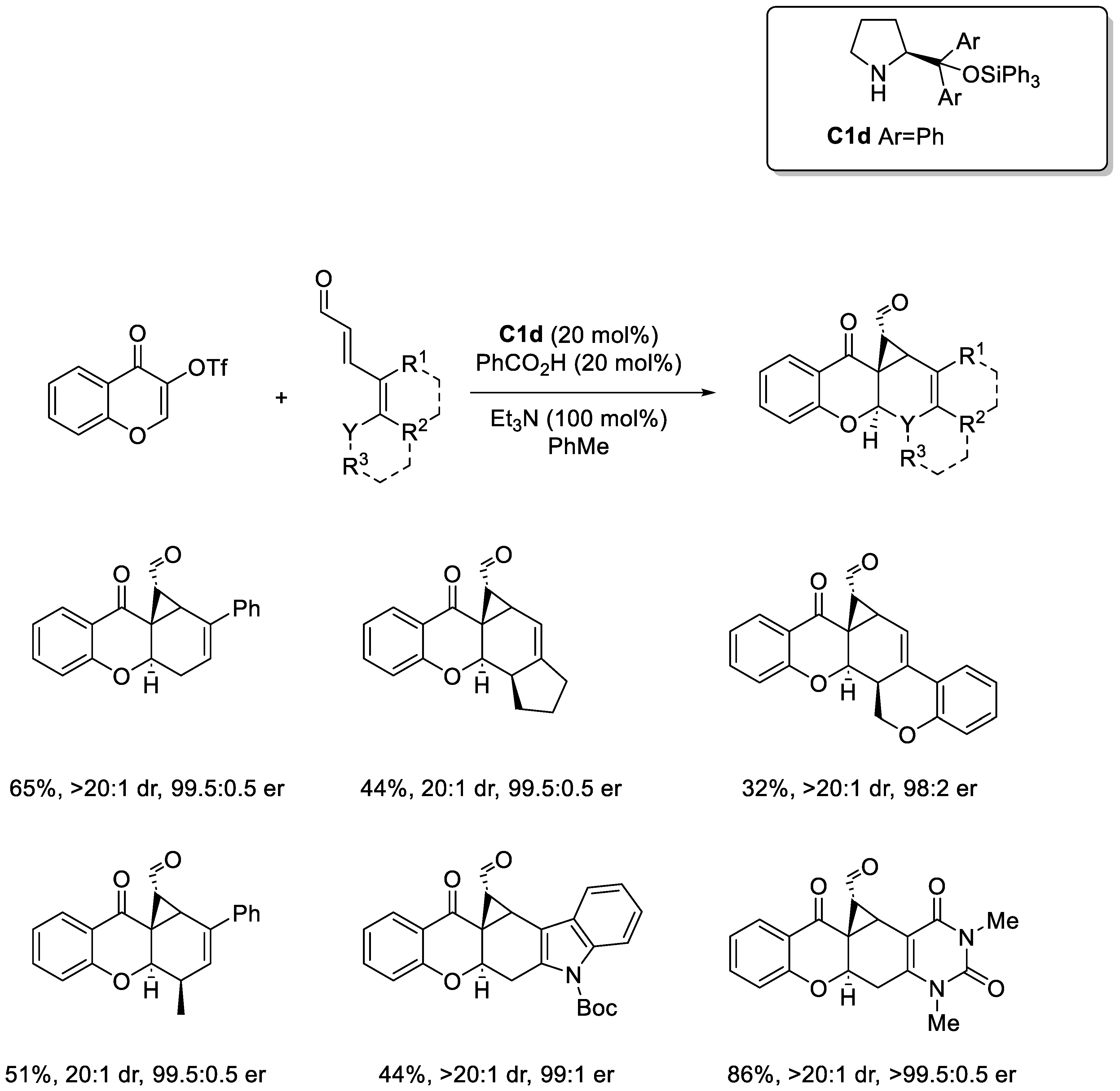
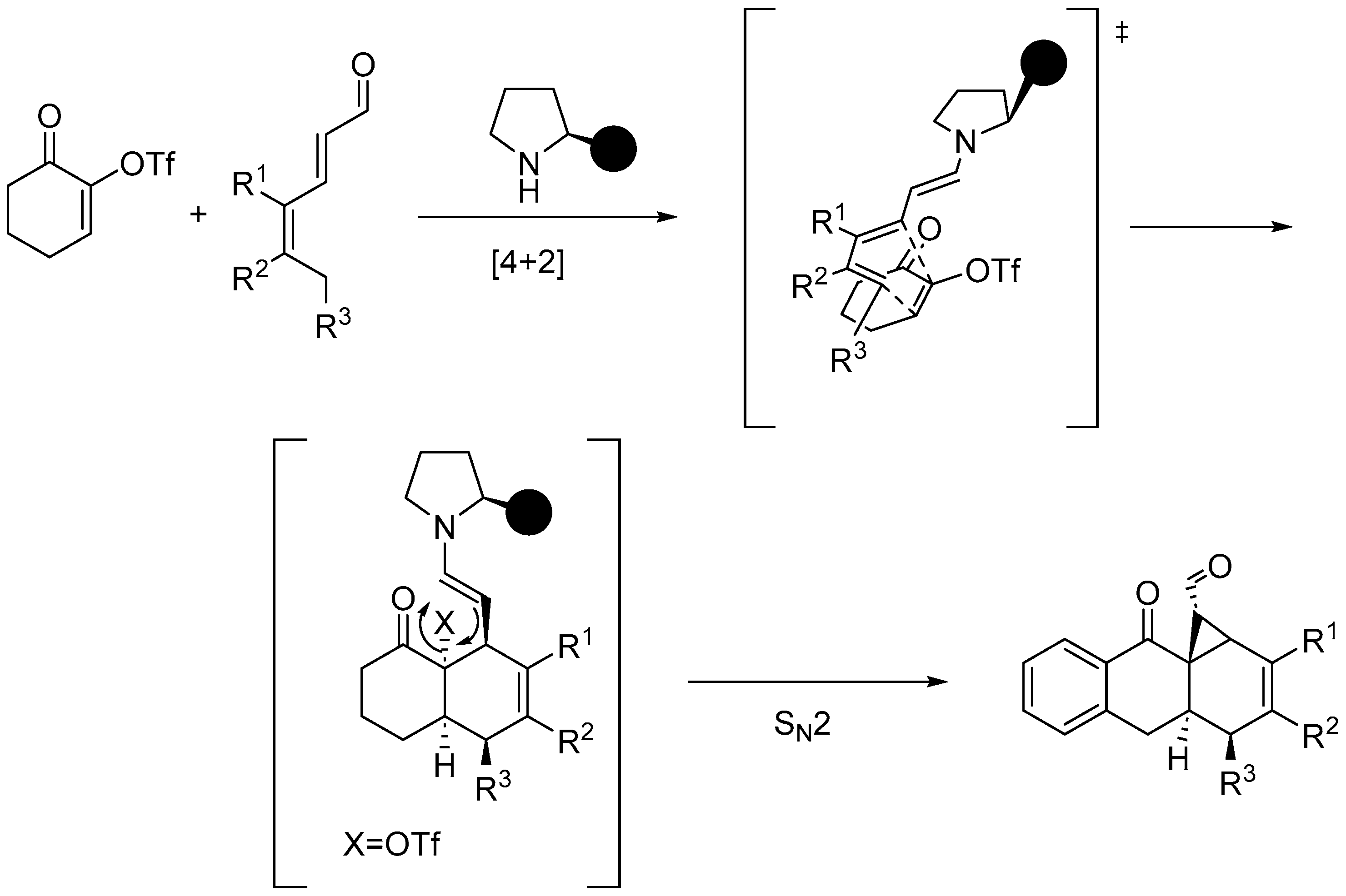

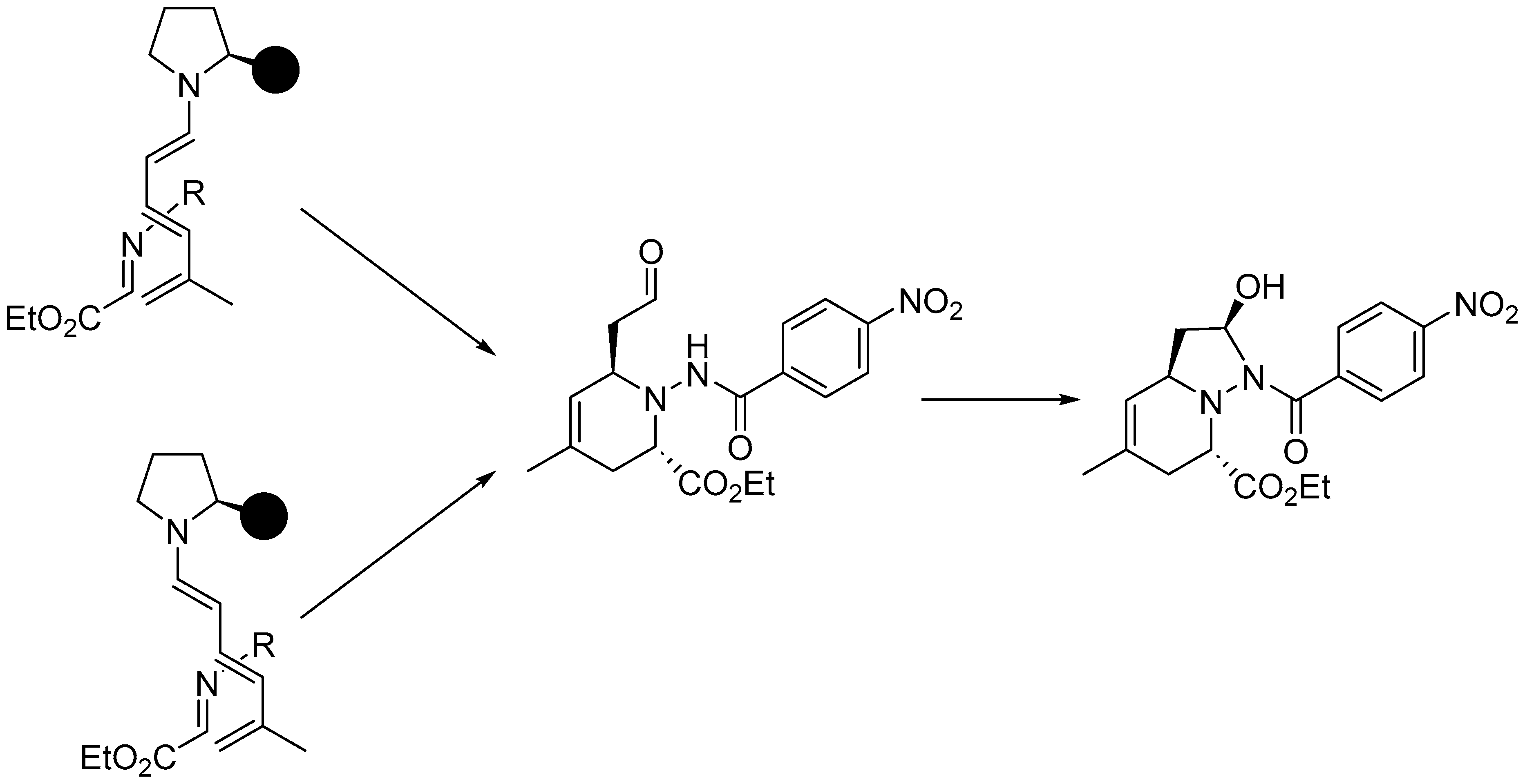
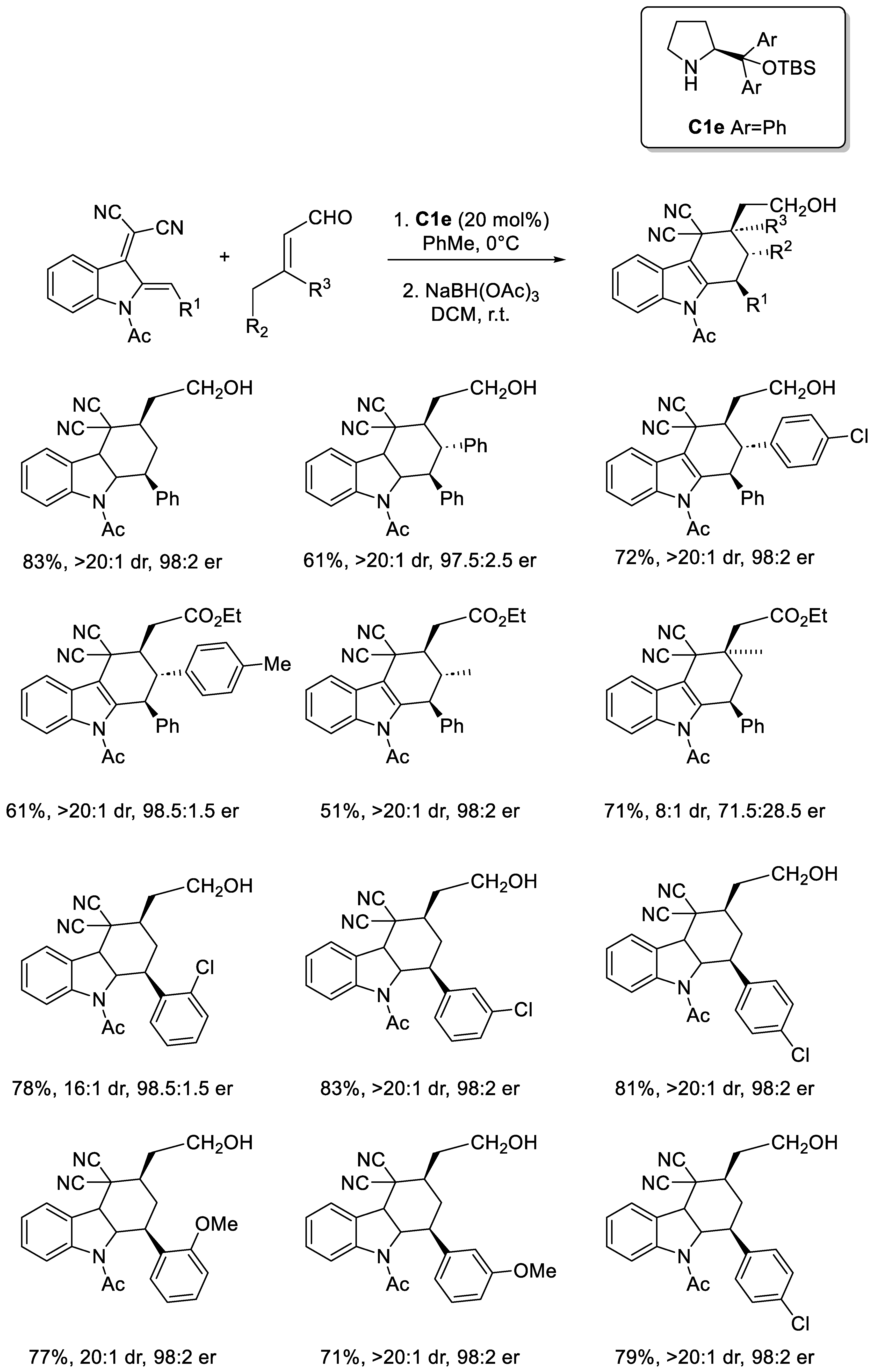
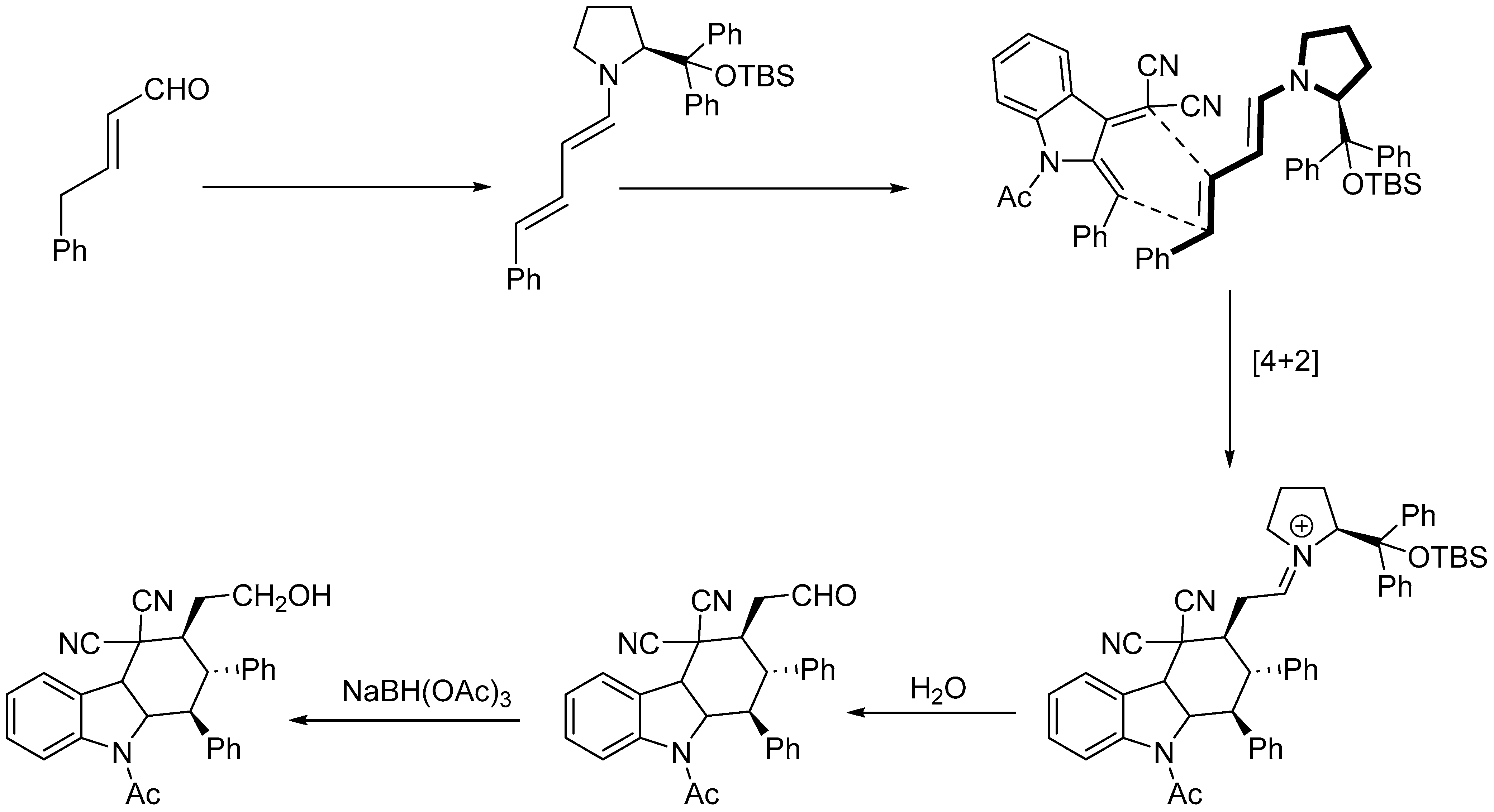
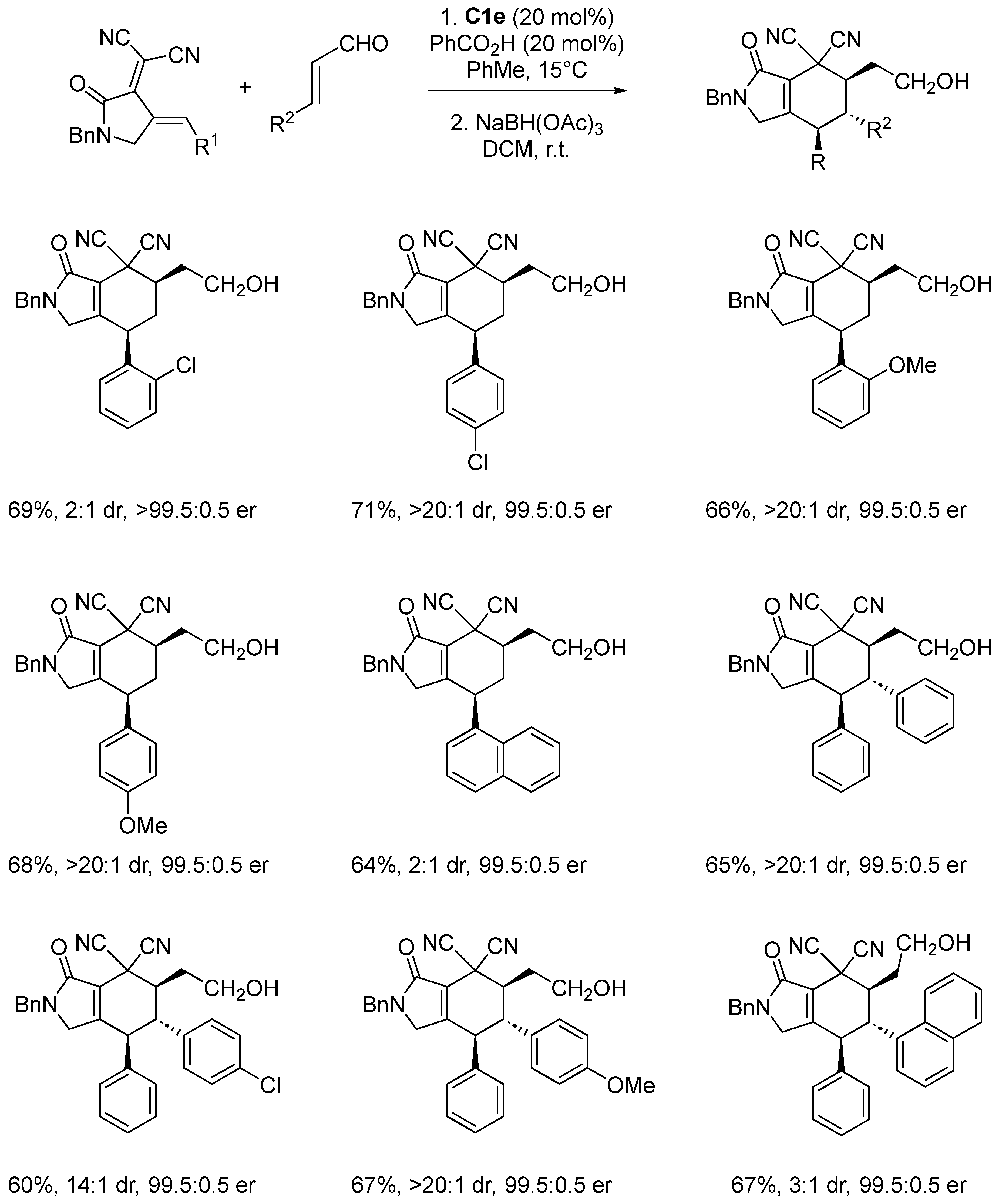
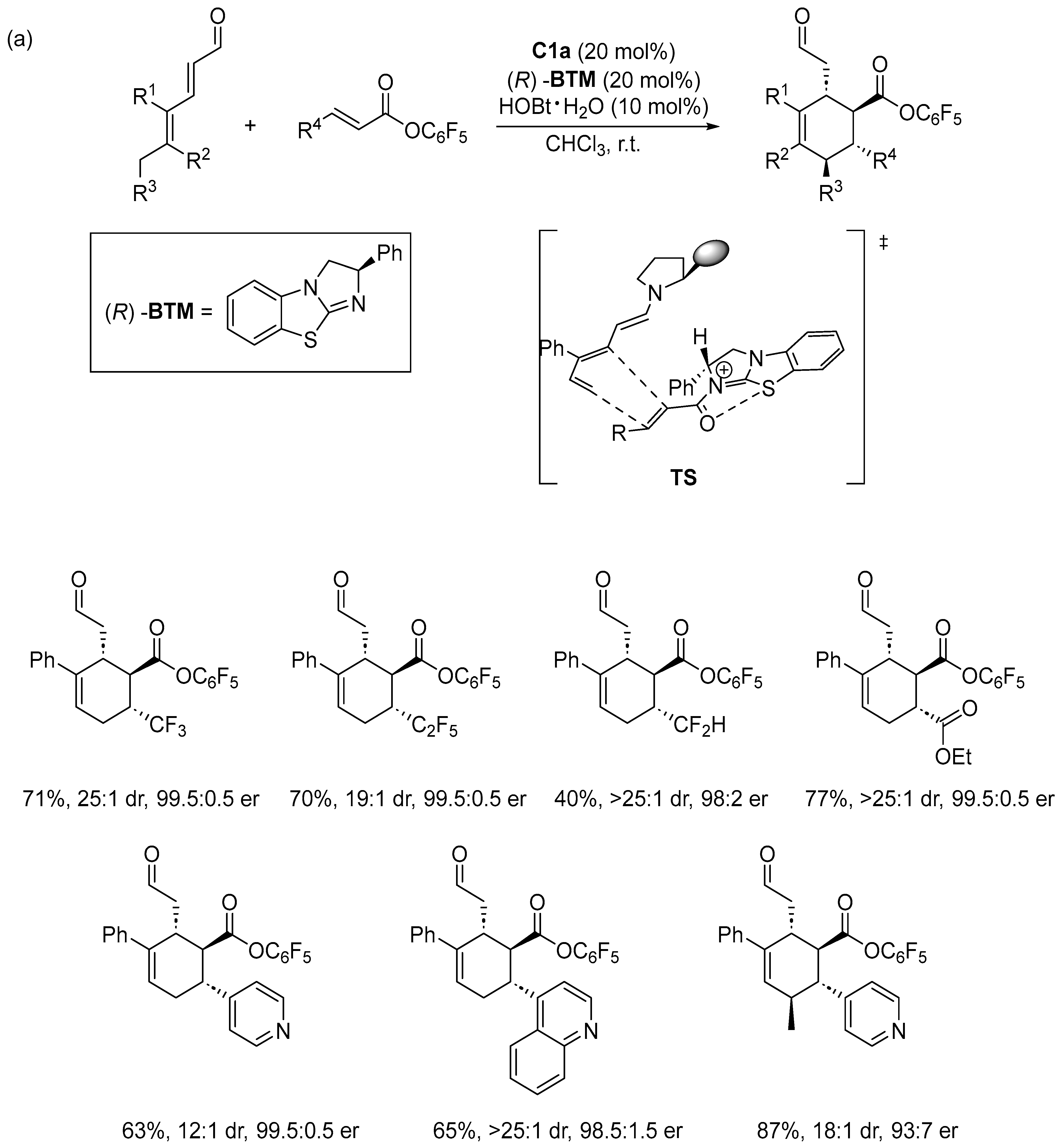
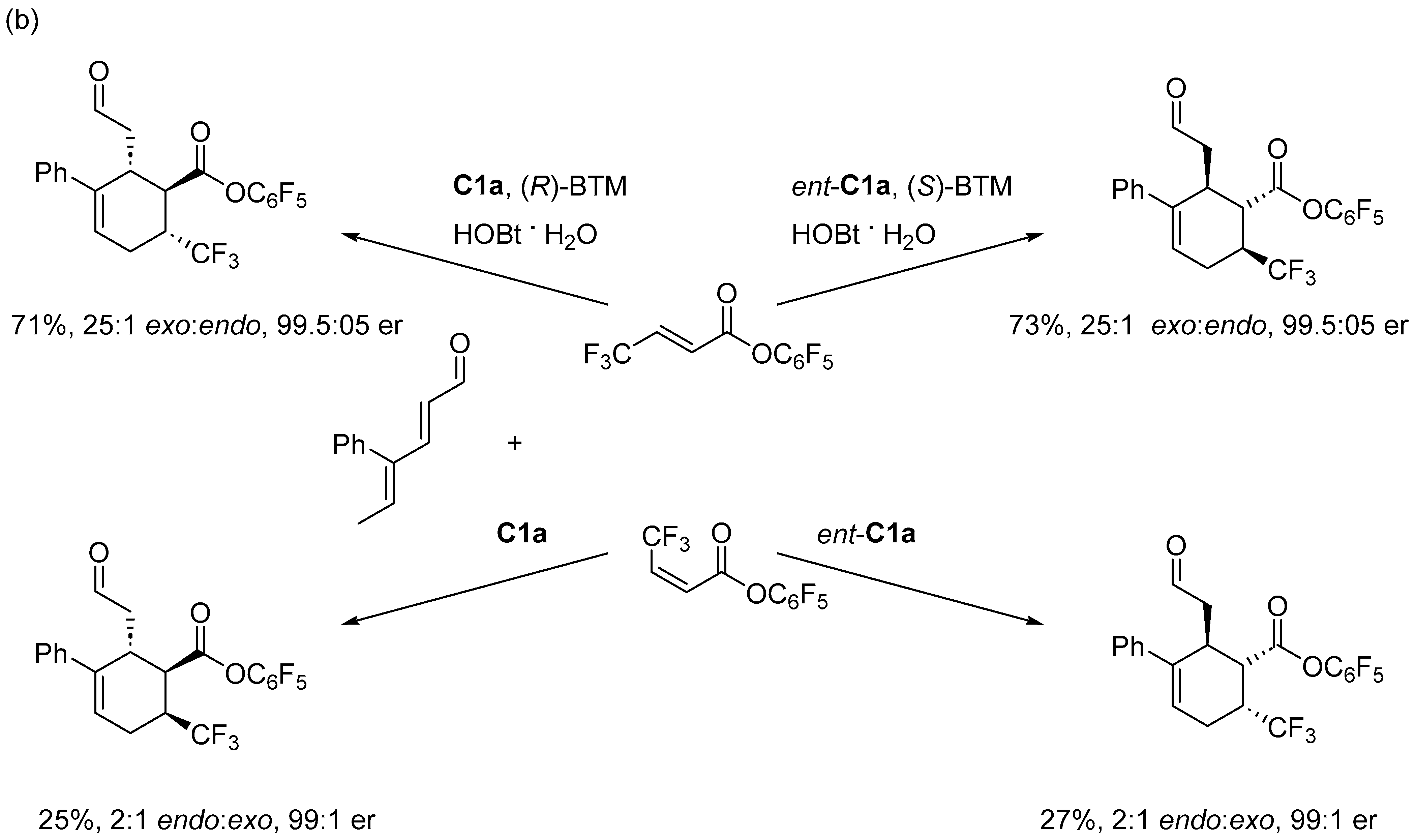
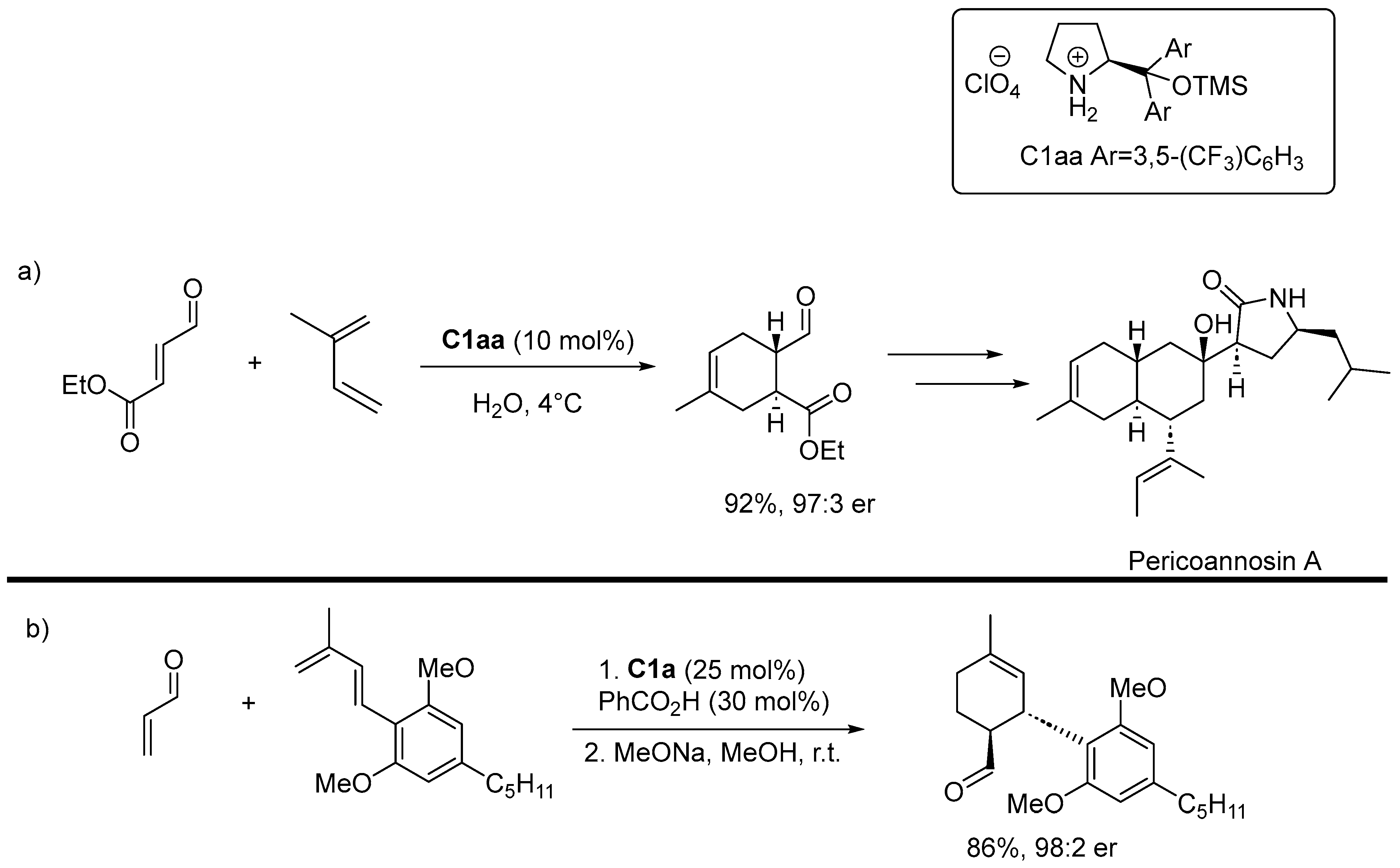
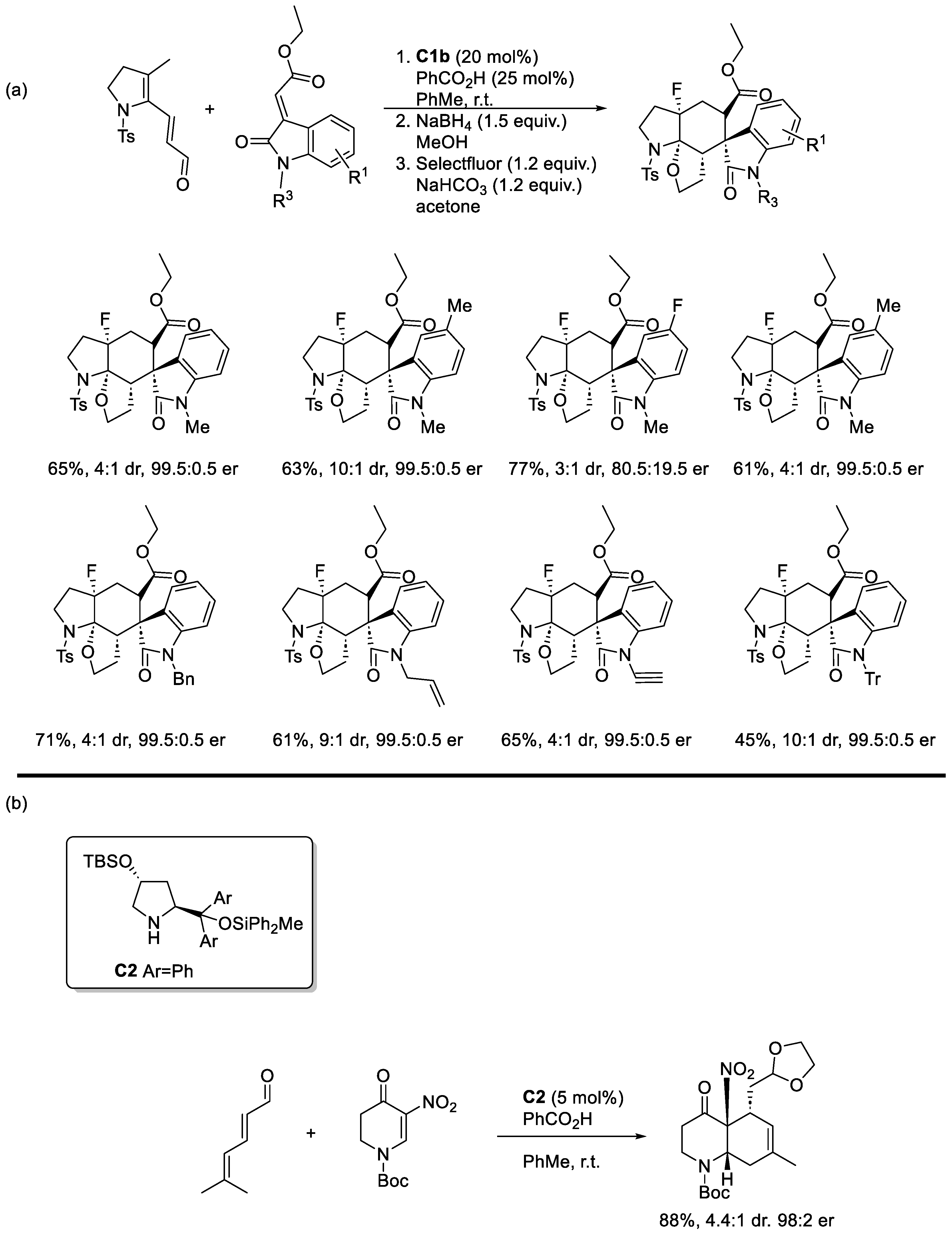
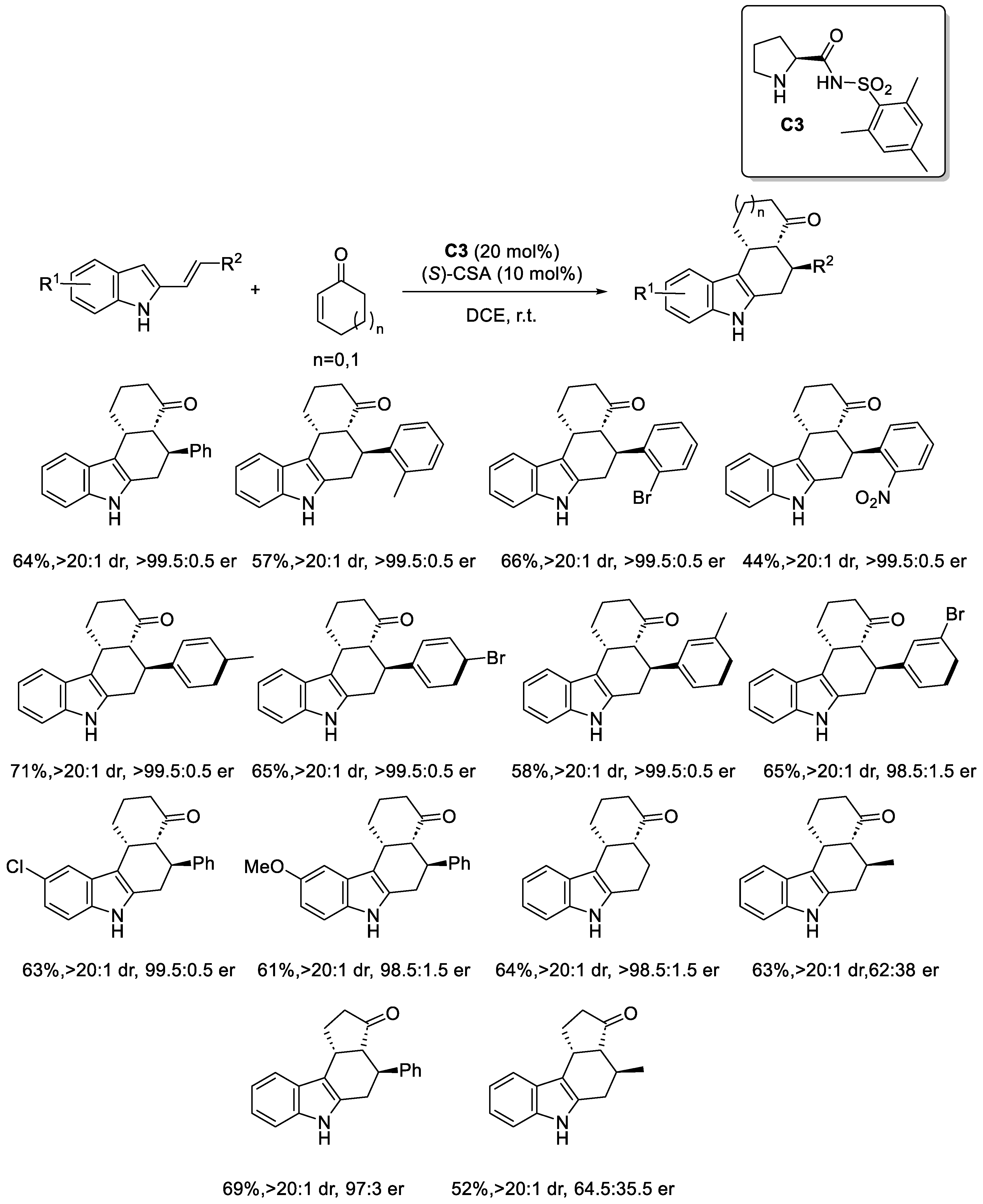
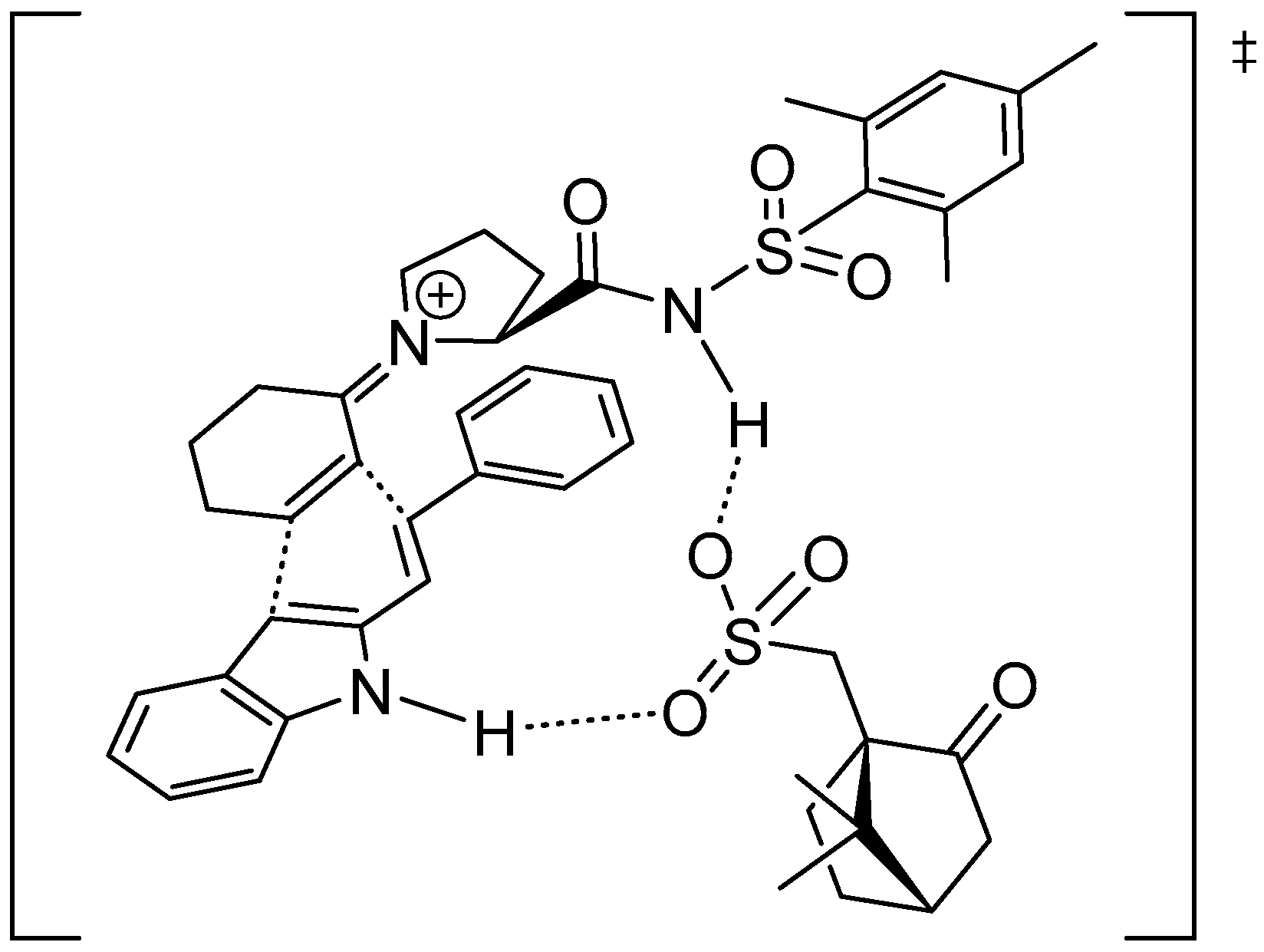

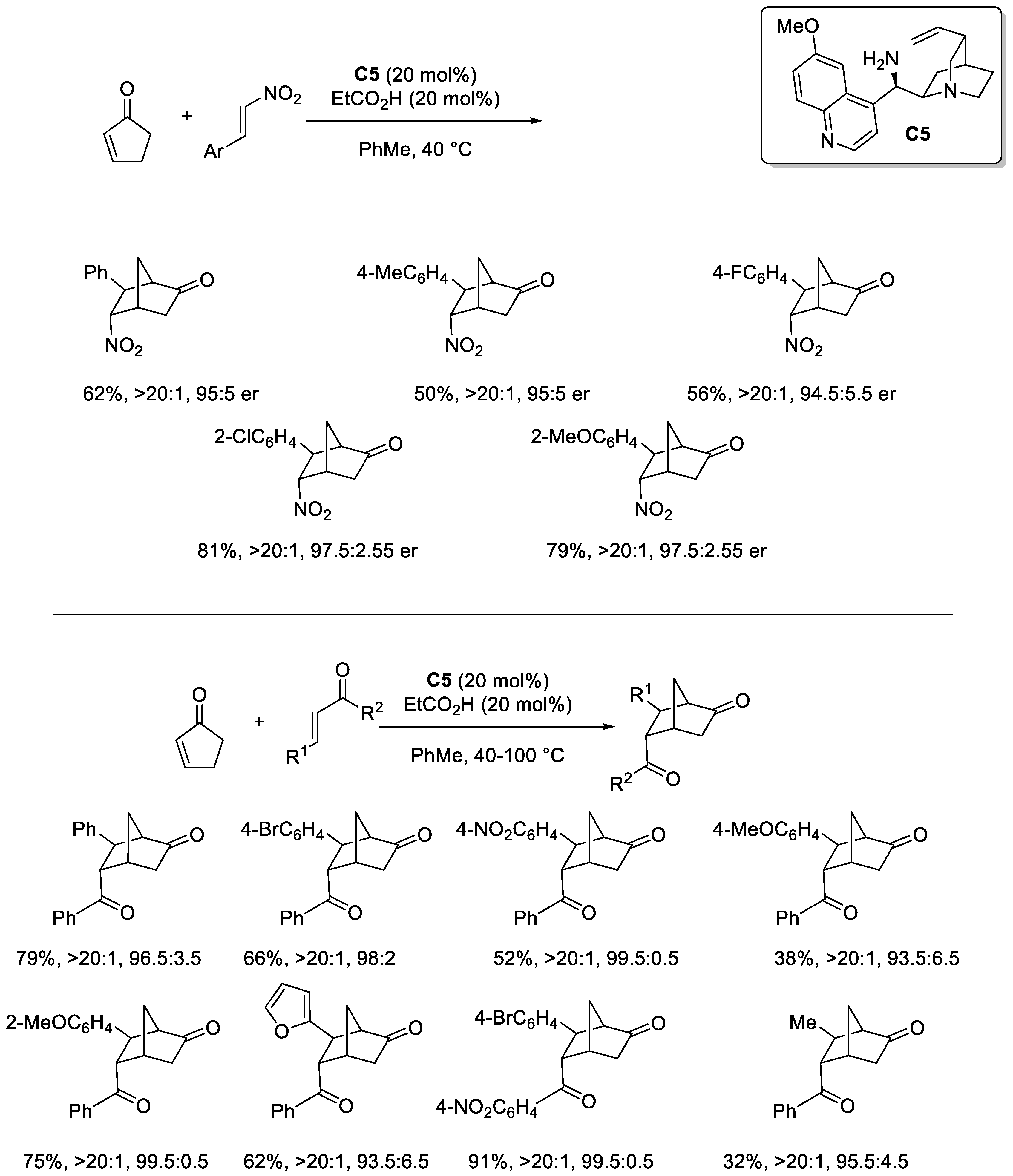
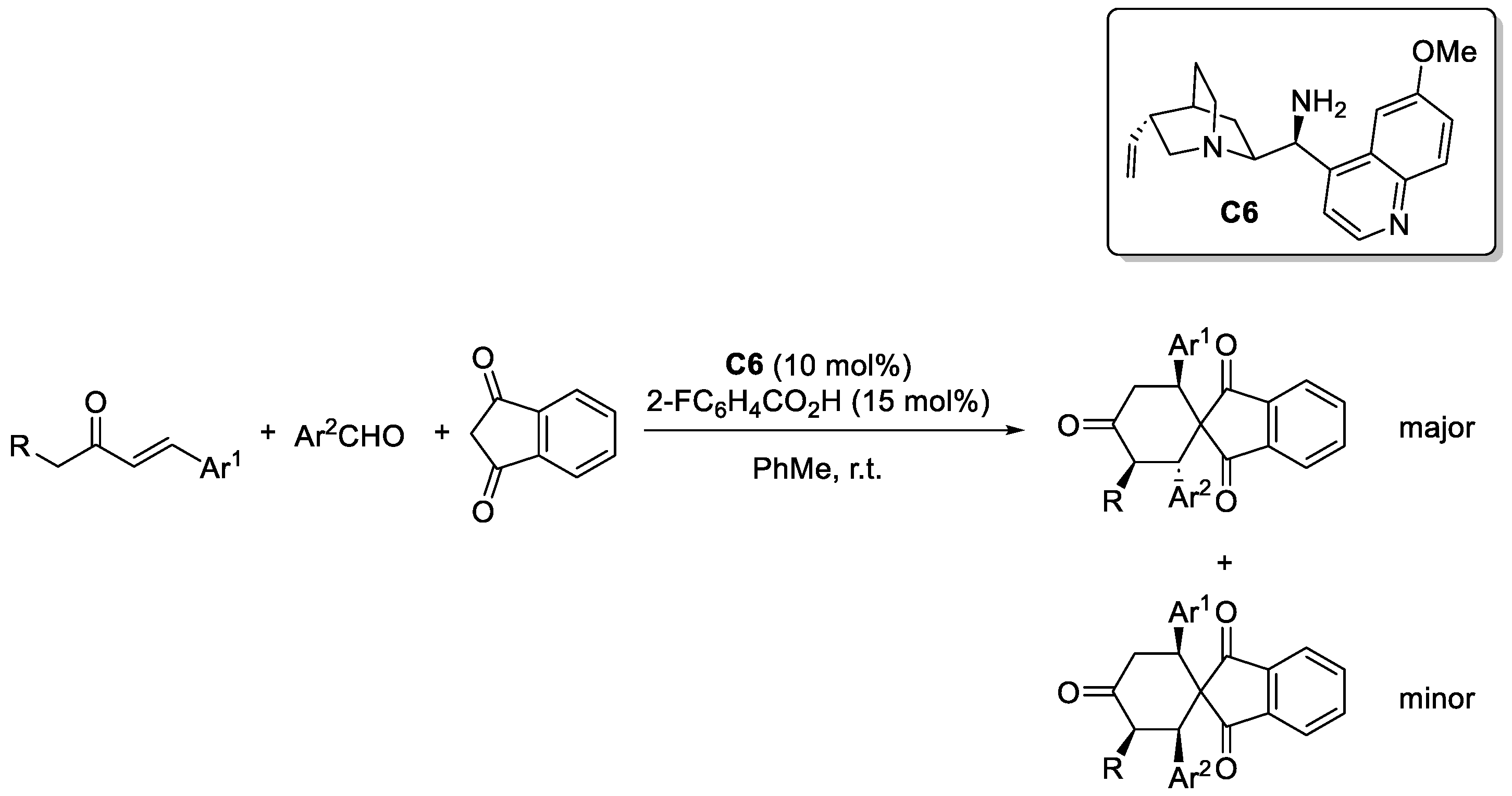
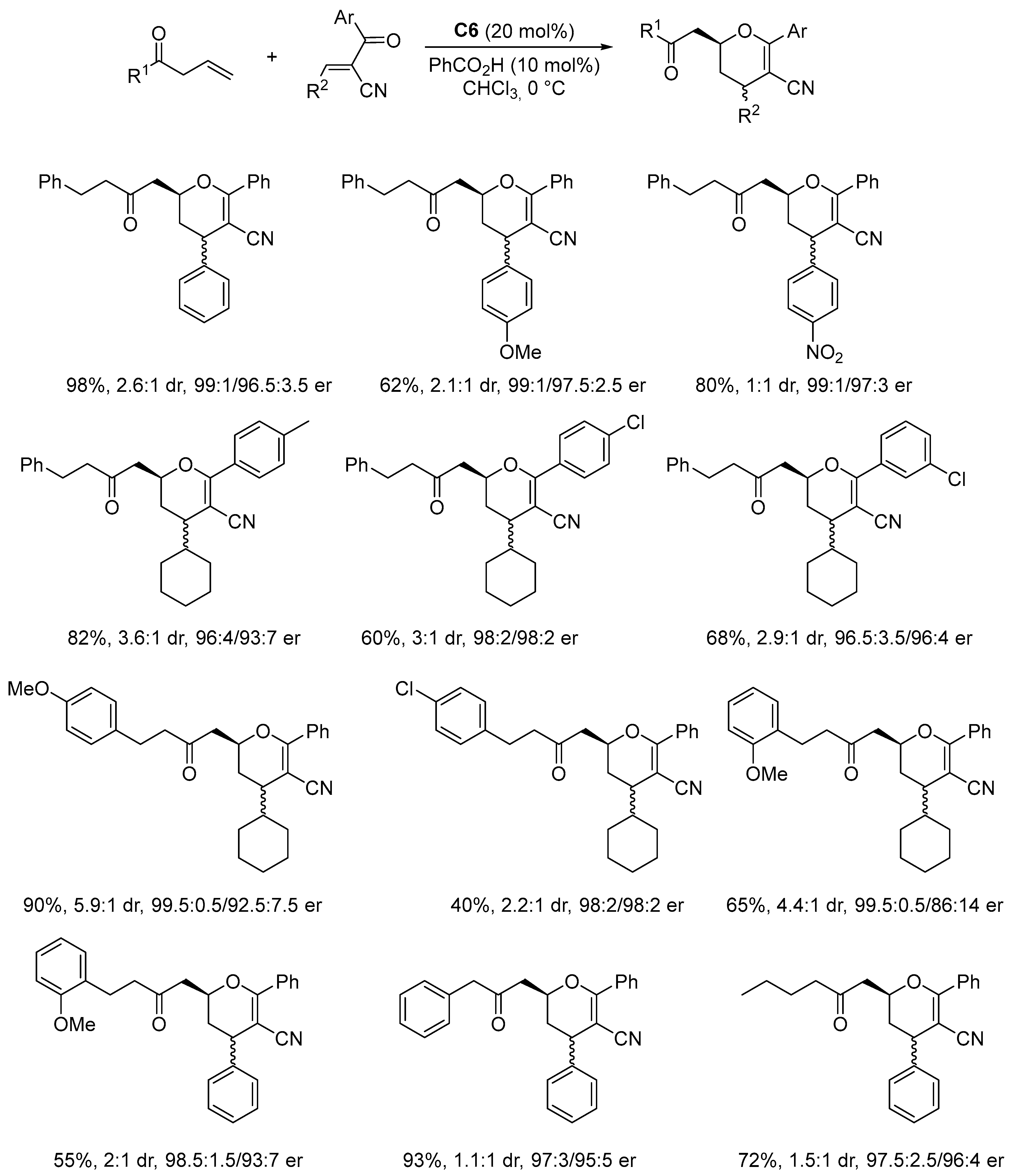
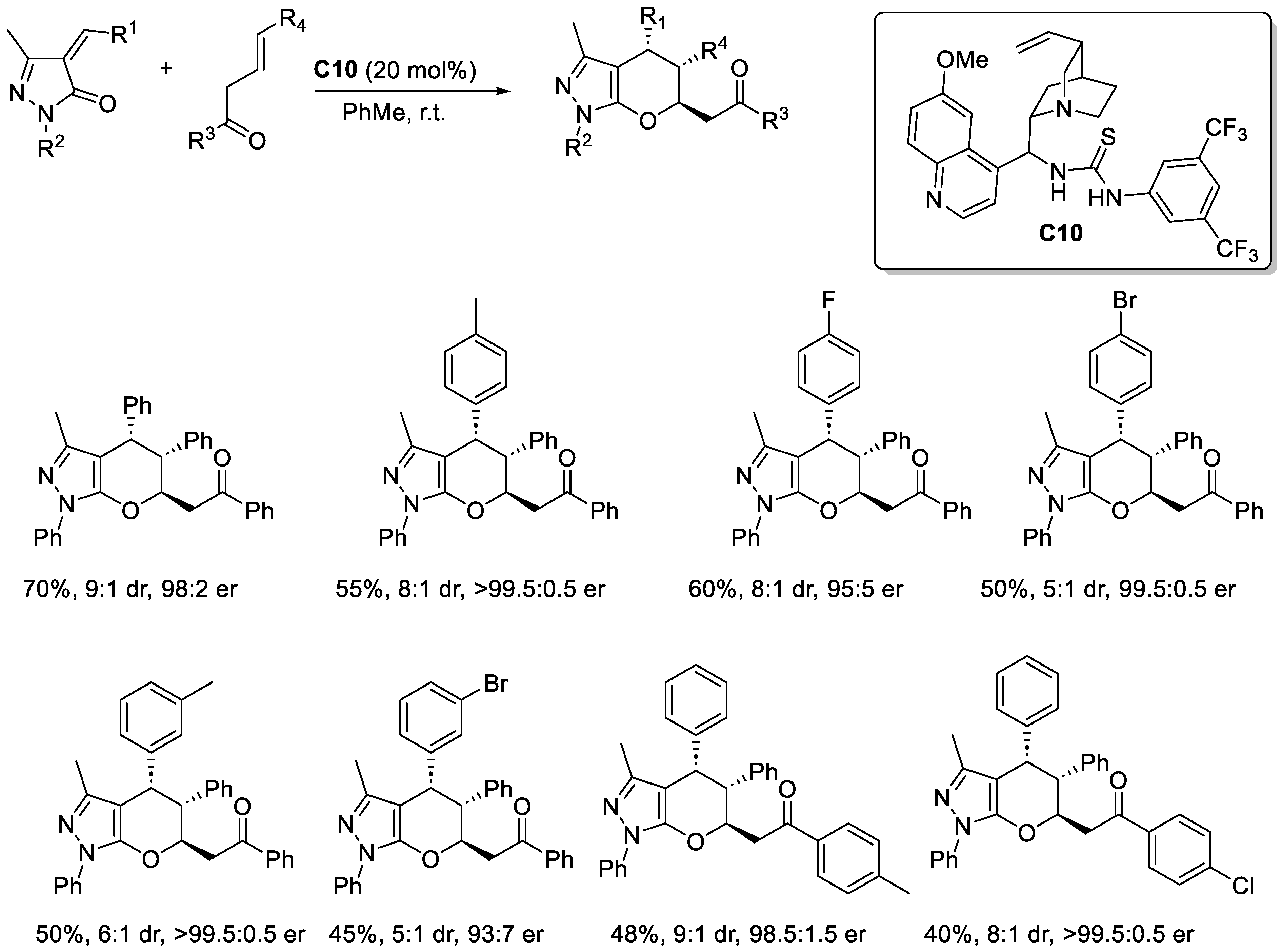
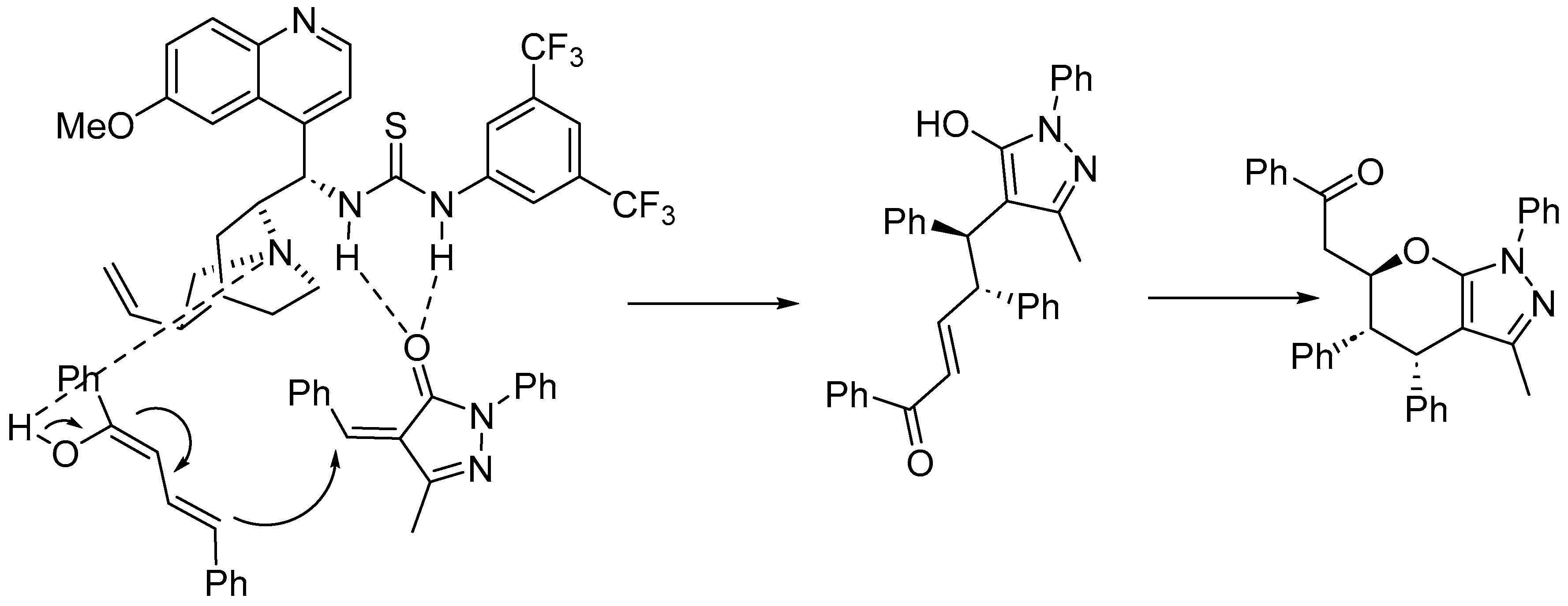
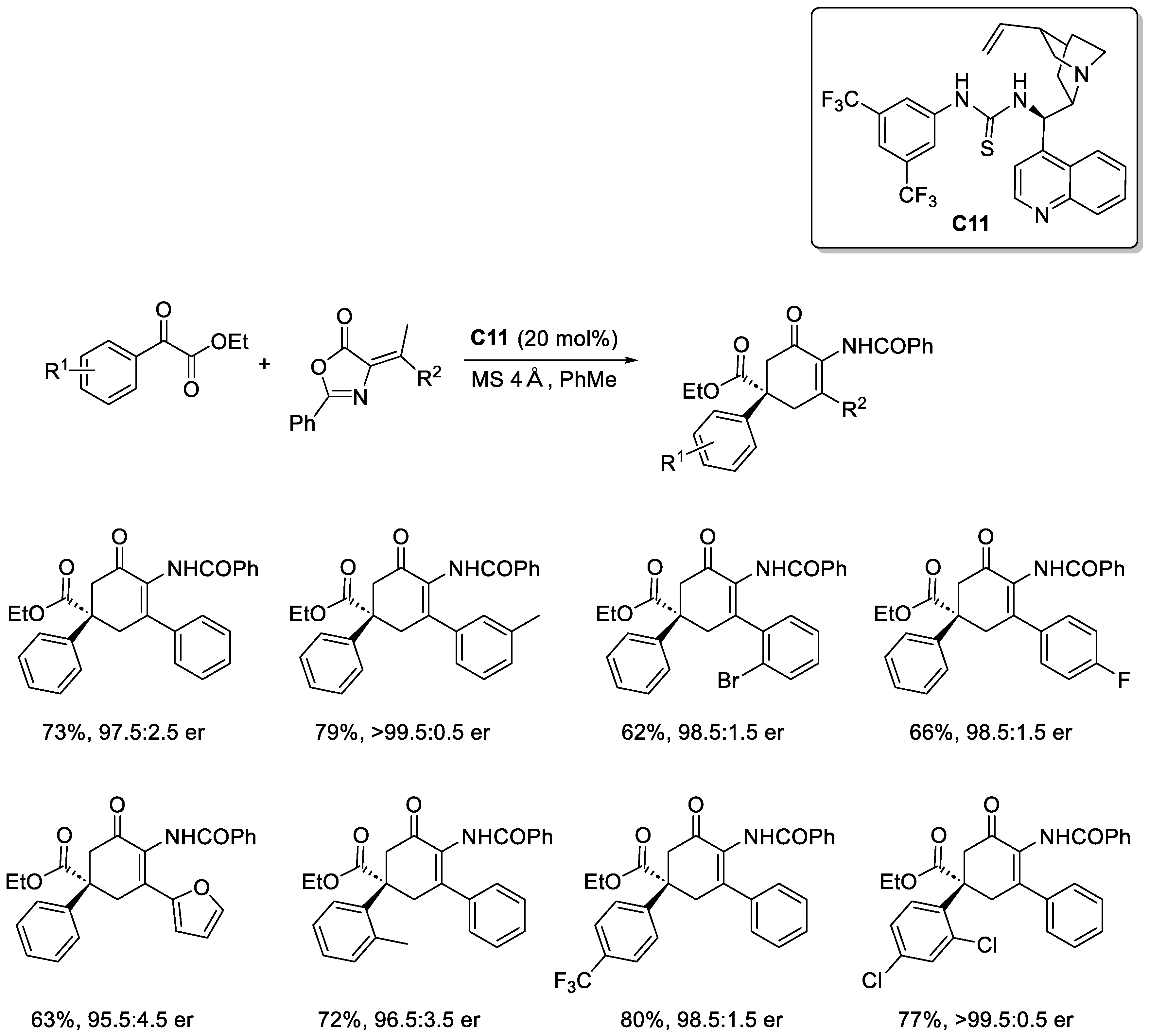

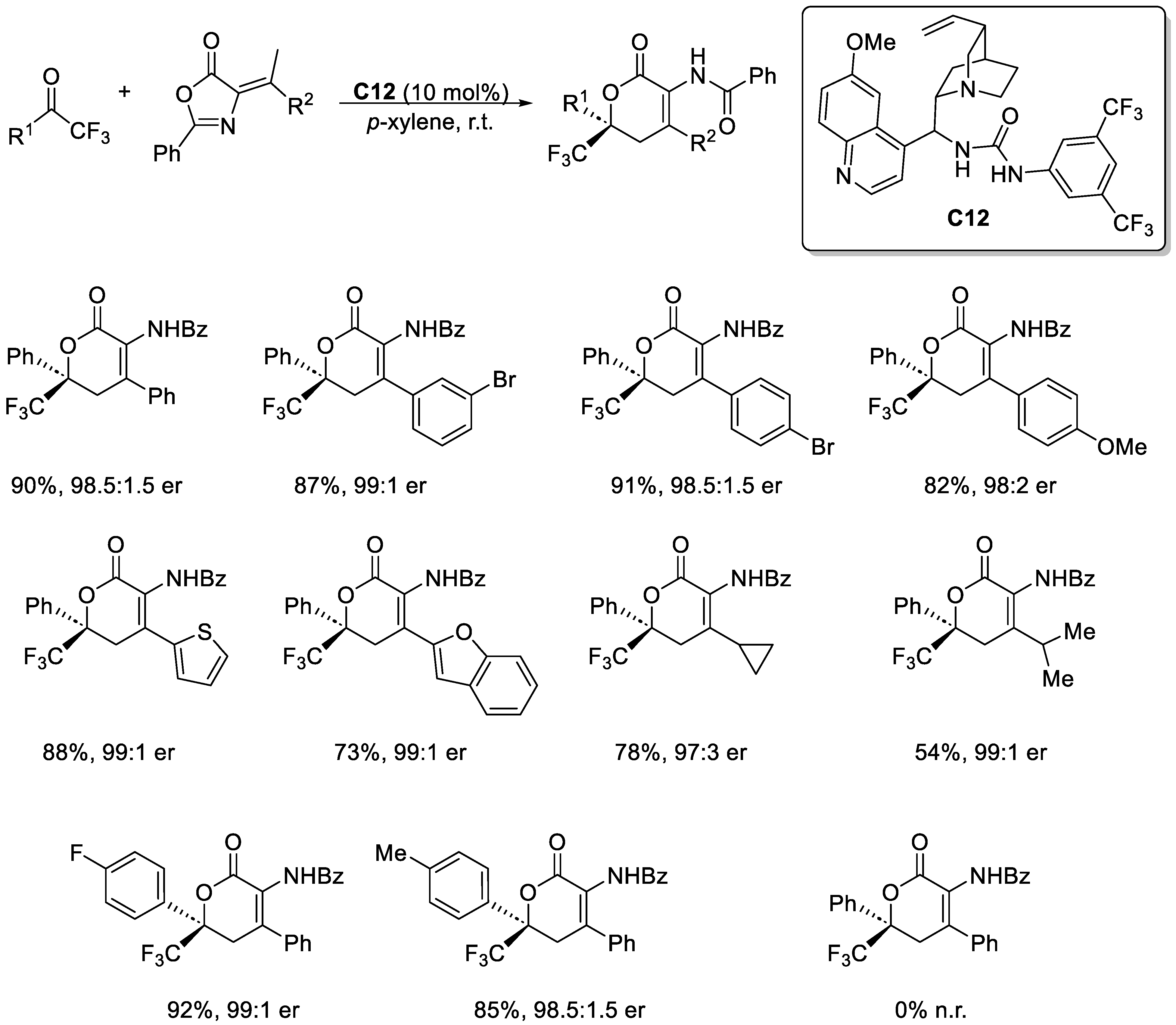

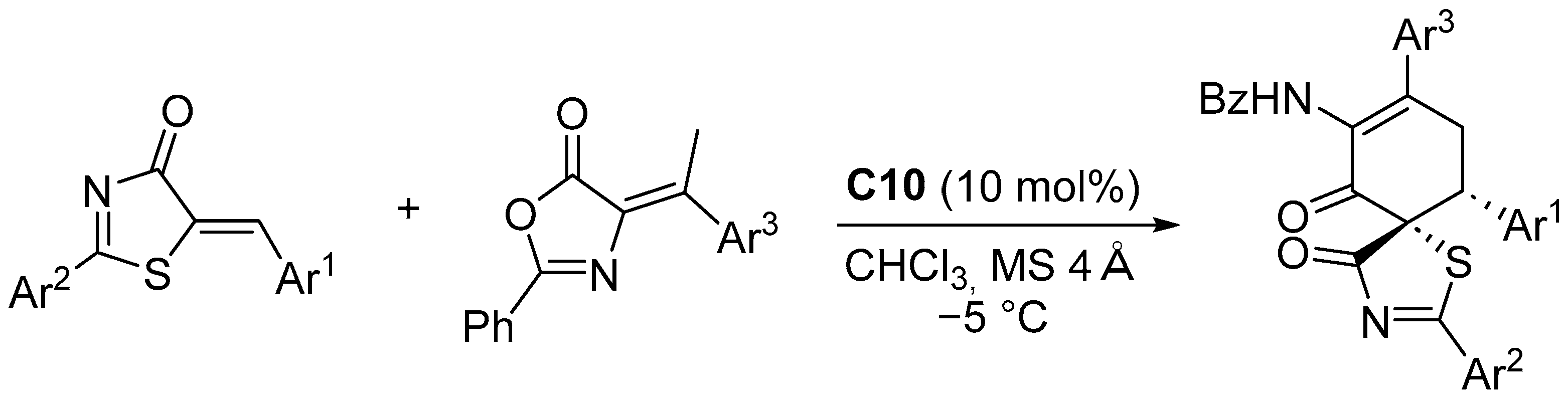
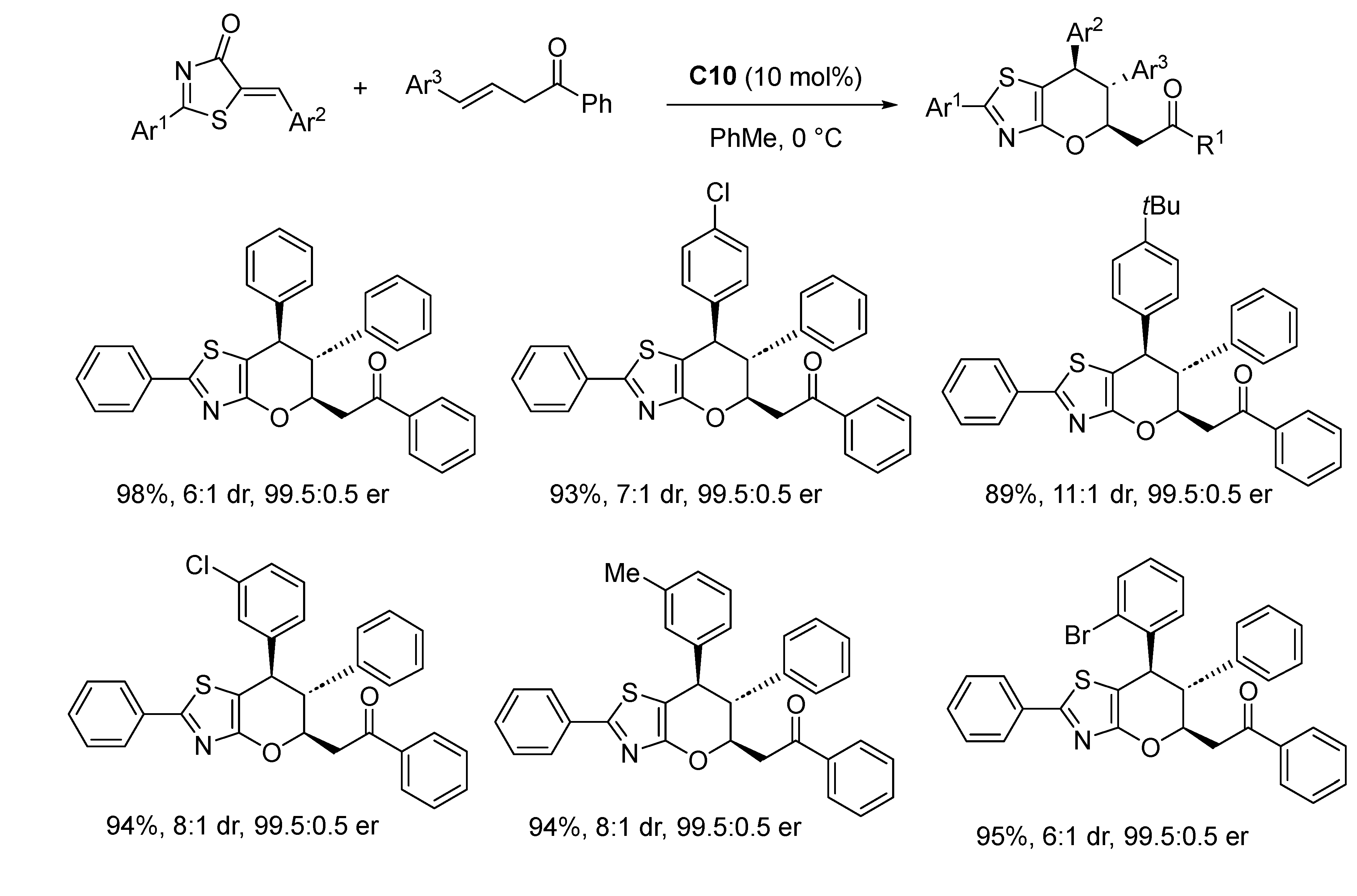
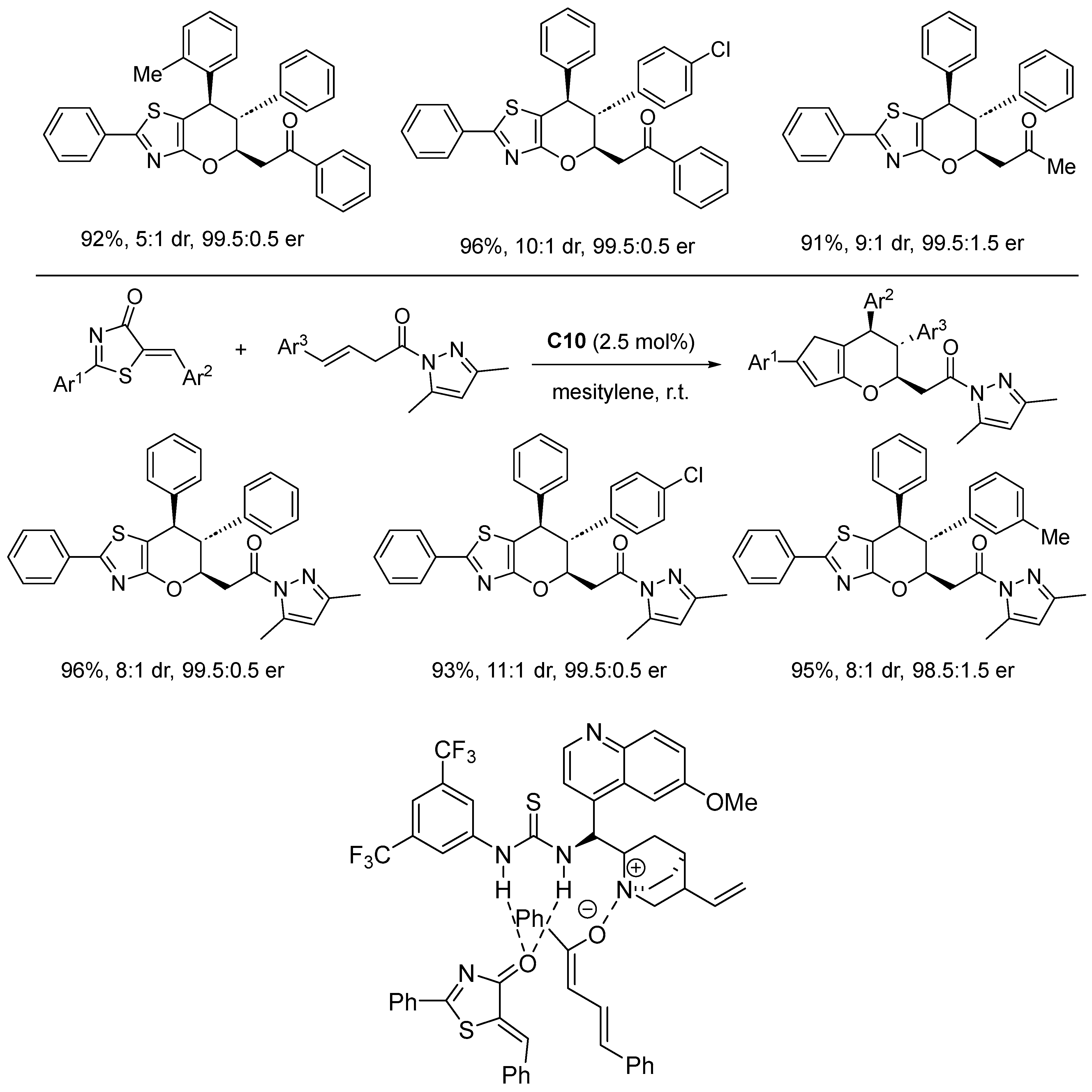

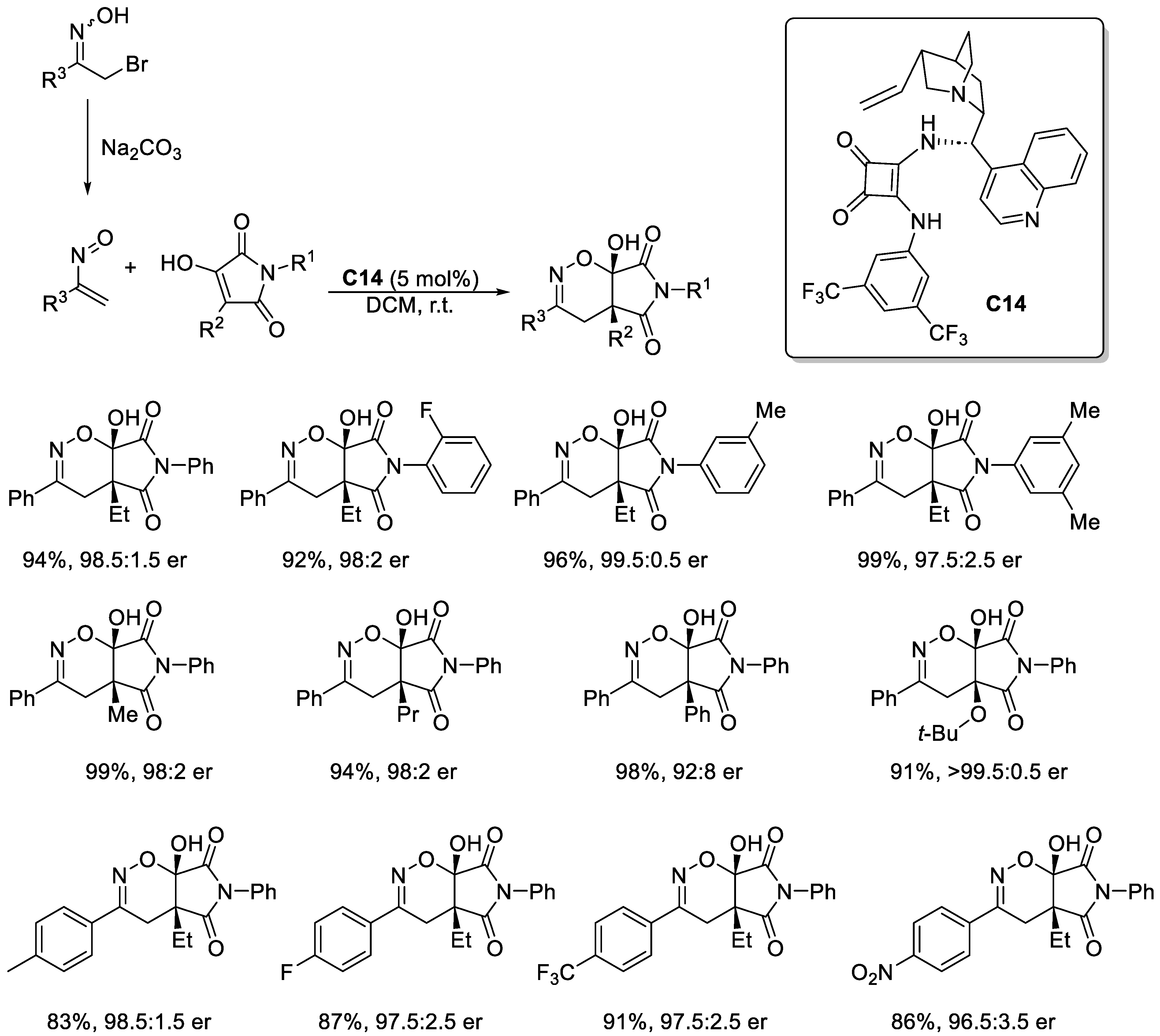
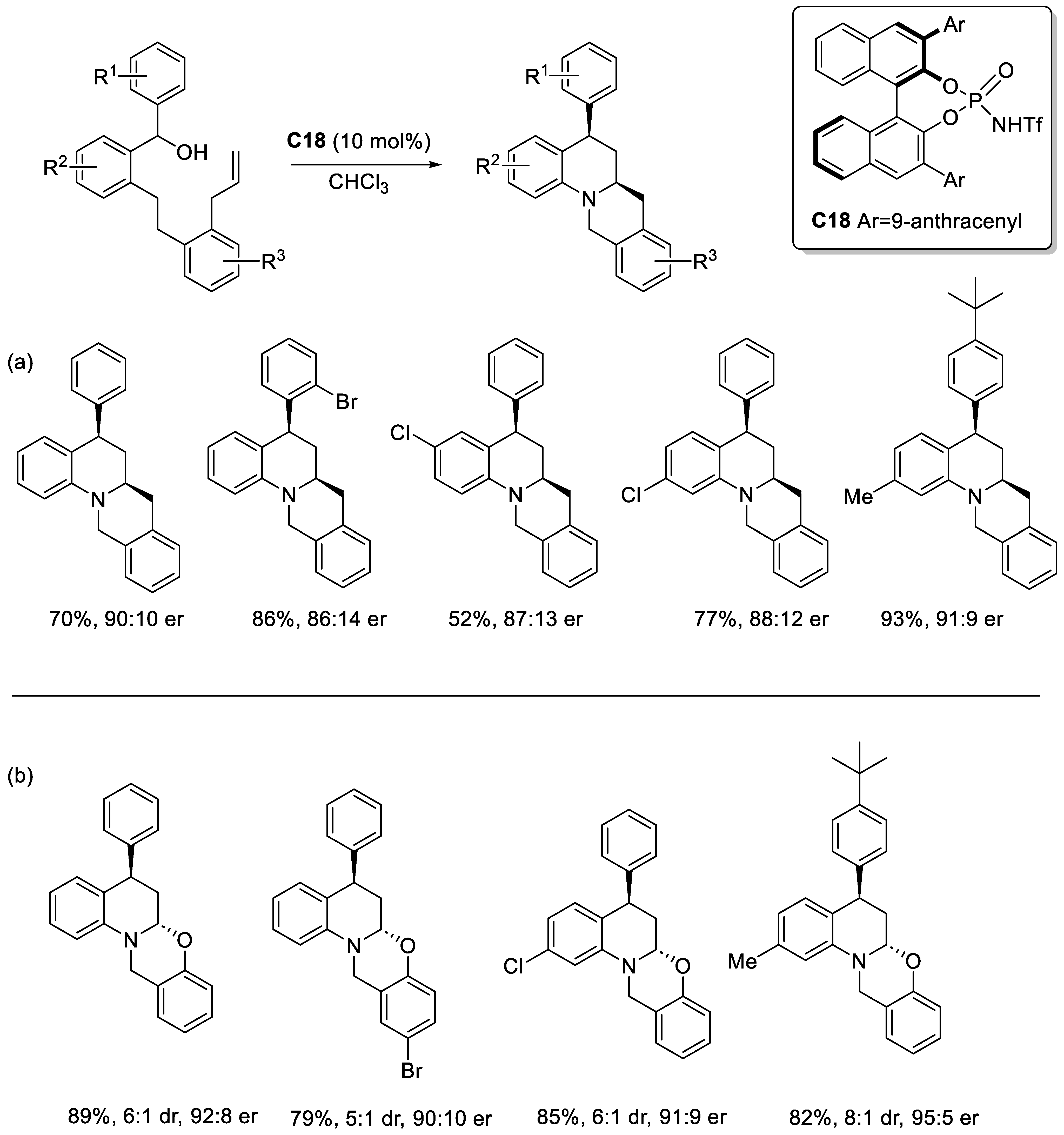
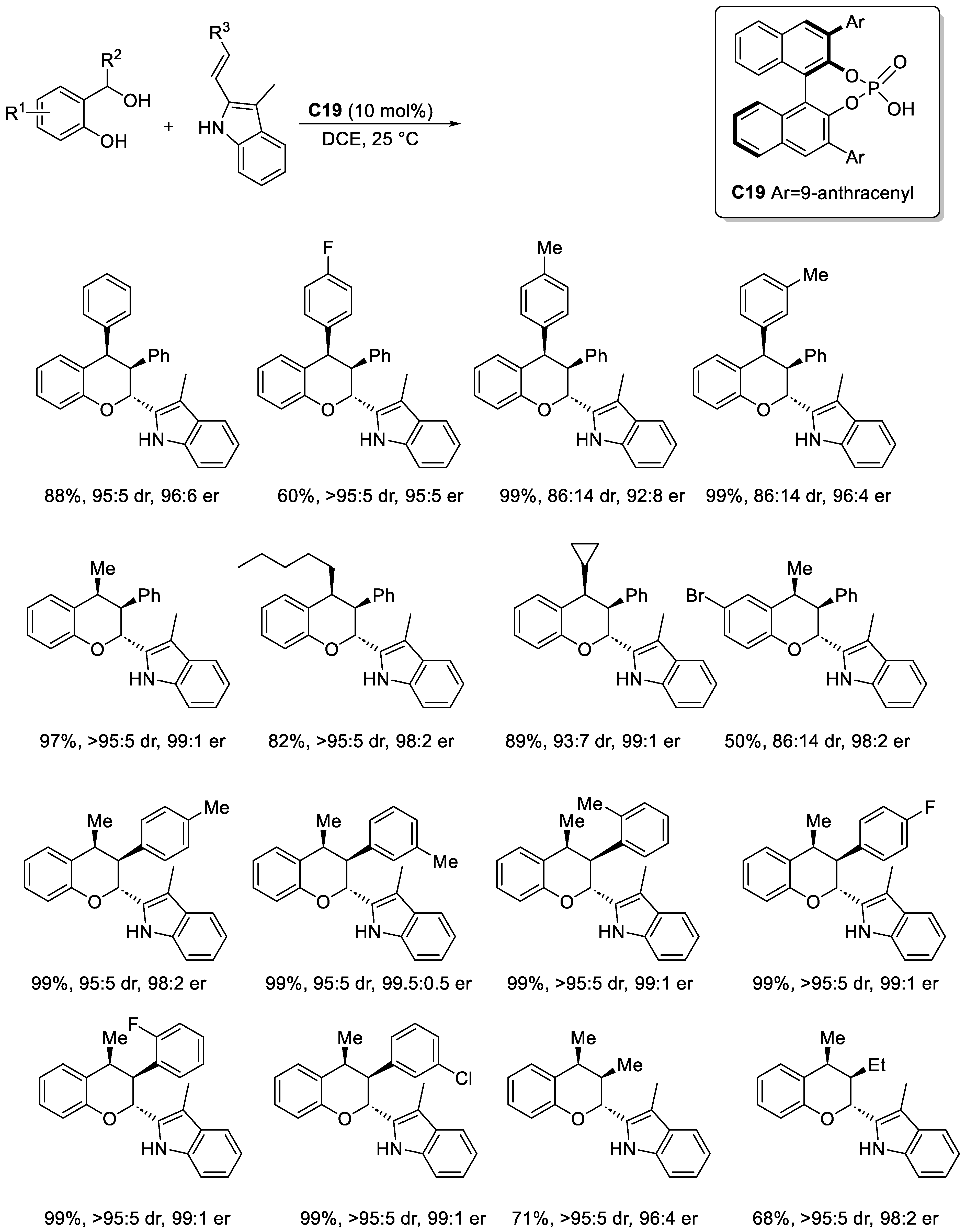
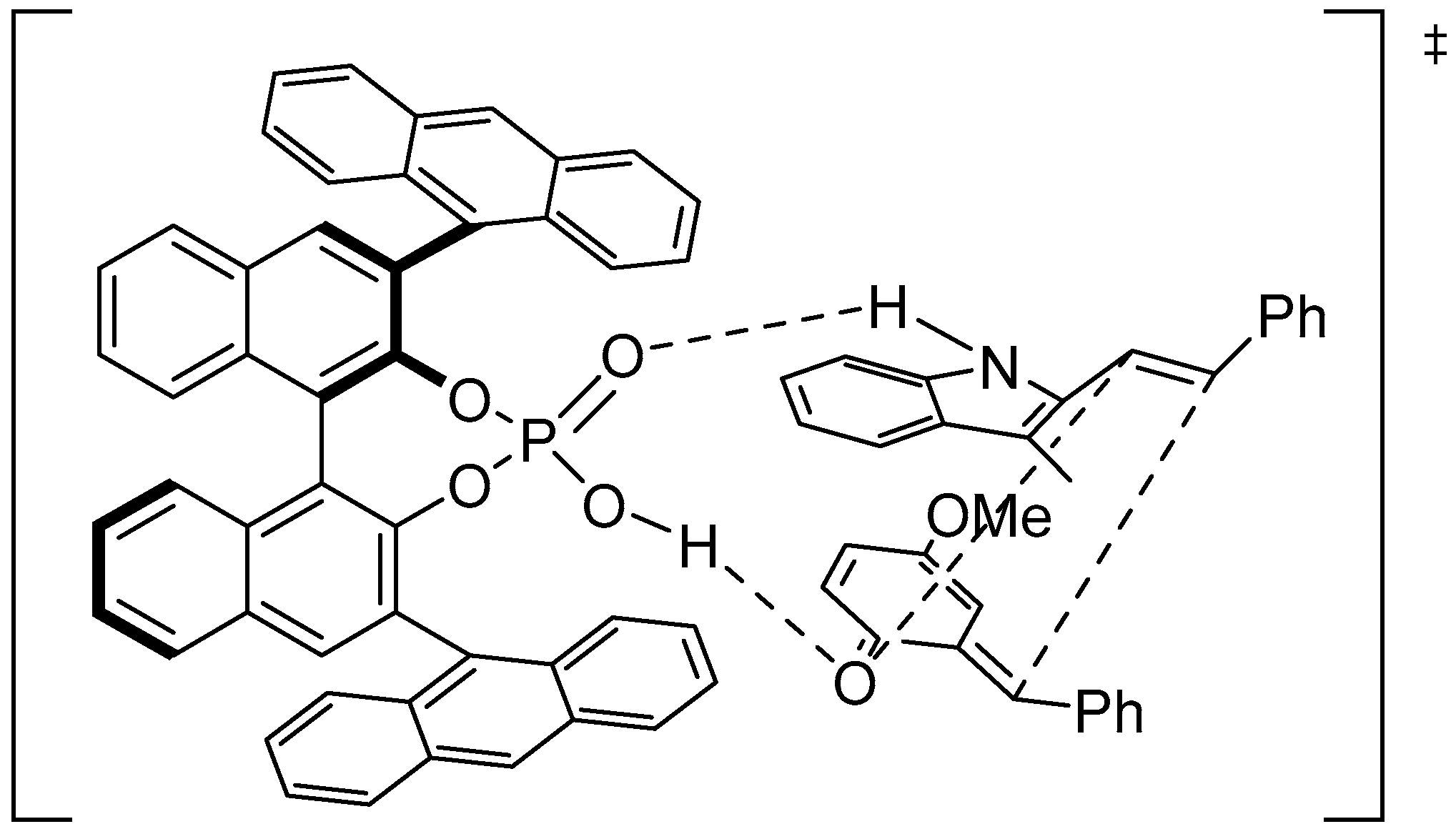
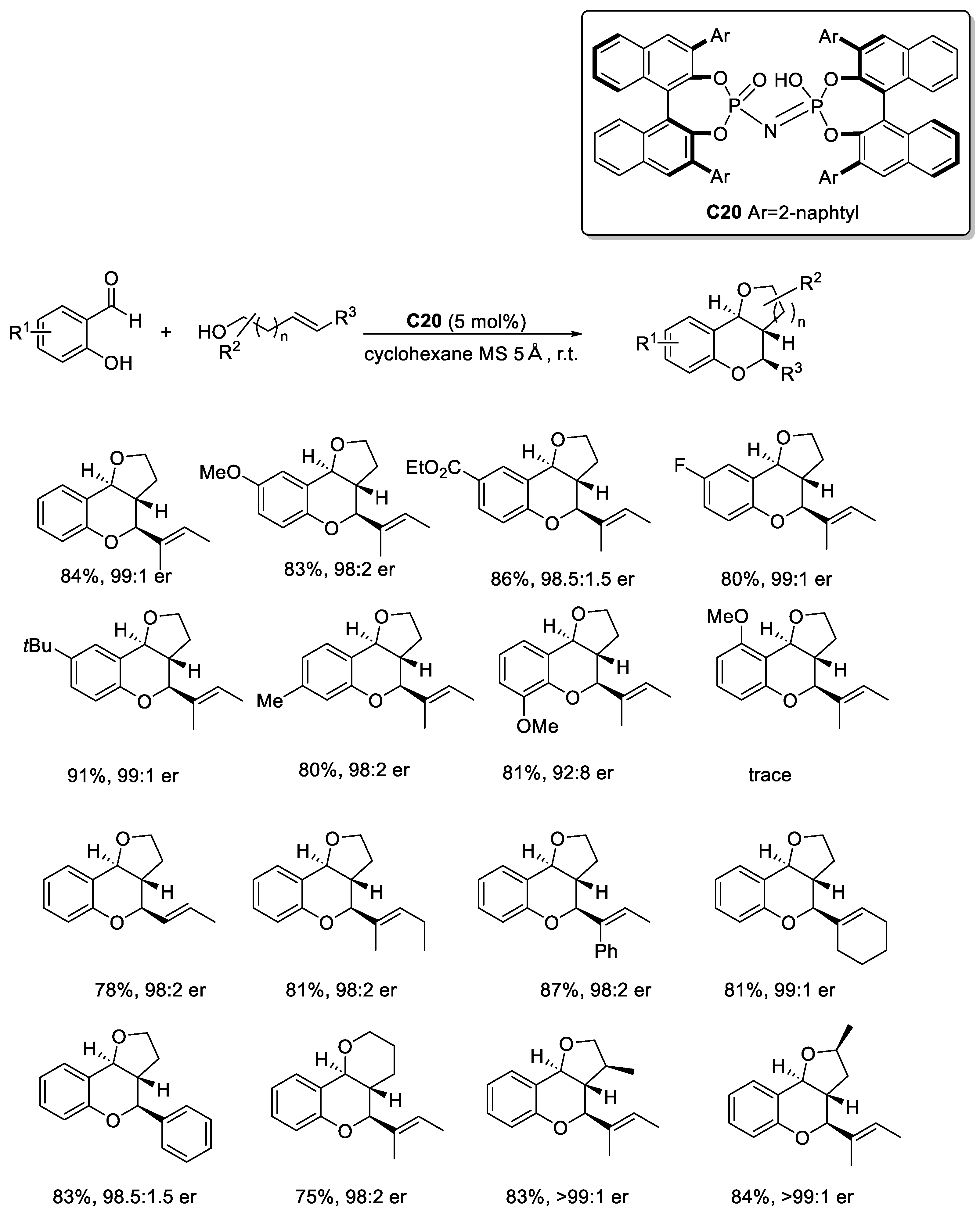

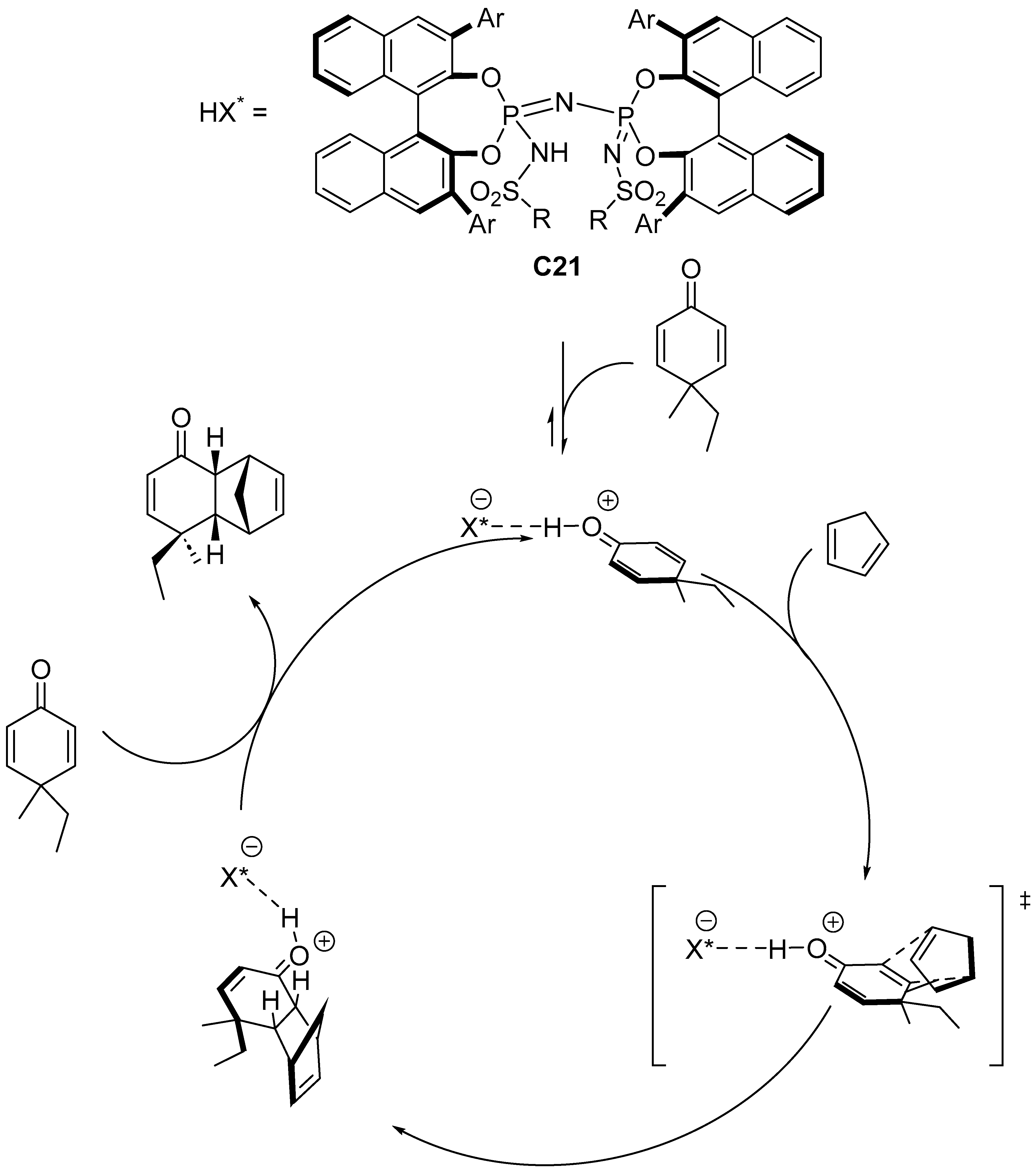
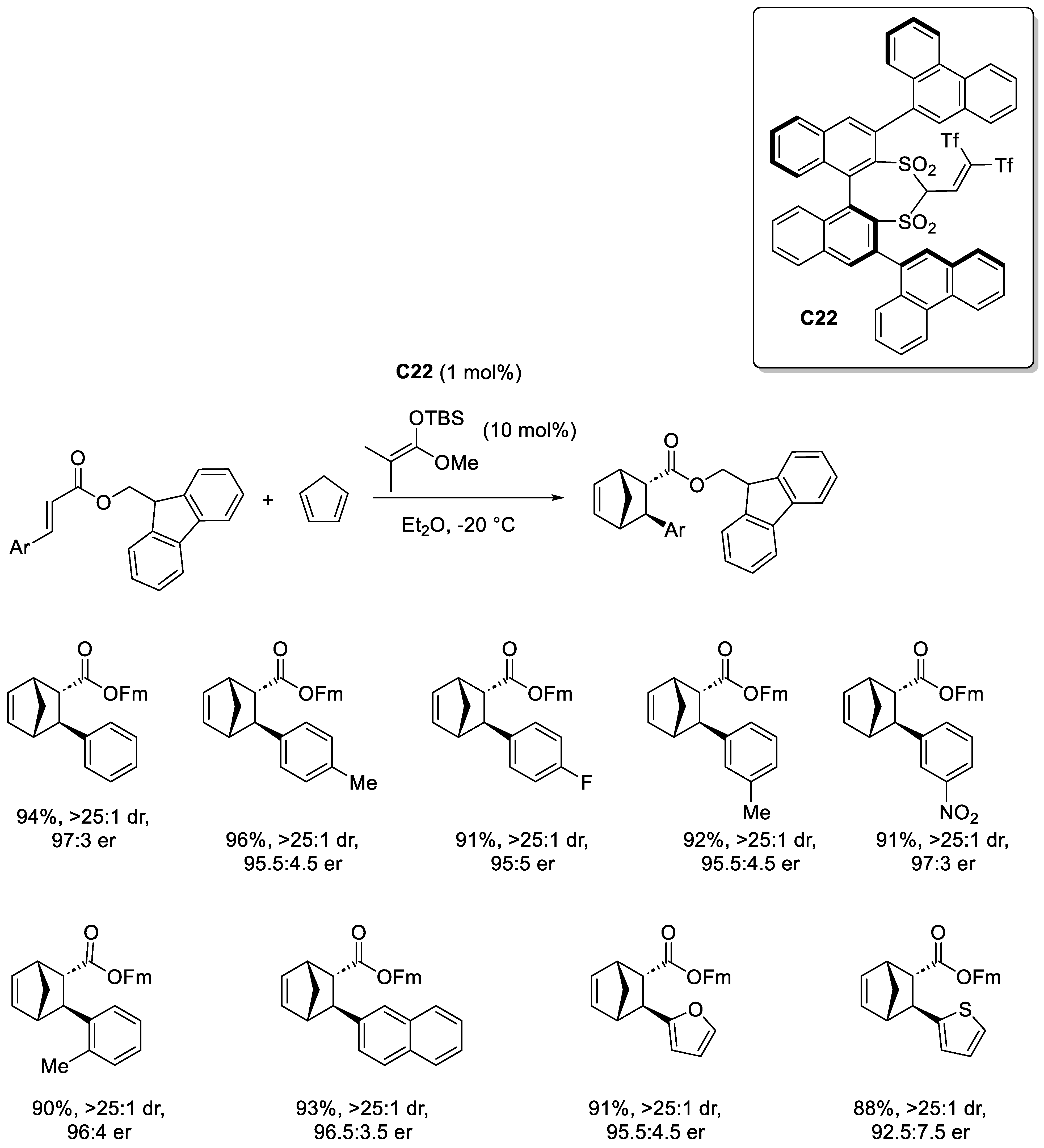
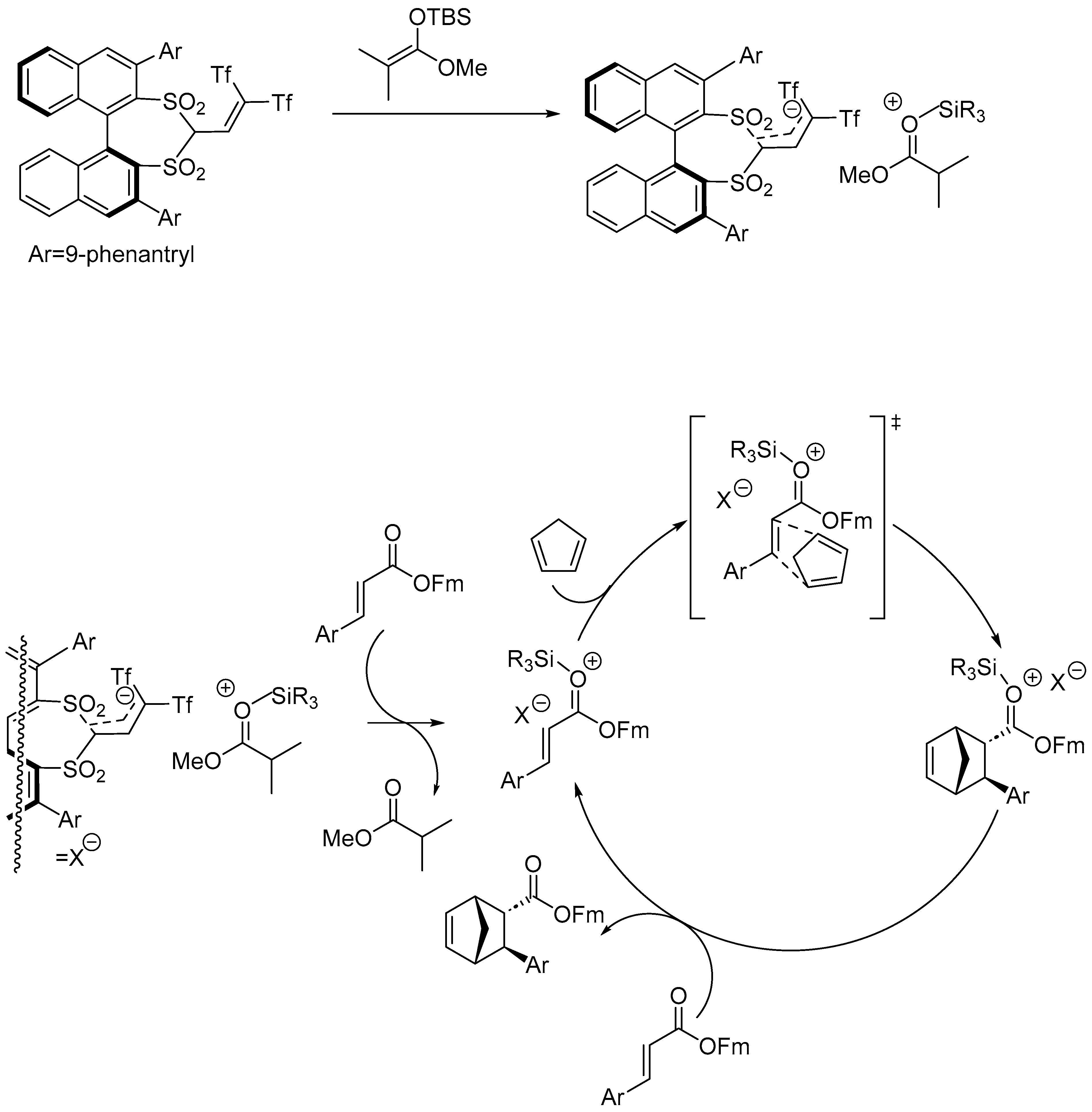
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, T. Recent Advances in the Enantioselective Organocatalytic [4+2] Cycloadditions. Molecules 2025, 30, 1978. https://doi.org/10.3390/molecules30091978
Bauer T. Recent Advances in the Enantioselective Organocatalytic [4+2] Cycloadditions. Molecules. 2025; 30(9):1978. https://doi.org/10.3390/molecules30091978
Chicago/Turabian StyleBauer, Tomasz. 2025. "Recent Advances in the Enantioselective Organocatalytic [4+2] Cycloadditions" Molecules 30, no. 9: 1978. https://doi.org/10.3390/molecules30091978
APA StyleBauer, T. (2025). Recent Advances in the Enantioselective Organocatalytic [4+2] Cycloadditions. Molecules, 30(9), 1978. https://doi.org/10.3390/molecules30091978