

Design, Synthesis, and Biological Evaluation of Naphthoquinone Salts as Anticancer Agents
 , , ,
, , ,  , ,
, ,  and
and 
Abstract
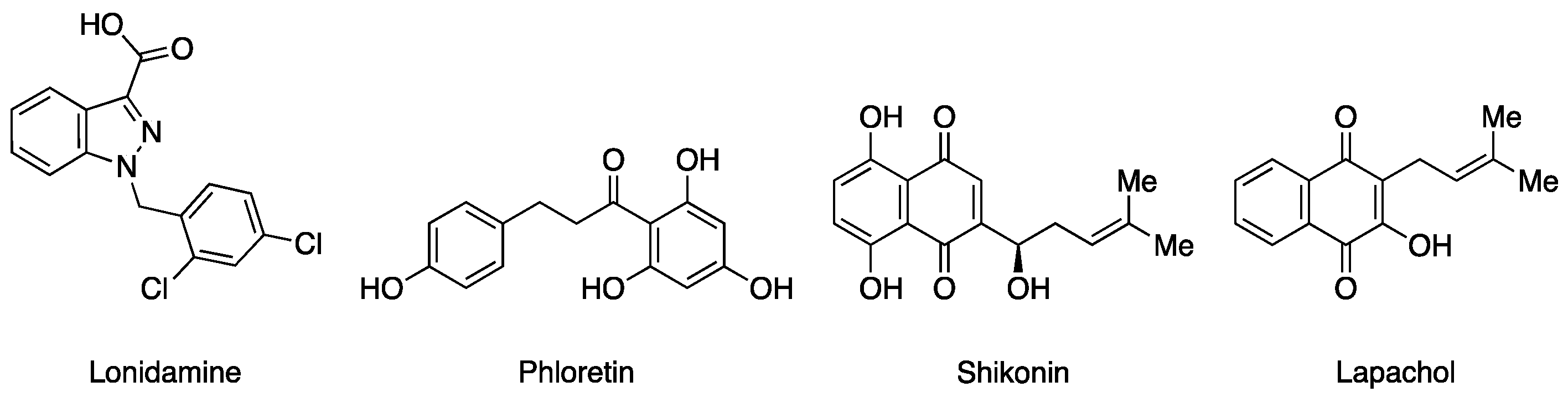
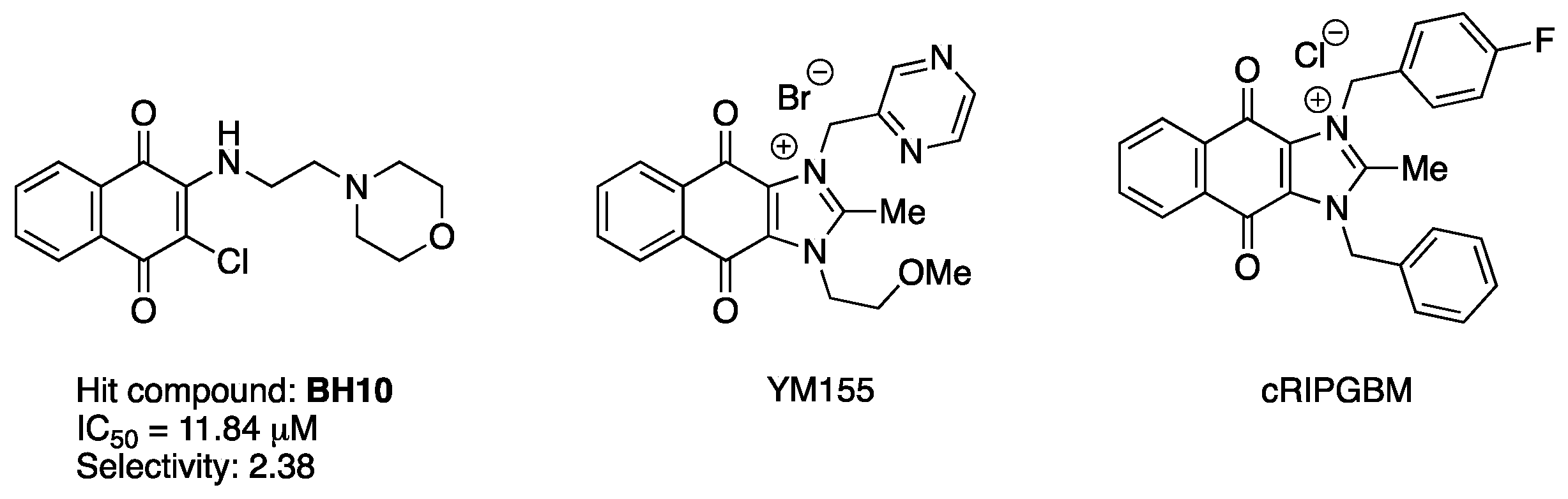
1. Introduction
2. Results and Discussion
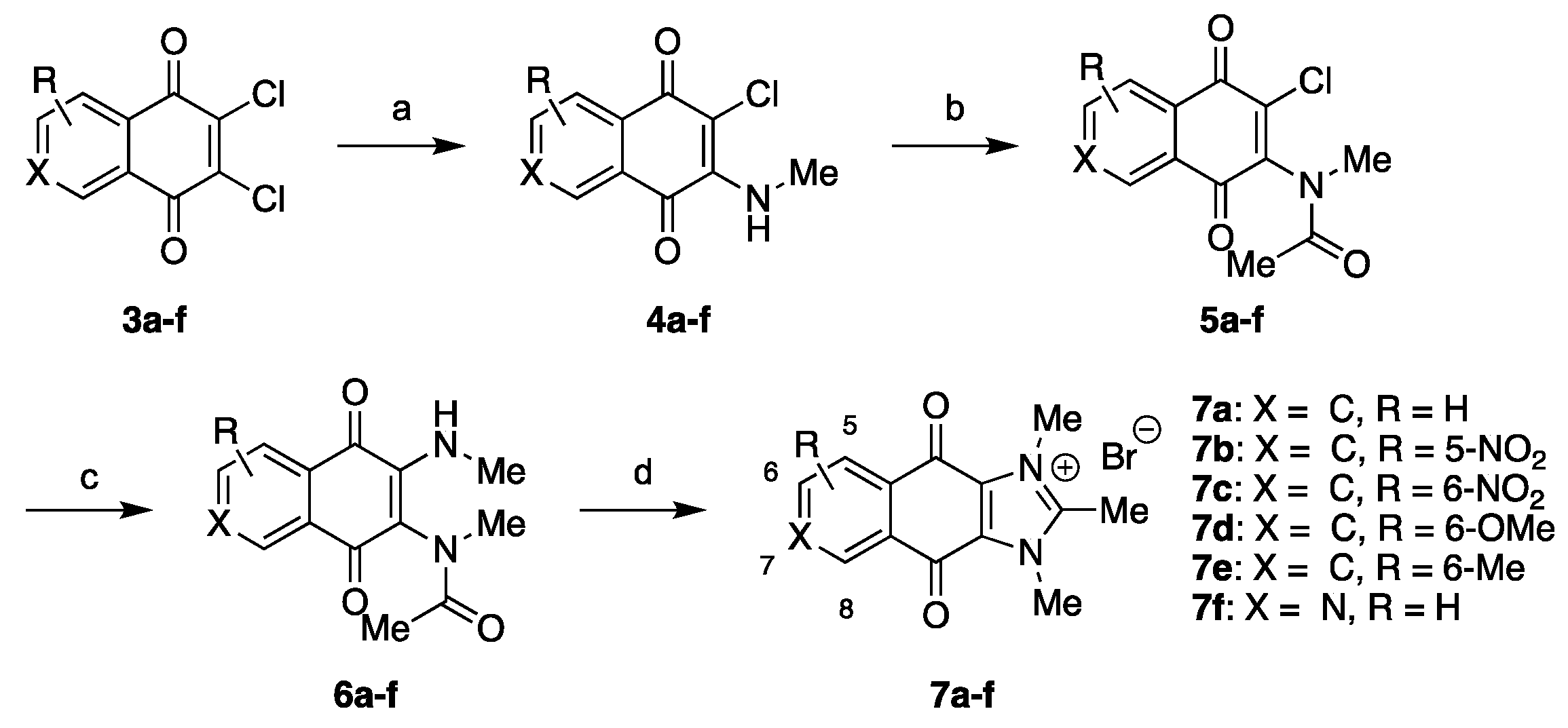
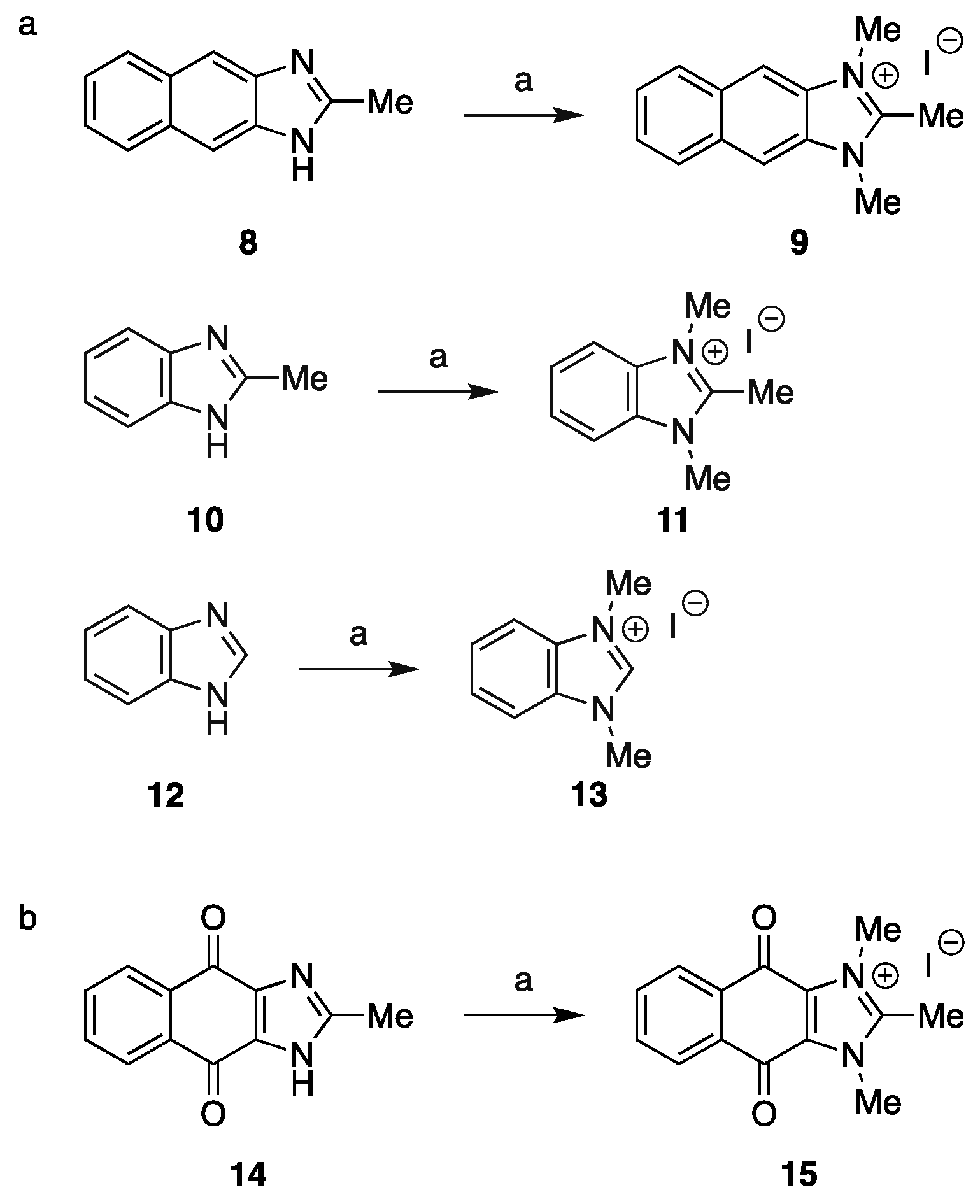
2.1. Chemistry
2.2. Structure–Activity Relationship (SAR) Study
2.3. Molecular Docking Study
3. Experimental
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis of Intermediates
3.1.3. Synthesis of Final Compounds
3.2. In Vitro Cell Viability Assay
3.3. In Silico Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deng, D.; Yan, N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Uzlova, E.V.; Zimatkin, S.M. Cellular ATP Synthase. Biol. Bull. Rev. 2021, 11, 134–142. [Google Scholar] [CrossRef]
- Parashar, A.; Jacob, V.D.; Gideon, D.A.; Manoj, K.M. Hemoglobin catalyzes ATP-synthesis in human erythrocytes: A murburn model. J. Biomol. Struct. Dyn. 2022, 40, 8783–8795. [Google Scholar] [CrossRef] [PubMed]
- Muhleip, A.; McComas, S.E.; Amunts, A. Structure of a mitochondrial ATP synthase with bound native cardiolipin. eLife 2019, 8, e51179. [Google Scholar] [CrossRef]
- Lizak, B.; Szarka, A.; Kim, Y.; Choi, K.S.; Nemeth, C.E.; Marcolongo, P.; Benedetti, A.; Banhegyi, G.; Margittai, E. Glucose Transport and Transporters in the Endomembranes. Int. J. Mol. Sci. 2019, 20, 5898. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Huang, Z.; Li, B.; Nice, E.C.; Huang, C.; Wei, L.; Zou, B. Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers 2022, 14, 4568. [Google Scholar] [CrossRef]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva, F.P., Jr. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207. [Google Scholar] [CrossRef] [PubMed]
- Giorgioni, G.; Ruggieri, S.; Di Stefano, A.; Sozio, P.; Cinque, B.; Di Marzio, L.; Santoni, G.; Claudi, F. Glycosyl and polyalcoholic prodrugs of lonidamine. Bioorganic Med. Chem. Lett. 2008, 18, 2445–2450. [Google Scholar] [CrossRef]
- Nakhate, K.T.; Badwaik, H.; Choudhary, R.; Sakure, K.; Agrawal, Y.O.; Sharma, C.; Ojha, S.; Goyal, S.N. Therapeutic Potential and Pharmaceutical Development of a Multitargeted Flavonoid Phloretin. Nutrients 2022, 14, 3638. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Krohn, K.; Ahmad, V.U.; Miana, G.A.; Green, I.R. Lapachol: An overview. Arkivoc 2007, 2, 145–171. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Wong, C.C.; Cheng, K.W.; Rigas, B. Preclinical predictors of anticancer drug efficacy: Critical assessment with emphasis on whether nanomolar potency should be required of candidate agents. J. Pharmacol. Exp. Ther. 2012, 341, 572–578. [Google Scholar] [CrossRef]
- Dancey, J.E.; Chen, H.X. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 649–659. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Vichaya, E.G.; Chiu, G.S.; Krukowski, K.; Lacourt, T.E.; Kavelaars, A.; Dantzer, R.; Heijnen, C.J.; Walker, A.K. Mechanisms of chemotherapy-induced behavioral toxicities. Front. Neurosci. 2015, 9, 131. [Google Scholar] [CrossRef]
- Patel, F.; Spassieva, S.D. Side Effects in Cancer Therapy: Are Sphingolipids to Blame? Adv. Cancer Res. 2018, 140, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Chehelgerdi, M.; Chehelgerdi, M.; Allela, O.Q.B.; Pecho, R.D.C.; Jayasankar, N.; Rao, D.P.; Thamaraikani, T.; Vasanthan, M.; Viktor, P.; Lakshmaiya, N.; et al. Progressing nanotechnology to improve targeted cancer treatment: Overcoming hurdles in its clinical implementation. Mol. Cancer 2023, 22, 169. [Google Scholar] [CrossRef] [PubMed]
- GERBER, D.E. Targeted Therapies: A New Generation of Cancer Treatments. Am. Fam. Phys. 2008, 77, 311–319. [Google Scholar]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Sawyers, C.L. The cancer biomarker problem. Nature 2008, 452, 548–552. [Google Scholar] [CrossRef]
- Byrne, F.L.; Olzomer, E.M.; Marriott, G.R.; Quek, L.-E.; Katen, A.; Su, J.; Kumar, N.; Hoehn, K.L. Phenotypic screen for oxygen consumption rate identifies an anti-cancer naphthoquinone that induces mitochondrial oxidative stress. Redox Biol. 2020, 28, 101374. [Google Scholar] [CrossRef]
- Cheng, Y.; Jones, J.P.; Yu, T.T.; Olzomer, E.M.; Su, J.; Katen, A.; Black, D.S.; Hart-Smith, G.; Childress, E.S.; Wilkins, M.R.; et al. Design, synthesis and biological evaluation of glucose metabolism inhibitors as anticancer agents. Bioorganic Chem. 2024, 151, 107665. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Jia, D.; Bhatt, V.; Akel, M.; Roberge, J.; Guo, J.Y.; Langenfeld, J. Ym155 localizes to the mitochondria leading to mitochondria dysfunction and activation of AMPK that inhibits BMP signaling in lung cancer cells. Sci. Rep. 2022, 12, 13135. [Google Scholar] [CrossRef]
- Lucki, N.C.; Villa, G.R.; Vergani, N.; Bollong, M.J.; Beyer, B.A.; Lee, J.W.; Anglin, J.L.; Spangenberg, S.H.; Chin, E.N.; Sharma, A.; et al. A cell type-selective apoptosis-inducing small molecule for the treatment of brain cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6435–6440. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, Z.; Dong, Y.; Gao, F.; Xia, X.; Wang, P.; Luo, Y.; Zhang, Z.; Yan, D.; Zhang, W. Pioneering 4,11-Dioxo-4,11-dihydro-1H-anthra[2,3-d]imidazol-3-ium Compounds as Promising Survivin Inhibitors by Targeting ILF3/NF110 for Cancer Therapy. J. Med. Chem. 2023, 66, 16843–16868. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Varghese Gupta, S. Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef]
- Acharya, P.C.; Marwein, S.; Mishra, B.; Ghosh, R.; Vora, A.; Tekade, R.K. Role of Salt Selection in Drug Discovery and Development. In Dosage Form Design Considerations; Academic Press: Cambridge, MA, USA, 2018; pp. 435–472. [Google Scholar] [CrossRef]
- Cheng, Y.; Yu, T.T.; Olzomer, E.; Beretta, M.; Katen, A.; Su, J.; Jones, J.P.; Black, D.S.; Hoehn, K.L.; Byrne, F.L.; et al. Design, synthesis and biological evaluation of naphthalene-1,4-diones analogues as anticancer agents. RSC Med. Chem. 2025. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Marques, L.B.; Ottoni, F.M.; Pinto, M.C.X.; Ribeiro, J.M.; de Sousa, F.S.; Weinlich, R.; de Victo, N.C.; Kisitu, J.; Holzer, A.K.; Leist, M.; et al. Lapachol acetylglycosylation enhances its cytotoxic and pro-apoptotic activities in HL60 cells. Toxicol. Vit. 2020, 65, 104772. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, Q.; Zheng, S.; Liu, T.; Yang, L.; Han, X.; Lu, X. Shikonin Inhibits Tumor Growth of ESCC by suppressing PKM2 mediated Aerobic Glycolysis and STAT3 Phosphorylation. J. Cancer 2021, 12, 4830–4840. [Google Scholar] [CrossRef]
- Rosbe, K.W.; Brann, T.W.; Holden, S.A.; Teicher, B.A.; Ill, E.F. Effect of lonidamine on the cytotoxicity of four alkylating agents in vitro. Cancer Chemother. Pharmacol. 1989, 25, 32–36. [Google Scholar] [CrossRef]
- Habtemariam, S. The Molecular Pharmacology of Phloretin: Anti-Inflammatory Mechanisms of Action. Biomedicines 2023, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- West, T.J.; Bi, J.; Martinez-Pena, F.; Curtis, E.J.; Gazaniga, N.R.; Mischel, P.S.; Lairson, L.L. A Cell Type Selective YM155 Prodrug Targets Receptor-Interacting Protein Kinase 2 to Induce Brain Cancer Cell Death. J. Am. Chem. Soc. 2023, 145, 8355–8363. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, G.; Sun, X.; Li, F.; Zhao, L.; Zhong, R.; Peng, Y. The Potential of Lonidamine in Combination with Chemotherapy and Physical Therapy in Cancer Treatment. Cancers 2020, 12, 3332. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Qiu, H.Y.; Zhu, X.; Luo, Y.L.; Lin, H.Y.; Tang, C.Y.; Qi, J.L.; Pang, Y.J.; Yang, R.W.; Lu, G.H.; Wang, X.M.; et al. Identification of New Shikonin Derivatives as Antitumor Agents Targeting STAT3 SH2 Domain. Sci. Rep. 2017, 7, 2863. [Google Scholar] [CrossRef]
- Ho, S.H.; Sim, M.Y.; Yee, W.L.; Yang, T.; Yuen, S.P.; Go, M.L. Antiproliferative, DNA intercalation and redox cycling activities of dioxonaphtho[2,3-d]imidazolium analogs of YM155: A structure-activity relationship study. Eur. J. Med. Chem. 2015, 104, 42–56. [Google Scholar] [CrossRef]
- Esteva-Font, C.; Phuan, P.W.; Anderson, M.O.; Verkman, A.S. A small molecule screen identifies selective inhibitors of urea transporter UT-A. Chem. Biol. 2013, 20, 1235–1244. [Google Scholar] [CrossRef]
- Salustiano, E.J.; Netto, C.D.; Fernandes, R.F.; da Silva, A.J.; Bacelar, T.S.; Castro, C.P.; Buarque, C.D.; Maia, R.C.; Rumjanek, V.M.; Costa, P.R. Comparison of the cytotoxic effect of lapachol, alpha-lapachone and pentacyclic 1,4-naphthoquinones on human leukemic cells. Investig. New Drugs 2010, 28, 139–144. [Google Scholar] [CrossRef]
- Blackburn, C. Solid-phase synthesis of 2-amino-3-chloro-5- and 8-nitro-1,4-naphthoquinones: A new and general colorimetric test for resin-bound amines. Tetrahedron Lett. 2005, 46, 1405–1409. [Google Scholar] [CrossRef]
- Naciuk, F.F.; Milan, J.C.; Andreao, A.; Miranda, P.C. Exploitation of a tuned oxidation with N-haloimides in the synthesis of caulibugulones A-D. J. Org. Chem. 2013, 78, 5026–5030. [Google Scholar] [CrossRef]
- Xie, W.; Xu, J.; Md Idros, U.; Katsuhira, J.; Fuki, M.; Hayashi, M.; Yamanaka, M.; Kobori, Y.; Matsubara, R. Metal-free reduction of CO2 to formate using a photochemical organohydride-catalyst recycling strategy. Nat. Chem. 2023, 15, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Jishkariani, D.; Sakhuja, R.; Hall, C.D.; Steel, P.J. Carbene-mediated transformations of 1-(benzylideneamino)benzimidazoles. J. Org. Chem. 2011, 76, 4082–4087. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mean IC50 (μM) | Selectivity Ratio | |
|---|---|---|---|
| HEC1A ± SD | MAD11 ± SD | ||
| BH10 | 11.84 ± 5.19 | 28.18 ± 12.20 | 2.38 |
| 2a | 21.12 ± 2.99 | >100 | >4.73 |
| 2b | >100 | >100 | - |
| 2c | 97.25 ± 8.20 | >100 | >1.23 |
| 2d | 58.77 ± 3.01 | >100 | >1.70 |
| 2e | >100 | >100 | - |
| 2f | >100 | >100 | - |
| 2g | 23.83 ± 4.17 | >100 | >4.20 |
| 2h | 95.53 ± 14.67 | >100 | - |
| 7a | 0.00953 ± 0.0007 | 0.06569 ± 0.00641 | 6.89 |
| 7b | 0.02297 ± 0.0033 | 0.9516 ± 0.2148 | 41.43 |
| 7c | 0.02882 ± 0.0014 | 0.06846 ± 0.0061 | 2.37 |
| 7d | 0.02562 ± 0.0021 | 0.19390 ± 0.0191 | 7.57 |
| 7e | 0.01492 ± 0.0012 | 0.1009 ± 0.0124 | 6.76 |
| 7f | 0.4724 ± 0.0378 | 4.8 ± 0.3274 | 10.16 |
| 9 | 23.88 ± 2.97 | 64.56 ± 13.98 | 2.70 |
| 11 | >100 | >100 | - |
| 13 | >100 | >100 | - |
| 15 | 0.01347 ± 0.0009 | 0.1539 ± 0.0109 | 11.43 |
| 17a | 11.17 ± 2.63 | 110.9 ± 45.0 | 9.93 |
| 17b | 6.79 ± 1.0 | >100 | >14.73 |
| 17c | 5.62 ± 1.43 | >100 | >17.79 |
| 19 | 6.35 ± 1.41 | 40.79 ± 14.12 | 6.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Yu, T.T.; Olzomer, E.M.; Hoehn, K.L.; Byrne, F.L.; Kumar, N.; Black, D.S. Design, Synthesis, and Biological Evaluation of Naphthoquinone Salts as Anticancer Agents. Molecules 2025, 30, 1938. https://doi.org/10.3390/molecules30091938
Cheng Y, Yu TT, Olzomer EM, Hoehn KL, Byrne FL, Kumar N, Black DS. Design, Synthesis, and Biological Evaluation of Naphthoquinone Salts as Anticancer Agents. Molecules. 2025; 30(9):1938. https://doi.org/10.3390/molecules30091938
Chicago/Turabian StyleCheng, Yao, Tsz Tin Yu, Ellen M. Olzomer, Kyle L. Hoehn, Frances L. Byrne, Naresh Kumar, and David StC Black. 2025. "Design, Synthesis, and Biological Evaluation of Naphthoquinone Salts as Anticancer Agents" Molecules 30, no. 9: 1938. https://doi.org/10.3390/molecules30091938
APA StyleCheng, Y., Yu, T. T., Olzomer, E. M., Hoehn, K. L., Byrne, F. L., Kumar, N., & Black, D. S. (2025). Design, Synthesis, and Biological Evaluation of Naphthoquinone Salts as Anticancer Agents. Molecules, 30(9), 1938. https://doi.org/10.3390/molecules30091938