
The Effect of Modifying the C9 Position of Fluorene with N-Donor Substituents on Selected Physicochemical Properties


Abstract

1. Introduction
2. Result and Discussion
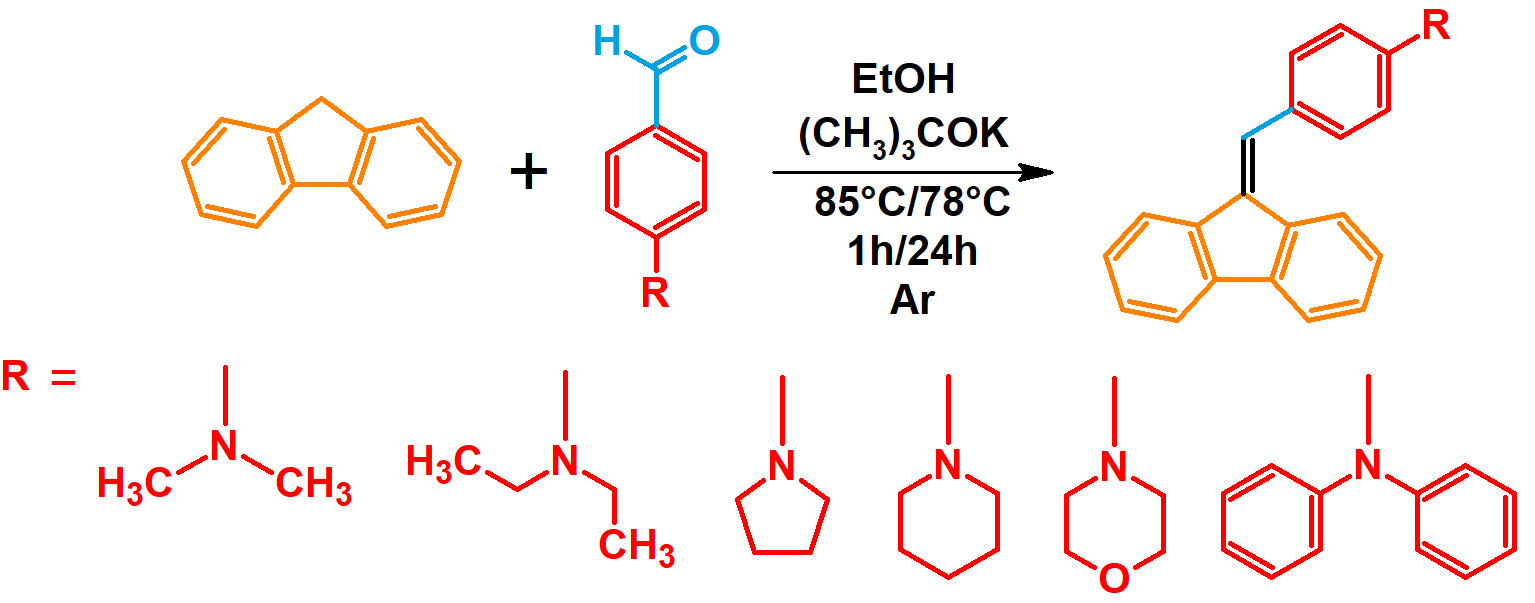
2.1. Synthesis
2.2. Optical Properties
2.3. DFT Calculations
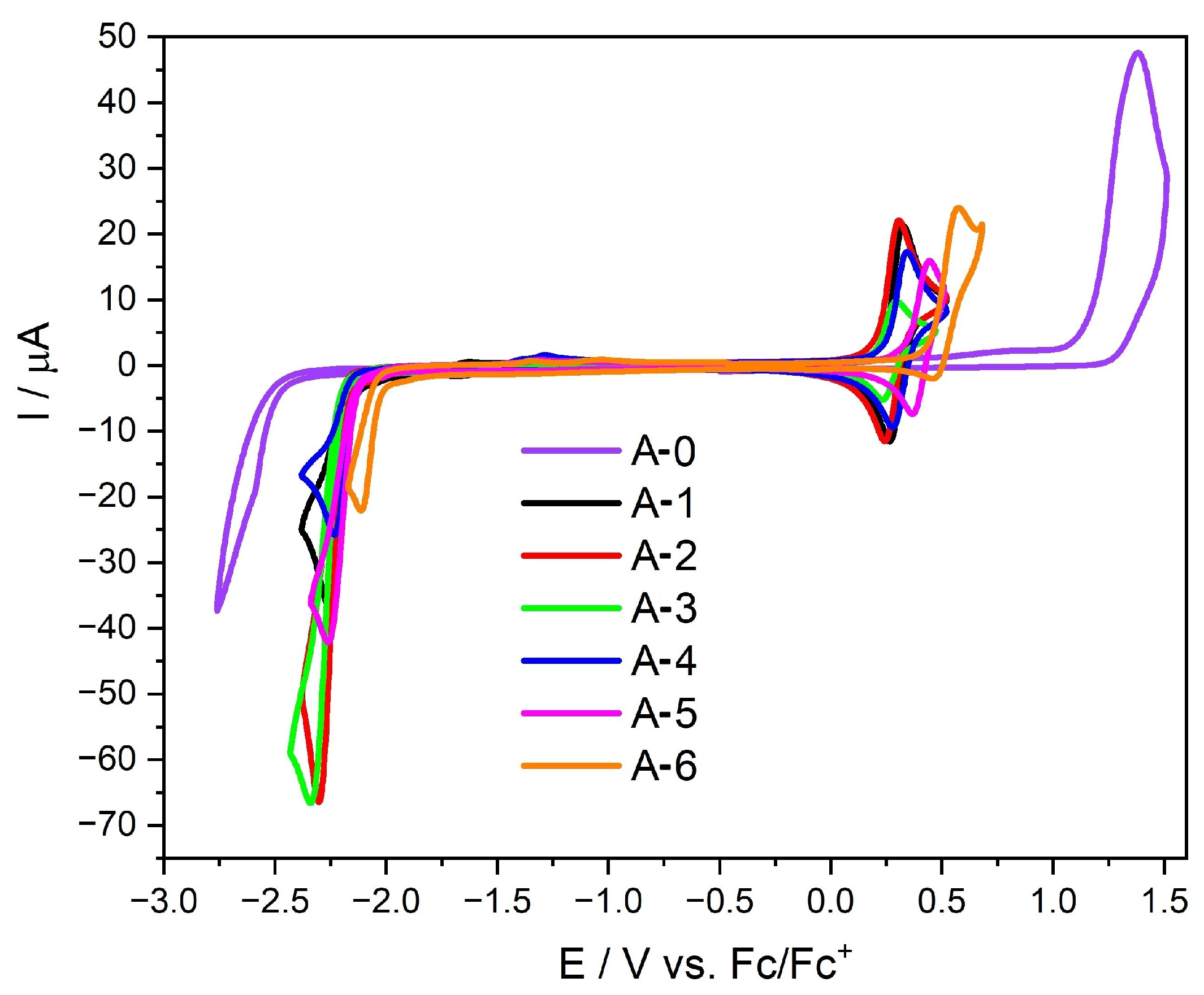
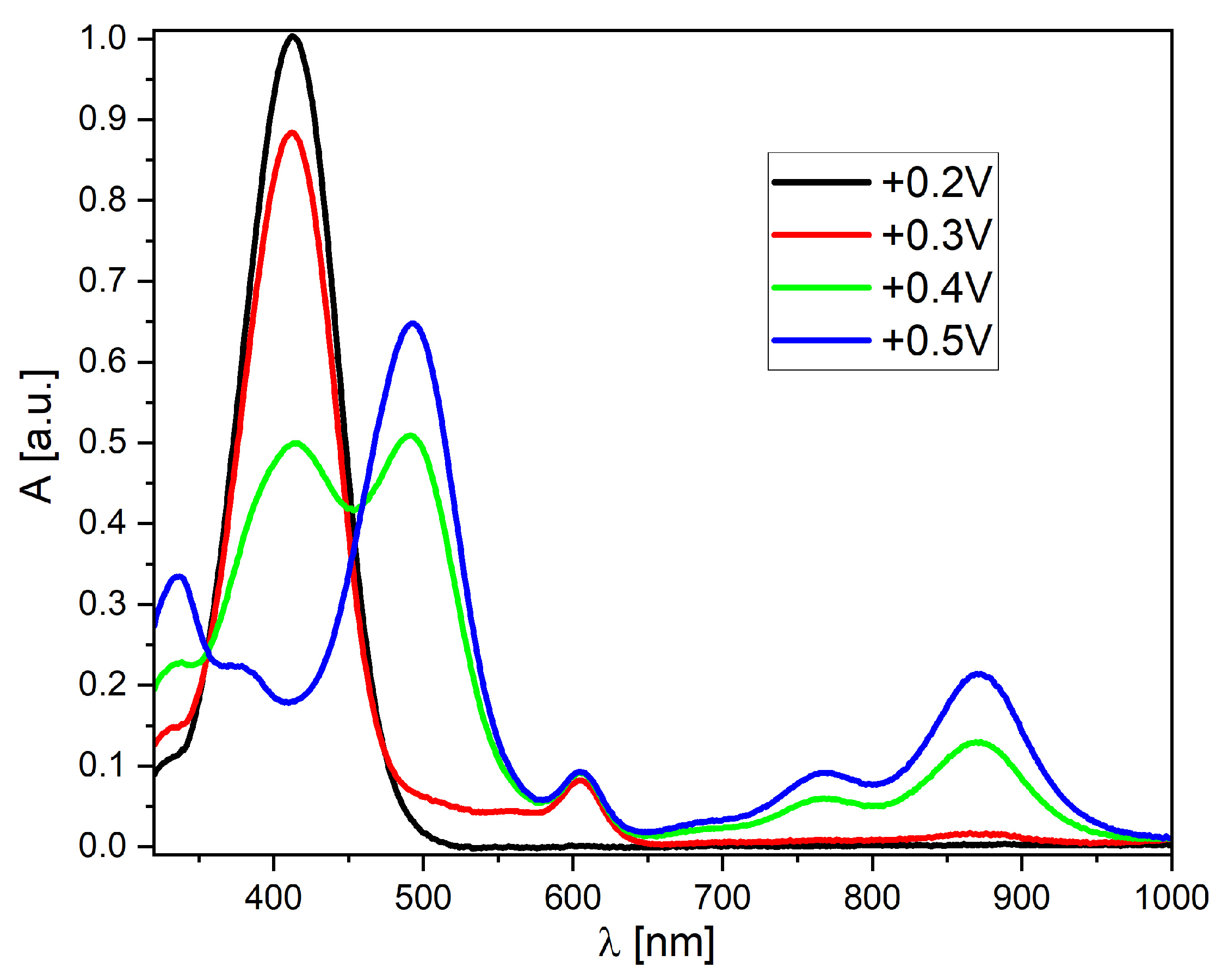
2.4. Electrochemical and Spectroelectrochemical Properties
3. Experimental Data—Materials, Methods, and Other Research Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, D.; Liu, Y.; Gao, H.; Chang, Q.; Cheng, X. α-Cyanostilbene and Fluorene Based Bolaamphiphiles: Synthesis, Self-Assembly, and AIEE Properties with Potential as White-Light Emissive Materials and Light-Emitting Liquid Crystal Displays. J. Mater. Chem. C 2020, 8, 17474–17481. [Google Scholar] [CrossRef]
- Islam, S.N.; Sil, A.; Patra, S.K. Achieving Yellow Emission by Varying the Donor/Acceptor Units in Rod-Shaped Fluorenyl-Alkynyl Based π-Conjugated Oligomers and Their Binuclear Gold (I) Alkynyl Complexes. Dalton Trans. 2017, 46, 5918–5929. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, H.; Ma, L.; Wu, W.; Liu, Y.; Zhao, J. Tuning the Photophysical Properties of N^N Pt(Ii) Bisacetylide Complexes with Fluorene Moiety and Its Applications for Triplet–Triplet-Annihilation Based Upconversion. J. Mater. Chem. 2012, 22, 5319. [Google Scholar] [CrossRef]
- Rouxel, C.; Charlot, M.; Mongin, O.; Krishna, T.R.; Caminade, A.-M.; Majoral, J.-P.; Blanchard-Desce, M. From Graftable Biphotonic Chromophores to Water-Soluble Organic Nanodots for Biophotonics: The Importance of Environmental Effects. Chem. A Eur. J. 2012, 18, 16450–16462. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, B.; Zhang, S.; Jiang, C.; Zou, Y.; Li, S.; Yang, Y.; Yao, Z.; Wan, X.; Chen, Y. Side Chain Isomerization Enables High Efficiency and Thickness Tolerant Organic Solar Cells. J. Mater. Chem. A 2023, 11, 700–707. [Google Scholar] [CrossRef]
- Zhang, D.; Wu‡, T.; Xu, P.; Ou, Y.; Sun, A.; Ma, H.; Cui, B.; Sun, H.; Ding, L.; Hua, Y. Importance of Terminated Groups in 9,9-Bis(4-Methoxyphenyl)-Substituted Fluorene-Based Hole Transport Materials for Highly Efficient Organic–Inorganic Hybrid and All-Inorganic Perovskite Solar Cells. J. Mater. Chem. A 2019, 7, 10319–10324. [Google Scholar] [CrossRef]
- Jegorovė, A.; Daškevičienė, M.; Kantminienė, K.; Jankauskas, V.; Čepas, R.J.; Gruodis, A.; Getautis, V.; Genevičius, K. New Fluorene-Based Bipolar Charge Transporting Materials. RSC Adv. 2024, 14, 2975–2982. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, Q.; Chen, Q.; Wang, Y.; Zhou, Y.; Song, B.; Yuan, N.; Ding, J.; Li, Y. High Efficiency Planar P-i-n Perovskite Solar Cells Using Low-Cost Fluorene-Based Hole Transporting Material. Adv. Funct. Mater. 2019, 29, 1900484. [Google Scholar] [CrossRef]
- Pasini, M.; Destri, S.; Porzio, W.; Botta, C.; Giovanella, U. Electroluminescent Poly(Fluorene-Co-Thiophene-S,S-Dioxide): Synthesis, Characterisation and Structure-Property Relationships. J. Mater. Chem. 2003, 13, 807–813. [Google Scholar] [CrossRef]
- Szlapa-Kula, A.; Ledwon, P.; Krawiec, A.; Kula, S. Dibenzofulvene Derivatives as Promising Materials for Photovoltaic and Organic Electronics. Energies 2023, 16, 8027. [Google Scholar] [CrossRef]
- Tigreros, A.; Rivera-Nazario, D.M.; Ortiz, A.; Martin, N.; Insuasty, B.; Echegoyen, L.A. Fluoren-9-Ylidene-Based Dyes for Dye-Sensitized Solar Cells. Eur. J. Org. Chem. 2015, 2015, 5537–5545. [Google Scholar] [CrossRef]
- Capodilupo, A.-L.; Giannuzzi, R.; Corrente, G.A.; De Marco, L.; Fabiano, E.; Cardone, A.; Gigli, G.; Ciccarella, G. Synthesis and Photovoltaic Performance of Dibenzofulvene-Based Organic Sensitizers for DSSC. Tetrahedron 2016, 72, 5788–5797. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Huang, G.-W.; Chang, Y.-J.; Wen, J.-J. Branched Dibenzofulvene-Based Organic Dyes for Dye-Sensitized Solar Cells under One Sun and Dim Light. J. Mater. Sci: Mater. Electron. 2019, 30, 12981–12991. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Li, Y.-H.; Chung, C.-L.; Hsu, H.-L.; Chen, C.-P. Triphenylamine Dibenzofulvene–Derived Dopant-Free Hole Transporting Layer Induces Micrometer-Sized Perovskite Grains for Highly Efficient near 20% for p-i-n Perovskite Solar Cells. Prog. Photovolt. Res. Appl. 2020, 28, 49–59. [Google Scholar] [CrossRef]
- Lin, H.-C.; Chen, L.-Y.; Lu, C.-C.; Lai, J.-Y.; Chen, Y.-C.; Hung, Y.-J. Ambipolar Carrier Transport Properties of Triphenylamine/Dibenzofulvene Derivative and Its Application for Efficient n-i-p Perovskite Solar Cells. Org. Electron. 2021, 95, 106200. [Google Scholar] [CrossRef]
- Lin, S.-C.; Cheng, T.-H.; Chen, C.-P.; Chen, Y.-C. Structural Effect on Triphenylamine Dibenzofulvene Based Interfacial Hole Transporting Materials for High-Performance Inverted Perovskite Solar Cells. Mater. Chem. Phys. 2022, 288, 126385. [Google Scholar] [CrossRef]
- Mandati, S.; Juneja, N.; Katerski, A.; Jegorovė, A.; Grzibovskis, R.; Vembris, A.; Dedova, T.; Spalatu, N.; Magomedov, A.; Karazhanov, S.; et al. 4.9% Efficient Sb2S3 Solar Cells from Semitransparent Absorbers with Fluorene-Based Thiophene-Terminated Hole Conductors. ACS Appl. Energy Mater. 2023, 6, 3822–3833. [Google Scholar] [CrossRef]
- Sholihah, N.; Cheng, H.-C.; Wang, J.-C.; Ni, J.-S.; Yu, Y.-Y.; Chen, C.-P.; Chen, Y.-C. Passivation of Inverted Perovskite Solar Cells by Trifluoromethyl-Group-Modified Triphenylamine Dibenzofulvene Hole Transporting Interfacial Layers. J. Phys. Chem. C 2023, 127, 6167–6178. [Google Scholar] [CrossRef]
- Kotowicz, S.; Tavgeniene, D.; Beresneviciute, R.; Zaleckas, E.; Krucaite, G.; Katarzyna Pająk, A.; Korzec, M.; Grzegorz Małecki, J.; Lipiński, M.; Grigalevicius, S.; et al. Effect of Substituent Structure in Fluorene Based Compounds: Experimental and Theoretical Study. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2023, 300, 122832. [Google Scholar] [CrossRef]
- Sánchez, J.G.; Aktas, E.; Martínez-Ferrero, E.; Capodilupo, A.L.; Corrente, G.A.; Beneduci, A.; Palomares, E. Increasing the Stability of Perovskite Solar Cells with Dibenzofulvene-Based Hole Transporting Materials. Electrochim. Acta 2022, 432, 141190. [Google Scholar] [CrossRef]
- Wu, N.; Zhang, X.; Liu, X.; Wang, Y.; Han, M.; Ghadari, R.; Wu, Y.; Ding, Y.; Cai, M.; Dai, S. Efficient Furan-Bridged Dibenzofulvene-Triphenylamine Hole Transporting Materials for Perovskite Solar Cells. Mater. Adv. 2023, 4, 515–522. [Google Scholar] [CrossRef]
- Nagar, M.R.; Choudhury, A.; Tavgeniene, D.; Beresneviciute, R.; Blazevicius, D.; Jankauskas, V.; Kumar, K.; Banik, S.; Ghosh, S.; Grigalevicius, S.; et al. Solution-Processable Phenothiazine and Phenoxazine Substituted Fluorene Cored Nanotextured Hole Transporting Materials for Achieving High-Efficiency OLEDs. J. Mater. Chem. C 2022, 10, 3593–3608. [Google Scholar] [CrossRef]
- Lu, B.; Chen, S.; Wang, J.; Xu, J.; Duan, X.; Pei, M. Soluble and Green-Light-Emitting Oligo(9-Fluorenylideneacetic Acid): Electrosynthesis and Characterization. Chin. J. Chem. 2012, 30, 1177–1184. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, L.; Gao, X.; Zhang, L.; Zhang, Q.; Chen, J. Highly Efficient Green PLED Based on Triphenlyaminesilole-Carbazole-Fluorene Copolymers with TPBI as the Hole Blocking Layer. Dye. Pigment. 2016, 127, 155–160. [Google Scholar] [CrossRef]
- Gopikrishna, P.; Das, D.; Adil, L.R.; Iyer, P.K. Saturated and Stable White Electroluminescence from Linear Single Polymer Systems Based on Polyfluorene and Mono-Substituted Dibenzofulvene Derivatives. J. Phys. Chem. C 2017, 121, 18137–18143. [Google Scholar] [CrossRef]
- Giangregorio, M.M.; Gambino, S.; Fabiano, E.; Leoncini, M.; Cardone, A.; Corrente, G.A.; Beneduci, A.; Accorsi, G.; Gigli, G.; Losurdo, M.; et al. Synthesis and Investigation of Electro-Optical Properties of H-Shape Dibenzofulvene Derivatives. Molecules 2022, 27, 1091. [Google Scholar] [CrossRef]
- Wong, M.Y.; Leung, L.M. A Comprehensive Study of Substituent Effects on Poly(Dibenzofulvene)s. New J. Chem. 2017, 41, 512–520. [Google Scholar] [CrossRef]
- Zhang, S.; Qin, L.; Lu, B.; Xu, J. Low-Potential Electrosynthesis of Novel Electroactive Poly(9-Fluorenemethanol) and Its Electrochromic and Blue-Light-Emitting Properties. Electrochim. Acta 2013, 90, 452–460. [Google Scholar] [CrossRef]
- He, Y.; Guo, W.; Pei, M.; Zhang, G. Electrosynthesis and Characterization of Poly(N-(9-Fluorenylmethoxycarbony)-Glycine). Chin. J. Chem. 2012, 30, 985–991. [Google Scholar] [CrossRef]
- Nakano, T.; Yade, T.; Fukuda, Y.; Yamaguchi, T.; Okumura, S. Free-Radical Polymerization of Dibenzofulvene Leading to a π-Stacked Polymer: Structure and Properties of the Polymer and Proposed Reaction Mechanism. Macromolecules 2005, 38, 8140–8148. [Google Scholar] [CrossRef]
- Nakano, T.; Yade, T. Synthesis, Structure, and Photophysical and Electrochemical Properties of a π-Stacked Polymer. J. Am. Chem. Soc. 2003, 125, 15474–15484. [Google Scholar] [CrossRef]
- Asiri, A.M.; Ahmed, S.A.; El-Daly, S.A.; Hussein, M.A.; Al-Soliemy, A.M.; Osman, O.I.; Shaaban, M.R.; Althagafi, I.I. Synthesis, Spectral Characteristics and DFT Studies of the New Dye 2,7-Diacetyl-9-((Dimethylamino)Methylene)-9H-Fluorene (DMMF) in Different Solvents. J. Fluoresc. 2015, 25, 1303–1314. [Google Scholar] [CrossRef]
- Corrente, G.A.; Fabiano, E.; Manni, F.; Chidichimo, G.; Gigli, G.; Beneduci, A.; Capodilupo, A.-L. Colorless to All-Black Full-NIR High-Contrast Switching in Solid Electrochromic Films Prepared with Organic Mixed Valence Systems Based on Dibenzofulvene Derivatives. Chem. Mater. 2018, 30, 5610–5620. [Google Scholar] [CrossRef]
- Majeed, S.; Waseem, M.T.; Junaid, H.M.; Khan, G.S.; Nawazish, S.; Mahmood, T.; Khan, A.M.; Shahzad, S.A. Aggregation Induced Emission Based Fluorenes as Dual-Channel Fluorescent Probes for Rapid Detection of Cyanide: Applications of Smartphones and Logic Gates. RSC Adv. 2022, 12, 18897–18910. [Google Scholar] [CrossRef]
- Gopikrishna, P.; Iyer, P.K. Monosubstituted Dibenzofulvene-Based Luminogens: Aggregation-Induced Emission Enhancement and Dual-State Emission. J. Phys. Chem. C 2016, 120, 26556–26568. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Aldred, M.P.; Chen, Z.-Q.; Chen, T.; Meng, X.; Zhu, M.-Q. Efficient Green-Red Piezofluorochromism of Bisanthracene-Modified Dibenzofulvene. RSC Adv. 2015, 5, 1079–1082. [Google Scholar] [CrossRef]
- Sicard, L.; Quinton, C.; Peltier, J.-D.; Tondelier, D.; Geffroy, B.; Biapo, U.; Métivier, R.; Jeannin, O.; Rault-Berthelot, J.; Poriel, C. Spirobifluorene Regioisomerism: A Structure–Property Relationship Study. Chem. A Eur. J. 2017, 23, 7719–7727. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J. Ab Initio Study of Ionic Solutions by a Polarizable Continuum Dielectric Model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum Solvation Models: A New Approach to the Problem of Solute’s Charge Distribution and Cavity Boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Bujak, P.; Kulszewicz-Bajer, I.; Zagorska, M.; Maurel, V.; Wielgus, I.; Pron, A. Polymers for Electronics and Spintronics. Chem. Soc. Rev. 2013, 42, 8895. [Google Scholar] [CrossRef]
- Majeed, S.; Junaid, H.M.; Waseem, M.T.; Khan, Z.A.; Khan, A.M.; Shahzad, S.A. Mechanochromic and AIE Active Fluorescent Probes for Solution and Vapor Phase Detection of Picric Acid: Application of Logic Gate. J. Photochem. Photobiol. A Chem. 2022, 432, 114057. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | A-1 | A-2 | A-3 | A-4 | A-5 | A-6 |
| Structure |  | 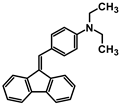 | 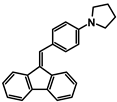 | 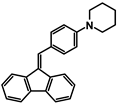 | 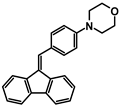 | 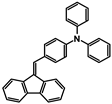 |
| Yield | 85% | 85% | 13% | 90% | 76% | 60% |
| Photo |  |  |  |  |  |  |
| Compound | Structure | Solvent | λabs [nm] | ε [L·mol−l·cm−l] | PLsolid [nm] | Egopt 1 [eV] |
|---|---|---|---|---|---|---|
| A-0 | 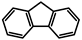 | Tol | 301 | 4774 | 409 | 4.05 |
| CHCl3 | 301 | 10,703 | ||||
| DCM | 301 | 8810 | ||||
| ACN | 299 | 8460 | ||||
| MeOH | 300 | 13,096 | ||||
| A-1 | 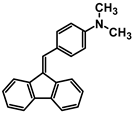 | Tol | 397 | 24,394 | - | 2.69 |
| CHCl3 | 398 | 23,942 | ||||
| DCM | 401 | 21,838 | ||||
| ACN | 399 | 23,744 | ||||
| MeOH | 394 | 20,512 | ||||
| A-2 | 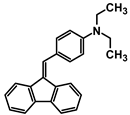 | Tol | 407 | 32,090 | - | 2.62 |
| CHCl3 | 410 | 27,062 | ||||
| DCM | 414 | 20,086 | ||||
| ACN | 411 | 28,248 | ||||
| MeOH | 406 | 30,596 | ||||
| A-3 | 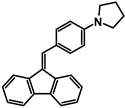 | Tol | 408 | 25,568 | 570 | 2.63 |
| CHCl3 | 409 | 25,358 | ||||
| DCM | 413 | 20,472 | ||||
| ACN | 411 | 25,220 | ||||
| MeOH | 405 | 14,128 | ||||
| A-4 | 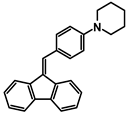 | Tol | 388 | 12,004 | 572 | 2.68 |
| CHCl3 | 386 | 22,802 | ||||
| DCM | 393 | 19,328 | ||||
| ACN | 390 | 24,950 | ||||
| MeOH | 380 | 22,630 | ||||
| A-5 | 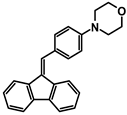 | Tol | 377 | 27,160 | 601 | 2.78 |
| CHCl3 | 373 | 23,708 | ||||
| DCM | 377 | 16,460 | ||||
| ACN | 376 | 21,624 | ||||
| MeOH | 370 | 25,572 | ||||
| A-6 | 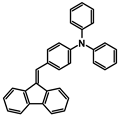 | Tol | 400 | 25,118 | 555 | 2.68 |
| CHCl3 | 401 | 23,092 | ||||
| DCM | 401 | 23,184 | ||||
| ACN | 394 | 22,694 | ||||
| MeOH | 393 | 27,010 |
| Code | Eox [V] | Ered [V] | IP 1 [eV] | EA 1 [eV] | Eg(cv) 2 [eV] | IP 3 [eV] | EA 3 [eV] | Eg 3 [eV] | Eg 4 [eV] |
|---|---|---|---|---|---|---|---|---|---|
| A-0 | 1.30 | −2.48 | −6.40 | −2.62 | 3.78 | −6.22 | −1.32 | 4.90 | 4.05 |
| A-1 | 0.22 | −2.15 | −5.32 | −2.95 | 2.37 | −5.00 | −2.38 | 2.62 | 2.69 |
| A-2 | 0.18 | −2.17 | −5.28 | −2.93 | 2.35 | −4.72 | −2.59 | 2.13 | 2.62 |
| A-3 | 0.19 | −2.19 | −5.29 | −2.91 | 2.38 | −4.91 | −2.35 | 2.55 | 2.63 |
| A-4 | 0.24 | −2.13 | −5.34 | −2.97 | 2.37 | −5.12 | −2.40 | 2.72 | 2.68 |
| A-5 | 0.32 | −2.11 | −5.42 | −2.99 | 2.42 | −5.22 | −2.42 | 2.80 | 2.78 |
| A-6 | 0.42 | −2.02 | −5.52 | −3.08 | 2.44 | −5.09 | −2.49 | 2.60 | 2.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalarus, P.; Szlapa-Kula, A.; Filapek, M.; Kula, S. The Effect of Modifying the C9 Position of Fluorene with N-Donor Substituents on Selected Physicochemical Properties. Molecules 2025, 30, 1924. https://doi.org/10.3390/molecules30091924
Kalarus P, Szlapa-Kula A, Filapek M, Kula S. The Effect of Modifying the C9 Position of Fluorene with N-Donor Substituents on Selected Physicochemical Properties. Molecules. 2025; 30(9):1924. https://doi.org/10.3390/molecules30091924
Chicago/Turabian StyleKalarus, Paweł, Agata Szlapa-Kula, Michał Filapek, and Sławomir Kula. 2025. "The Effect of Modifying the C9 Position of Fluorene with N-Donor Substituents on Selected Physicochemical Properties" Molecules 30, no. 9: 1924. https://doi.org/10.3390/molecules30091924
APA StyleKalarus, P., Szlapa-Kula, A., Filapek, M., & Kula, S. (2025). The Effect of Modifying the C9 Position of Fluorene with N-Donor Substituents on Selected Physicochemical Properties. Molecules, 30(9), 1924. https://doi.org/10.3390/molecules30091924