

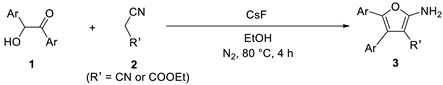
Metal-Free Catalytic Synthesis of Tetrasubstituted Furans from α-Hydroxy Ketones and Cyano Compounds

Abstract
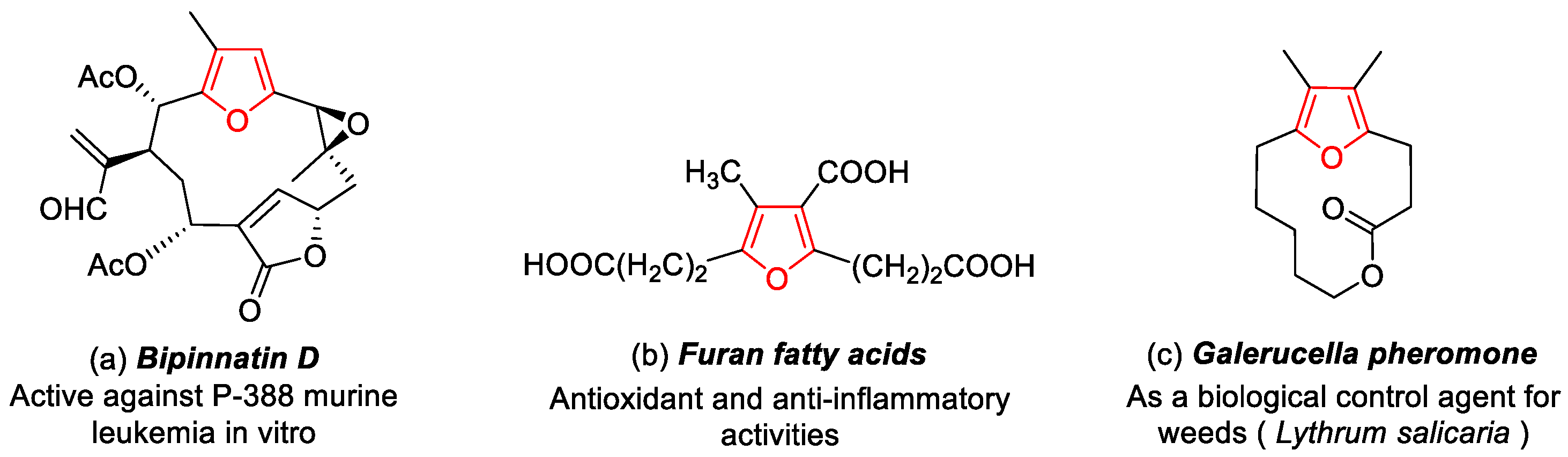
1. Introduction
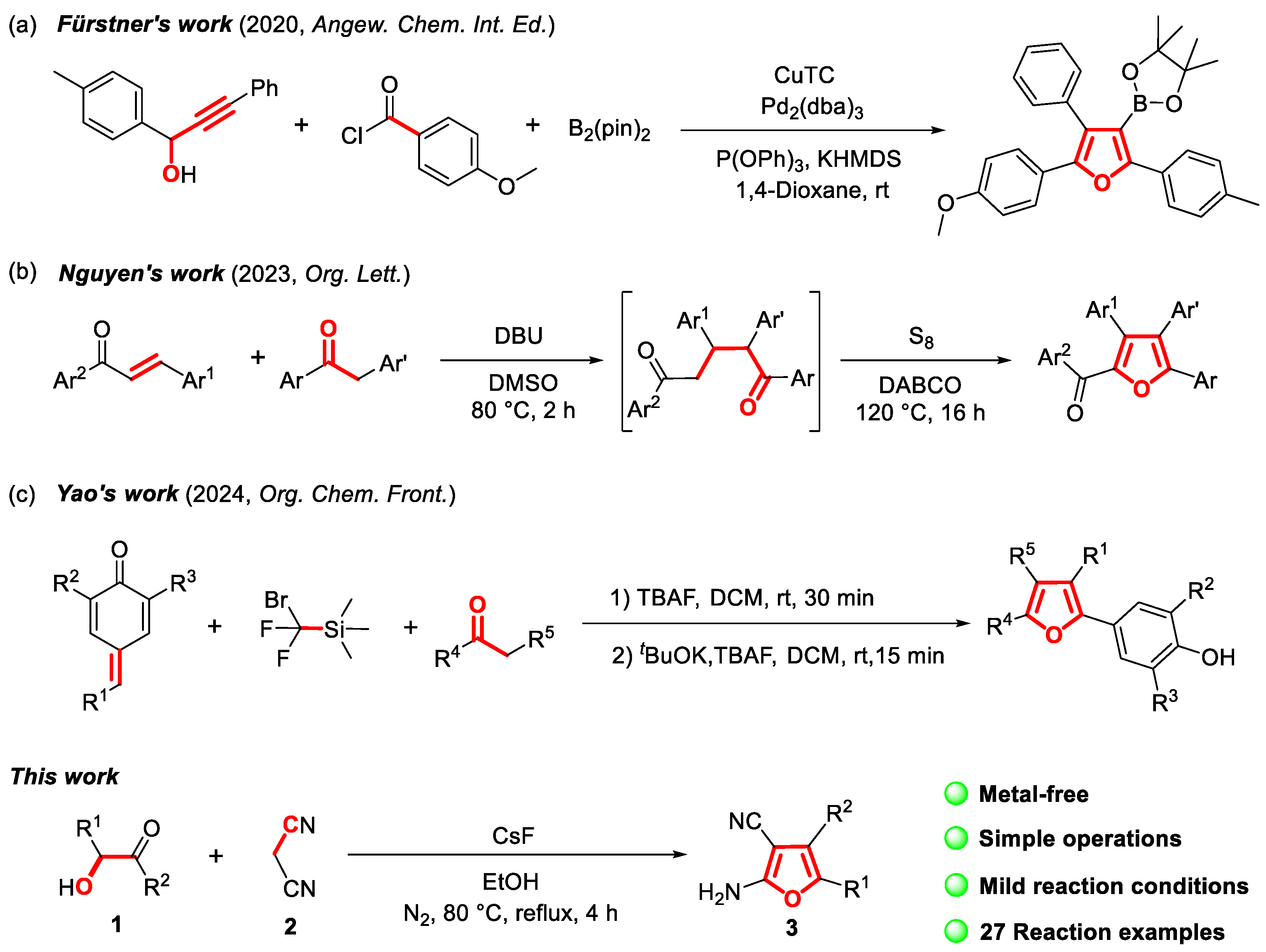
2. Results and Discussion
2.1. Optimization of Reaction Conditions
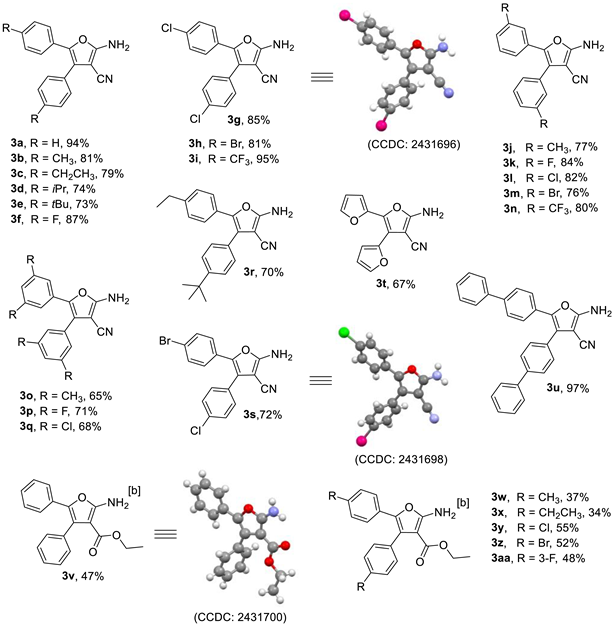
2.2. Scope of Substrates
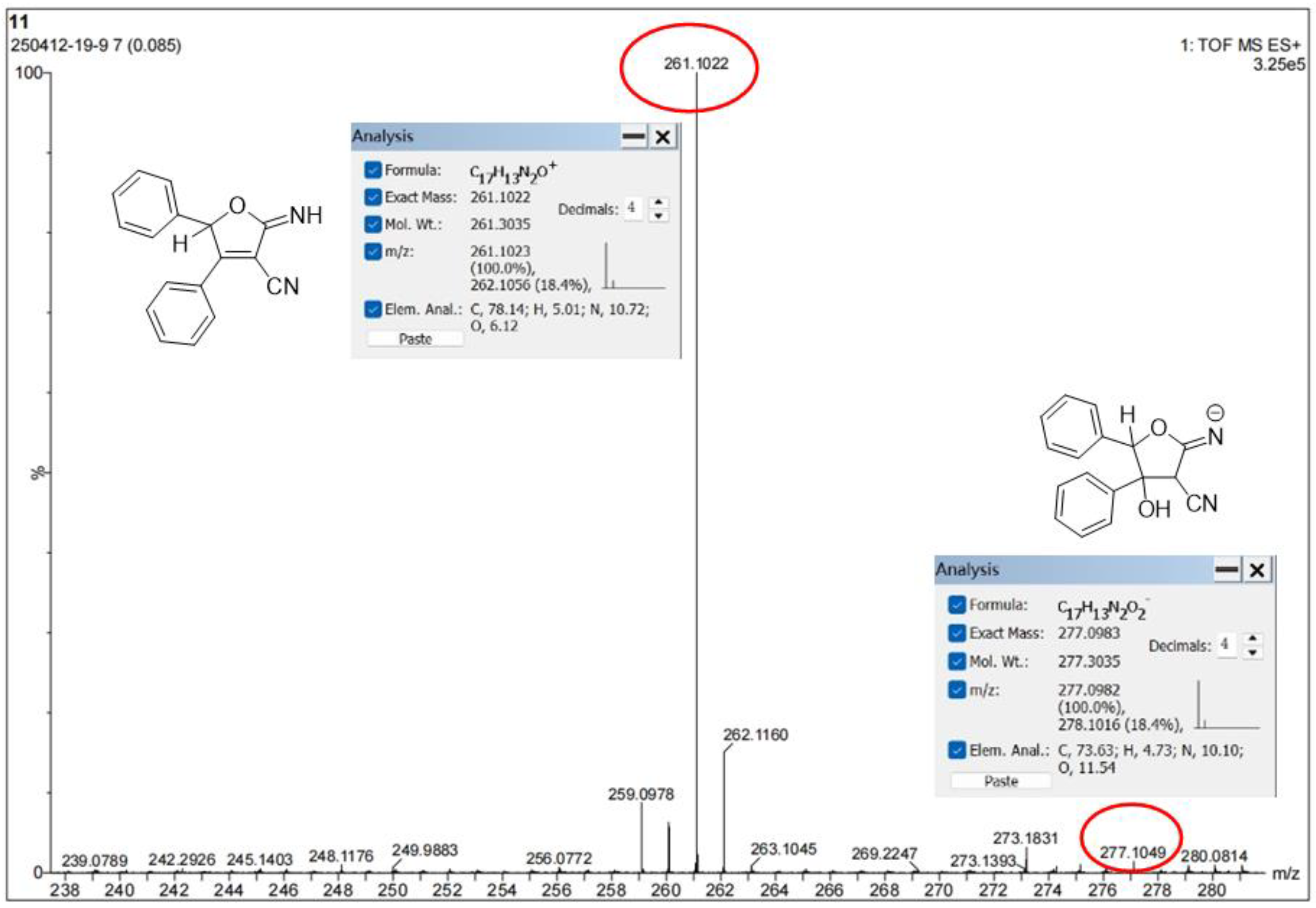
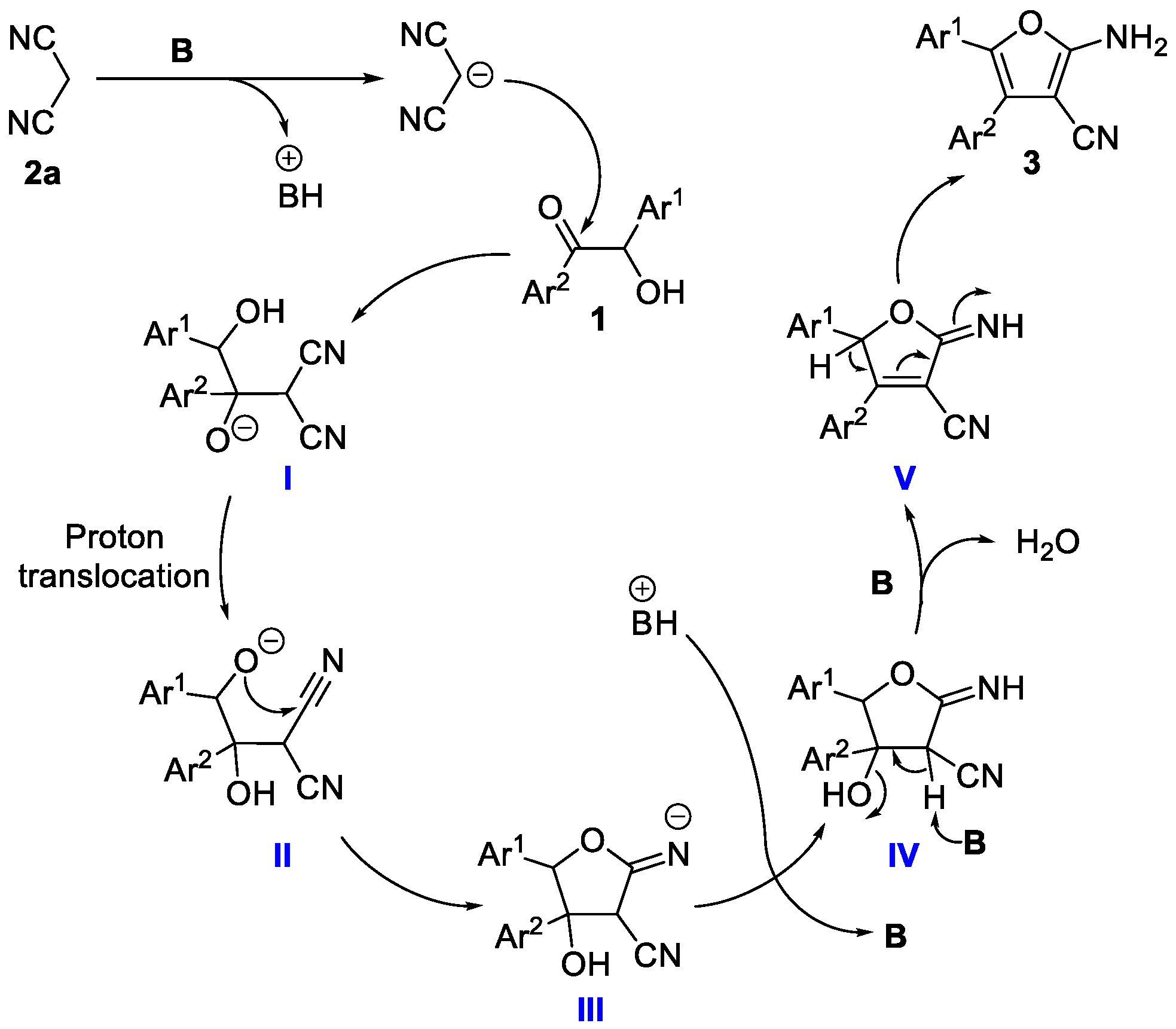
2.3. Mechanism Investigation
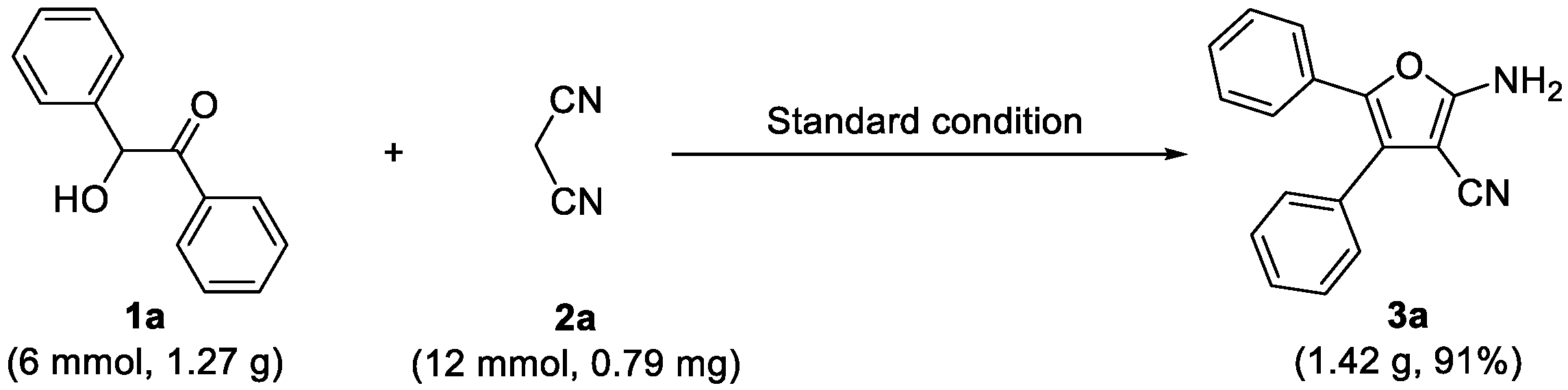
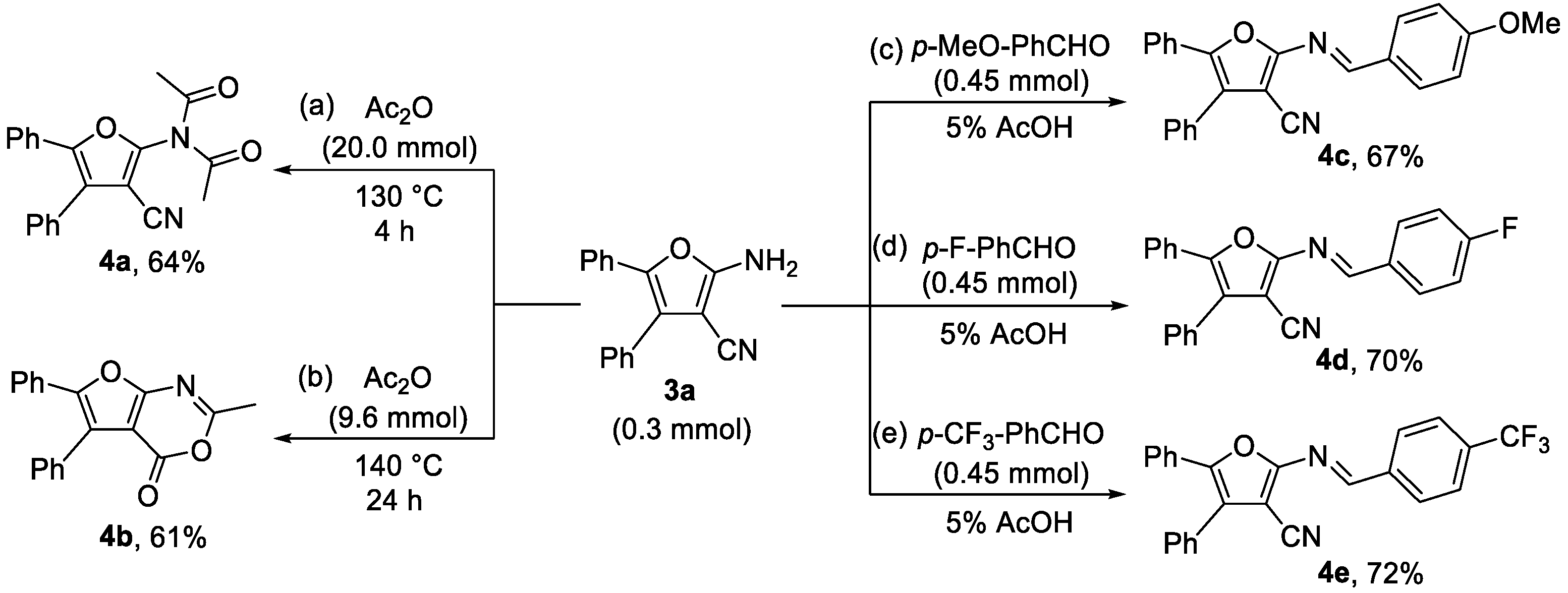
2.4. Gram Scale Reaction and Derivatization Applications
3. Materials and Methods
3.1. General Information
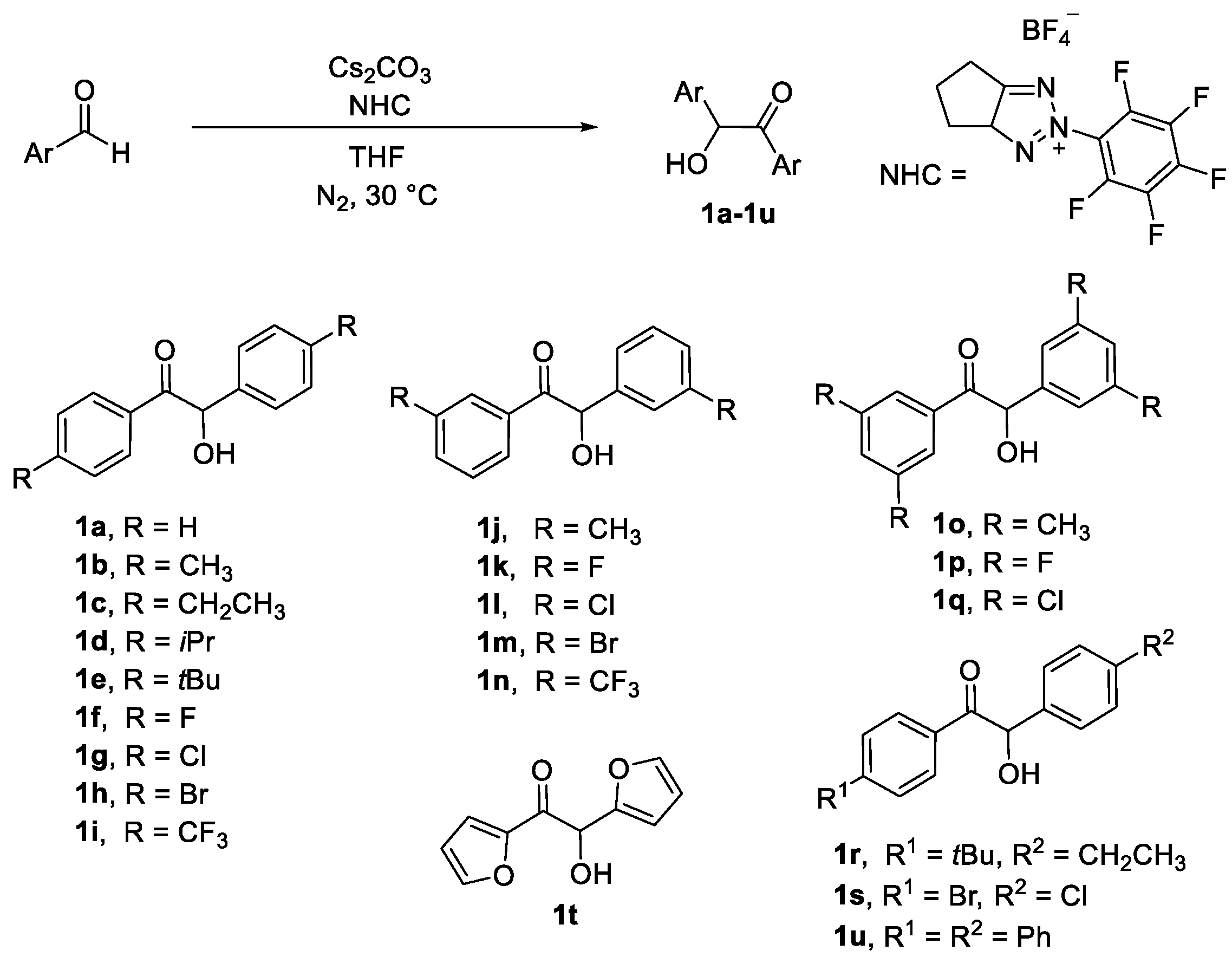
3.2. Experimental Procedure for α-Hydroxy Ketones 1a–1u
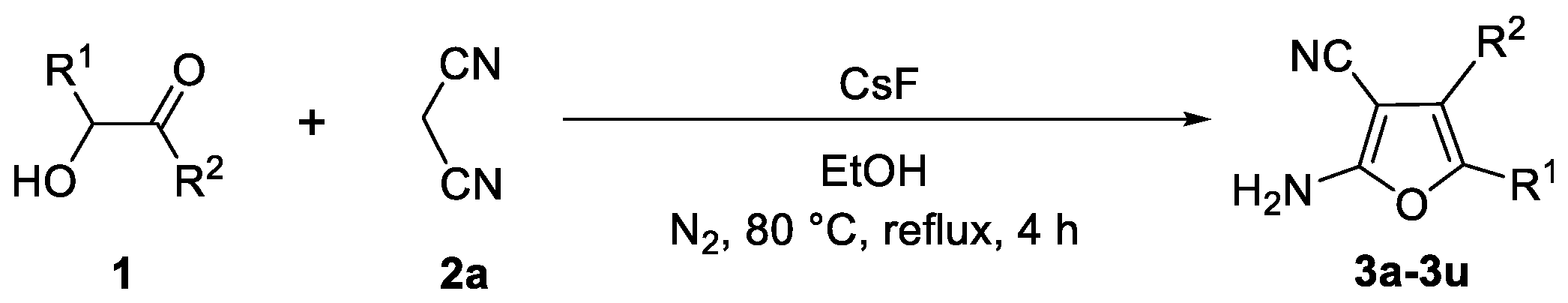
3.3. Experimental Procedure for Compounds 3a–3u
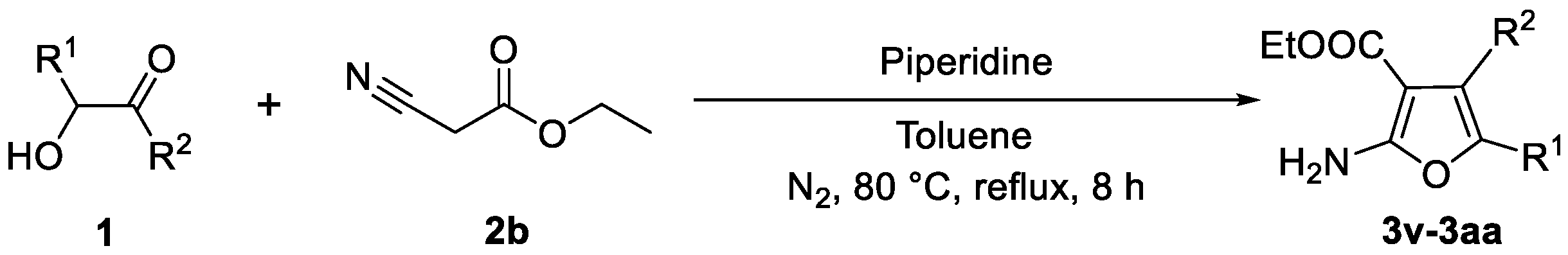
3.4. Experimental Procedure for Compounds 3v–3aa
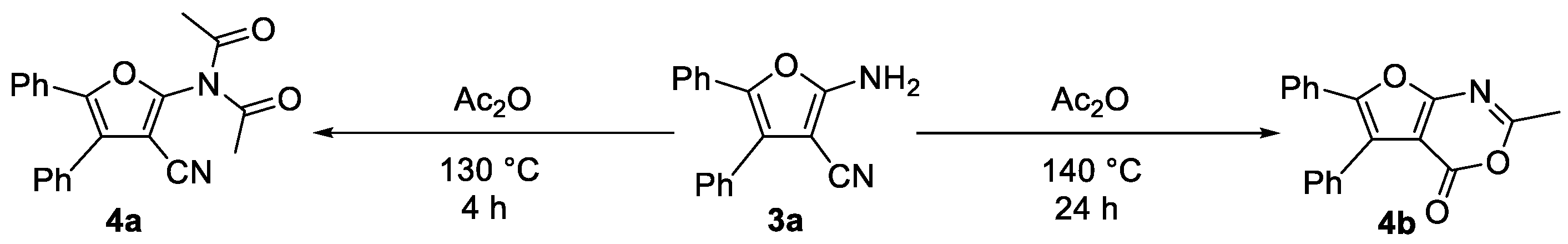
3.5. Experimental Procedure for Compounds 4a and 4b
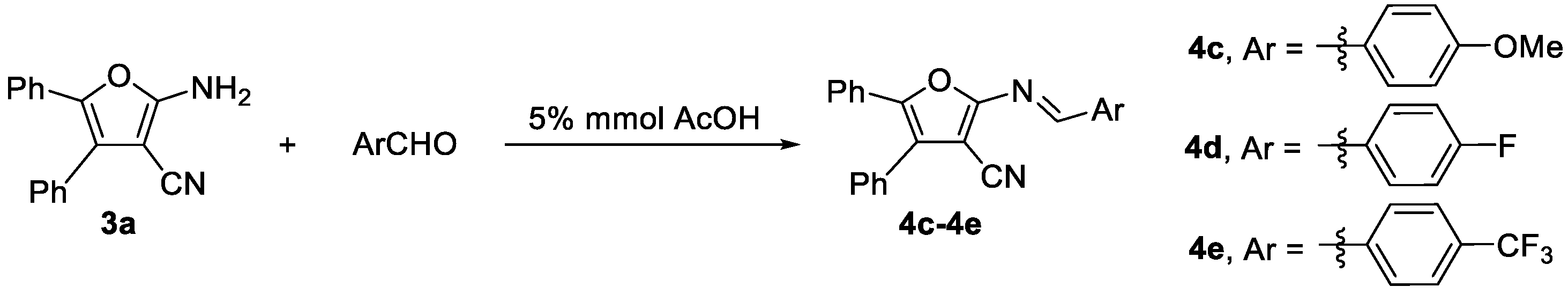
3.6. Experimental Procedure for Compounds 4c–4e
3.7. Characterization Data for All Products 3a–3aa and 4a–4e
- (1) 2-Amino-4,5-diphenylfuran-3-carbonitrile (3a): White solid (72.2 mg, yield 93%), m.p. 205.2–205.9 °C (204.0–206.0 °C [51]); 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.72 (s, 2H, NH2), 7.48–7.44 (m, 2H, ArH), 7.43–7.40 (m, 1H, ArH), 7.39–7.37 (m, 2H, ArH), 7.27–7.22 (m, 4H, ArH), 7.19–7.16 (m, 1H, ArH).
- (2) 2-Amino-4,5-di-p-tolylfuran-3-carbonitrile (3b): White solid (71.7 mg, yield 81%), m.p. 214.8–216.6 °C (216.0 °C [52]); 1H NMR (600 MHz, CDCl3), δ, ppm: 7.33 (d, 2H, J = 7.8 Hz, ArH), 7.27 (d, 2H, J = 7.8 Hz, ArH), 7.21 (d, 2H, J = 7.8 Hz, ArH), 7.06 (d, 2H, J = 7.8 Hz, ArH), 4.99 (s, 2H, NH2), 2.40 (s, 3H, CH3), 2.32 (s, 3H, CH3).
- (3) 2-Amino-4,5-bis(4-ethylphenyl)furan-3-carbonitrile (3c): White solid (75.1 mg, yield 79%), m.p. 156.0–158.0 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.36 (d, 2H, J = 8.4 Hz, ArH), 7.29 (d, 2H, J = 8.4 Hz, ArH), 7.23 (d, 2H, J = 8.4 Hz, ArH), 7.08 (d, 2H, J = 8.4 Hz, ArH), 5.00 (s, 2H, NH2), 2.67–2.72 (q, J = 7.8 Hz, 2H, CH2), 2.58–2.63 (q, J = 7.8 Hz, 2H, CH2), 1.28 (t, J = 7.8 Hz, 3H, CH3), 1.21 (t, J = 7.8 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 161.8, 144.4, 143.7, 139.8, 129.0, 128.5, 128.0, 127.2, 125.4, 121.0, 115.5, 73.3, 28.8, 15.4; ESI-HRMS, m/z: Calcd for C21H21N2O [M + H]+: 317.1648, Found: 317.1644.
- (4) 2-Amino-4,5-bis(4-isopropylphenyl)furan-3-carbonitrile (3d): White solid (76.4 mg, yield 74%), m.p. 157.1–158.8 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.39 (d, 2H, J = 7.8 Hz, ArH), 7.32 (d, 2H, J = 7.8 Hz, ArH), 7.27 (d, 2H, J = 7.8 Hz, ArH), 7.12 (d, 2H, J = 7.8 Hz, ArH), 4.85 (b, 2H, NH2), 2.98–2.92 (m, 1H, CH), 2.90–2.84 (m, 1H, CH), 1.30 (d, J = 6.6 Hz, 6H, 2CH3), 1.23 (d, J = 7.2 Hz, 6H, 2CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 162.1, 154.5, 149.9, 135.2, 130.0, 128.9, 127.0, 126.6, 125.3, 113.7, 72.9, 34.0, 24.0; ESI-HRMS, m/z: Calcd for C23H24N2ONa [M+Na]+: 367.1786, Found: 367.1781.
- (5) 2-Amino-4,5-bis(4-(tert-butyl)phenyl)furan-3-carbonitrile (3e): White solid (81.2 mg, yield 73%), m.p. 178.4–180.4 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.42 (d, 2H, J = 8.4 Hz, ArH), 7.39 (d, 2H, J = 8.4 Hz, ArH), 7.34 (d, 2H, J = 8.4 Hz, ArH), 7.27 (d, 2H, J = 8.4 Hz, ArH), 5.03 (s, 2H, NH2), 1.36 (s, 9H, 3CH3), 1.30 (s, 9H, 3CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 161.9, 151.2, 150.5, 139.7, 128.7, 127.0, 125.9, 125.5, 125.0, 121.0, 115.6, 73.3, 34.8, 31.4; ESI-HRMS, m/z: Calcd for C25H29N2O [M + H]+: 373.2274, Found: 373.2272.
- (6) 2-Amino-4,5-bis(4-fluorophenyl)furan-3-carbonitrile (3f): White solid (77.3 mg, yield 87%), m.p. 210.3–211.9 °C (210.0–212.0 °C [51]); 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.73 (s, 2H, NH2), 7.44–7.40 (m, 2H, ArH), 7.33–7.29 (m, 2H, ArH), 7.25–7.22 (m, 2H, ArH), 7.16–7.12 (m, 2H, ArH).
- (7) 2-Amino-4,5-bis(4-chlorophenyl)furan-3-carbonitrile (3g): White solid (84.0 mg, yield 85%), m.p. 222.7–224.6 °C (223.0–225.0 °C [51]); 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.81 (s, 2H, NH2), 7.54 (d, 2H, J = 8.4 Hz, ArH), 7.41 (d, 2H, J = 8.4 Hz, ArH), 7.37 (d, 2H, J = 8.4 Hz, ArH), 7.21 (d, 2H, J = 8.4 Hz, ArH).
- (8) 2-Amino-4,5-bis(4-bromophenyl)furan-3-carbonitrile (3h): White solid (100.9 mg, yield 81%), m.p. 225.1–226.7 °C (226.0–227.0 °C [51]); 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.82 (s, 2H, NH2), 7.67 (d, 2H, J = 8.4 Hz, ArH), 7.49 (d, 2H, J = 8.4 Hz, ArH), 7.34 (d, 2H, J = 8.4 Hz, ArH), 7.14 (d, 2H, J = 8.4 Hz, ArH).
- (9) 2-Amino-4,5-bis(4-bromophenyl)furan-3-carbonitrile (3i): White solid (113.2 mg, yield 95%), m.p. 254.7–256.5 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 8.00 (s, 2H, NH2), 7.87 (d, 2H, J = 7.8 Hz, ArH), 7.66 (d, 2H, J = 8.4 Hz, ArH), 7.65 (d, 2H, J = 8.4 Hz, ArH), 7.38 (d, 2H, J = 7.8 Hz, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.5, 136.2, 135.6, 133.2, 130.3, 129.5 (q, J = 32.3 Hz), 127.4 (q, J = 31.5 Hz), 126.6 (q, J = 4.1 Hz), 126.4 (q, J = 267.8 Hz), 126.3 (q, J = 4.2 Hz), 124.6 (q, J = 269.3 Hz), 123.7, 121.9, 115.4, 70.1; 19F NMR (564 MHz, DMSO-d6), δ, ppm: −61.178, −61.811; ESI-HRMS, m/z: Calcd for C19H9F6N2O [M − H]−: 395.0625, Found: 395.0626.
- (10) 2-Amino-4,5-di-m-tolylfuran-3-carbonitrile (3j): White solid (66.6 mg, yield 77%), m.p. 157.7–159.4 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.31–7.26 (m, 2H, ArH), 7.24 (s, 1H, ArH), 7.23 (s, 1H, ArH), 7.18 (d, J = 7.8 Hz, 1H, ArH), 7.14–7.09 (m, 2H, ArH), 7.02 (d, J = 7.2 Hz, 1H, ArH), 5.04 (s, 2H, NH2), 2.36 (s, 3H, CH3), 2.27 (s, 3H, CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 161.9, 139.7, 138.6, 138.2, 131.1, 129.7, 129.2, 128.4, 126.2, 125.9, 122.5, 121.8, 115.3, 73.4, 21.6, 21.5; ESI-HRMS, m/z: Calcd for C19H17N2O [M + H]+: 289.1335, Found: 289.1331.
- (11) 2-Amino-4,5-bis(3-fluorophenyl)furan-3-carbonitrile (3k): White solid (74.6 mg, yield 84%), m.p. 201.3–202.9 °C (201.0–202.0 °C [51]); 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.87 (s, 2H, NH2), 7.56–7.51 (m, 1H, ArH), 7.34–7.27 (m, 2H, ArH), 7.24 (d, J = 7.2 Hz, 2H, ArH), 7.04–7.00 (m, 2H, ArH), 6.94 (d, J = 8.4 Hz, 1H, ArH).
- (12) 2-Amino-4,5-bis(3-chlorophenyl)furan-3-carbonitrile (3l): White solid (80.4, yield 82%), m.p. 154.7–156.4 °C (154.0–156.0 °C [51]); 1H NMR (600 MHz, CDCl3), δ, ppm: 7.41 (s, 1H, ArH), 7.40–7.35 (m, 3H, ArH), 7.32–7.30 (m, 1H, ArH), 7.20–7.17 (m, 1H, ArH), 7.16–7.12 (m, 2H, ArH), 5.13 (s, 2H, NH2).
- (13) 2-Amino-4,5-bis(3-bromophenyl)furan-3-carbonitrile (3m): White solid (94.8 mg, yield 76%), m.p. 185.4–187.0 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.89 (s, 2H, NH2), 7.65 (d, 2H, J = 8.4 Hz, ArH), 7.59 (s, 1H, ArH), 7.46–7.43 (m, 1H, ArH), 7.41 (d, J = 7.8 Hz, 1H, ArH), 7.38–7.35 (m, 2H, ArH), 7.24–7.20 (m, 1H, ArH), 7.14 (d, J = 8.4 Hz, 1H, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.2, 135.7, 133.6, 132.0, 131.8, 131.3, 130.1, 128.5, 127.0, 123.3, 122.6, 115.6, 69.8; ESI-HRMS, m/z: Calcd for C17H11N2OBr2 [M + H]+: 416.9238, Found: 416.9237.
- (14) 2-Amino-4,5-bis(3-(trifluoromethyl)phenyl)furan-3-carbonitrile (3n): White solid (95.0, yield 80%), m.p. 253.7–255.4 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.70–7.67 (m, 3H, ArH), 7.65 (d, J = 7.8 Hz, 1H, ArH), 7.61 (s, 1H, ArH), 7.60–7.57 (m, 1H, ArH), 7.46 (d, J = 7.2 Hz, 1H, ArH), 7.42 (d, J = 7.8 Hz, 1H, ArH), 7.34 (t, J = 7.8 Hz, 1H, ArH), 5.27 (s, 2H, NH2); 13C NMR (150 MHz, CDCl3), δ, ppm: 162.3, 138.4, 132.3, 131.5 (q, J = 247.4 Hz), 131.4 (q, J = 243.0 Hz), 129.8, 129.1, 127.9, 125.7 (q, J = 43.4 Hz), 125.6 (q, J = 42.5 Hz), 124.6, 124.2 (q, J = 3.9 Hz), 122.8, 121.8 (q, J = 4.5 Hz), 114.4, 72.9; 19F NMR (564 MHz, CDCl3), δ, ppm: −62.910, −63.205; ESI-HRMS, m/z: Calcd for C19H9F6N2O [M − H]−: 395.0625, Found: 395.0622.
- (15) 2-Amino-4,5-bis(4-bromophenyl)furan-3-carbonitrile (3o): White solid (61.6 mg, yield 65%), m.p. 170.2–172.1 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.64 (s, 2H, NH2), 7.03 (s, 1H, ArH), 6.99 (s, 2H, ArH), 6.89 (s, 2H, ArH), 6.80 (s, 1H, ArH), 2.26 (s, 6H, CH3), 2.12 (s, 6H, CH3); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.0, 138.3, 137.3, 131.6, 130.2, 128.9, 127.0, 123.7, 122.5, 116.2, 69.6, 21.4, 21.3; ESI-HRMS, m/z: Calcd for C21H21N2O [M + H]+: 317.1648, Found: 317.1644.
- (16) 2-Amino-4,5-bis(3,5-difluorophenyl)furan-3-carbonitrile (3p): White solid (70.9, yield 71%), m.p. 228.3–230.2 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 8.00 (s, 2H, NH2), 7.39 (s, 1H, ArH), 7.18 (s, 2H, ArH), 7.09 (s, 1H, ArH), 6.79 (s, 2H, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.1, 163.2 (d, J = 246.3 Hz), 163.1 (d, J = 246.9 Hz), 163.0 (d, J = 244.4 Hz), 162.9 (d, J = 244.1 Hz), 135.2 (d, J = 3.5 Hz), 134.4, 132.3, 123.0 (d, J = 3.2 Hz), 115.1, 112.9 (d, J = 25.4 Hz), 107.3 (d, J = 27.5 Hz), 105.2 (d, J = 25.7 Hz), 105.0 (d, J = 25.4 Hz), 103.0 (d, J = 26.0 Hz), 102.8 (d, J = 26.0 Hz), 70.1; 19F NMR (564 MHz, DMSO-d6), δ, ppm: −108.408, −108.771; ESI-HRMS, m/z: Calcd for C17H7F4N2O [M − H]−: 331.0500, Found: 331.0499.
- (17) 2-Amino-4,5-bis(3,5-dichlorophenyl)furan-3-carbonitrile (3q): White solid (80.8, yield 68%), m.p. 243.6–245.5 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 8.03 (s, 2H, NH2), 7.74 (s, 1H, ArH), 7.50 (s, 2H, ArH), 7.41 (s, 1H, ArH), 7.11 (s, 2H, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.3, 135.3, 135.0, 134.7, 134.3, 132.3, 129.1, 128.1, 126.6, 122.6, 115.1, 70.0; ESI-HRMS, m/z: Calcd for C17H8N2ONaCl4 [M+Na]+: 418.9288, Found: 418.9286.
- (18) 2-Amino-4-(4-(tert-butyl)phenyl)-5-(4-ethylphenyl)furan-3-carbonitrile (3r): White solid (72.2 mg, yield 70%), m.p. 188.0–190.0 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.43–7.40 (m, 1H, ArH), 7.39–7.35 (m, 2H, ArH), 7.33–7.30 (m, 2H, ArH), 7.28–7.26 (m, 1H, ArH), 7.25–7.22 (m, 1H, ArH), 7.10–7.07 (m, 1H, ArH), 5.00 (s, 2H, NH2), 2.72–2.59 (m, 2H, CH2), 1.36 (s, 3H, CH3), 1.31–1.27 (m, 9H, 3CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 162.0, 156.9, 151.2, 150.5, 144.4, 139.6, 129.8, 129.0, 128.0, 125.5, 125.0, 113.7, 73.2, 35.3, 31.3, 28.8, 15.4, ESI-HRMS, m/z: Calcd for C23H25N2O [M + H]+: 345.1961, Found: 345.1958.
- (19) 2-Amino-4-(4-bromophenyl)-5-(4-chlorophenyl)furan-3-carbonitrile (3s): White solid (80.6 mg, yield 72%), m.p. 235.2–236.9 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.83 (s, 2H, NH2), 7.68 (d, 1H, J = 8.4 Hz, ArH), 7.54 (d, 1H, J = 9.0 Hz, ArH), 7.50 (d, 1H, J = 7.2 Hz, ArH), 7.41 (d, 1H, J = 8.4 Hz, ArH), 7.37 (d, 1H, J = 7.2 Hz, ArH), 7.34 (d, 1H, J = 8.4 Hz, ArH), 7.23–7.20 (m, 1H, ArH), 7.16–7.13 (m, 1H, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.2, 136.3, 133.8, 132.7, 131.4, 130.7, 129.7, 128.8, 126.7, 122.4, 120.5, 115.7, 69.6; ESI-HRMS, m/z: Calcd for C17H9BrClN2O [M − H]−: 370.9592, Found: 370.9593.
- (20) 5′-Amino-[2,2′:3′,2″-terfuran]-4′-carbonitrile (3t): Brown solid (48.2 mg, yield 67%), m.p. 186.2–187.9 °C (187.0–188.0 °C [48]); 1H NMR (600 MHz, CDCl3), δ, ppm: 7.86 (s, 2H, NH2), 7.81 (d, J = 2.4 Hz, 1H, ArH), 7.75 (d, J = 2.4 Hz, 1H, ArH), 6.85 (d, J = 2.4 Hz, 1H, ArH), 6.74 (d, J = 2.4 Hz, 1H, ArH), 6.64–6.63 (m, 1H, ArH), 6.62–6.60 (m, 1H, ArH).
- (21) 4,5-Bi([1,1′-biphenyl]-4-yl)-2-aminofuran-3-carbonitrile (3u): White solid (119.9 mg, yield 97%), m.p. 237.1–239.1 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.80 (d, J = 8.4 Hz, 2H, ArH), 7.79 (s, 2H, NH2), 7.75 (d, J = 8.4 Hz, 2H, ArH), 7.63 (d, J = 7.2 Hz, 2H, ArH), 7.61 (d, J = 8.4 Hz, 2H, ArH), 7.52 (d, J = 8.4 Hz, 2H, ArH), 7.51–7.48 (m, 2H, ArH), 7.44–7.41 (m, 3H, ArH), 7.38 (d, J = 8.4 Hz, 2H, ArH), 7.33 (t, J = 7.2 Hz, 1H, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.2, 140.4, 139.8, 138.8, 137.1, 130.8, 130.0, 129.5, 129.0, 128.3, 127.7, 127.3, 127.1, 126.8, 125.2, 122.2, 116.1, 69.8; ESI-HRMS, m/z: Calcd for C29H21N2O [M + H]+: 413.1648, Found: 413.1643.
- (22) 1-(2-Amino-4,5-diphenylfuran-3-yl)propan-1-one (3v): White solid (43.3 mg, yield 47%), m.p. 161.2–163.0 °C (163.0–165.0 °C [53]); 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.40–7.34 (m, 3H, ArH), 7.29–7.28 (m, 2H, ArH), 7.27 (s, 2H, NH2), 7.20–7.16 (m, 2H, ArH), 7.11–7.07 (m, 3H, ArH), 3.92 (q, J = 7.2 Hz, 2H, OCH2), 0.91 (t, J = 7.2 Hz, 3H, CH3).
- (23) Ethyl 2-amino-4,5-di-p-tolylfuran-3-carboxylate (3w): White solid (37.2 mg, yield 37%), m.p. 147.0–148.8 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.21 (d, J = 7.8 Hz, 2H, ArH), 7.16 (d, J = 7.8 Hz, 4H, ArH), 7.15 (d, J = 7.2 Hz, 2H, ArH), 6.98 (d, J = 7.2 Hz, 2H, ArH), 4.06 (q, J = 7.2 Hz, 2H, OCH2), 2.39 (s, 3H, CH3), 2.26 (s, 3H, CH3), 1.03 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 163.5, 158.9, 140.7, 136.8, 130.1, 129.9, 129.4, 128.9, 128.7, 124.7, 128.3, 89.2, 59.2, 21.4, 14.0; ESI-HRMS, m/z: Calcd for C21H22NO3 [M + H]+: 336.1594, Found: 336.1589.
- (24) Ethyl 2-amino-4,5-bis(4-ethylphenyl)furan-3-carboxylate (3x): White solid (37.0 mg, yield 34%), m.p. 157.0–159.7 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.24 (d, J = 8.4 Hz, 2H, ArH), 7.19 (d, J = 8.4 Hz, 4H, ArH), 7.02 (d, J = 8.4 Hz, 2H, ArH), 4.03 (q, J = 7.2 Hz, 2H, OCH2), 2.70 (q, J = 7.8 Hz, 2H, CH2), 2.60 (q, J = 7.2 Hz, 2H, CH2), 1.27 (t, J = 7.8 Hz, 3H, CH3), 1.18 (t, J = 7.8 Hz, 3H, CH3), 0.99 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 163.6, 162.6, 146.8, 145.0, 134.7, 130.3, 128.9, 128.2, 127.7, 127.5, 125.8, 89.3, 61.7, 28.5, 15.0, 13.9; ESI-HRMS, m/z: Calcd for C23H26NO3 [M + H]+: 364.1907, Found: 364.1902.
- (25) Ethyl 2-amino-4,5-bis(4-chlorophenyl)furan-3-carboxylate (3y): White solid (61.9 mg, yield 55%), m.p. 170.0–172.0 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.36 (d, J = 8.4 Hz, 2H, ArH), 7.26 (s, 2H, NH2), 7.25 (d, J = 8.4 Hz, 2H, ArH), 7.15 (d, J = 9.0 Hz, 2H, ArH), 7.12 (d, J = 9.0 Hz, 2H, ArH), 4.06 (q, J = 7.2 Hz, 2H, OCH2), 1.03 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 165.2, 161.7, 138.6, 133.7, 132.0, 131.7, 130.0, 128.7, 128.6, 125.9, 92.2, 59.6, 14.1; ESI- HRMS, m/z: Calcd for C19H16Cl2NO3 [M + H]+: 376.0502, Found: 376.0496.
- (26) Ethyl 2-amino-4,5-bis(4-bromophenyl)furan-3-carboxylate (3z): White solid (72.2 mg, yield 52%), m.p. 155.2–156.8 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.51 (d, J = 8.4 Hz, 2H, ArH), 7.30 (d, J = 8.4 Hz, 2H, ArH), 7.19 (d, J = 8.4 Hz, 2H, ArH), 7.06 (d, J = 8.4 Hz, 2H, ArH), 5.71 (b, 2H, NH2), 4.06 (q, J = 7.2 Hz, 2H, OCH2), 1.03 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 165.2, 161.7, 138.6, 132.5, 132.0, 131.6, 131.5, 126.2, 121.9, 92.2, 59.6, 14.1; ESI-HRMS, m/z: Calcd for C19H16Br2NO3 [M + H]+: 463.9491, Found: 463.9461.
- (27) Ethyl 2-amino-4,5-bis(3-fluorophenyl)furan-3-carboxylate (3aa): White solid (49.4 mg, yield 48%), m.p. 157.2–158.8 °C; 1H NMR (600 MHz, CDCl3), δ, ppm: 7.37–7.33 (m, 1H, ArH), 7.14–7.05 (m, 4H, ArH), 6.95–6.91 (m, 2H, ArH), 6.82–6.78 (m, 1H, ArH), 5.74 (b, 2H, NH2), 4.05 (q, J = 7.2 Hz, 2H, OCH2), 1.00 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3), δ, ppm: 165.1, 162.7 (d, J = 243.2 Hz), 162.6 (d, J = 244.4 Hz), 161.6, 138.2, 135.5 (d, J = 8.6 Hz), 132.1 (d, J = 8.6 Hz), 129.9 (d, J = 8.6 Hz), 129.7 (d, J = 8.3 Hz), 125.8 (d, J = 2.7 Hz), 121.6, 120.0 (d, J = 2.9 Hz), 117.2 (d, J = 21.5 Hz), 114.6 (d, J = 20.1 Hz), 113.3 (d, J = 21.3 Hz), 111.2 (d, J = 24.2 Hz), 92.3, 59.4, 13.8; 19F NMR (564 MHz, CDCl3), δ, ppm: −112.718, −113.766; ESI-HRMS, m/z: Calcd for C19H16F2NO3 [M + H]+: 344.1093, Found: 344.1089.
- (28) N-Acetyl-N-(3-cyano-4,5-diphenylfuran-2-yl)acetamide (4a): White solid (66.1 mg, yield 64%), m.p. 157.6–159.5 °C (156.0–158.0 °C [48]); 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.55–7.49 (m, 5H, ArH), 7.46–7.43 (m, 2H, ArH), 7.40–7.39 (m, 3H, ArH), 2.43 (s, 6H, 2CH3).
- (30) (E)-2-((4-Methoxybenzylidene)amino)-4,5-diphenylfuran-3-carbonitrile (4c): Yellow solid (76.0 mg, yield 67%), m.p. 168.7–170.5 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 9.09 (s, 1H, CH), 8.04 (d, J = 9.0 Hz, 2H, ArH), 7.55–7.49 (m, 5H, ArH), 7.48–7.47 (m, 2H, ArH), 7.41–7.34 (m, 3H, ArH), 7.15 (d, J = 9.0 Hz, 2H, ArH), 3.88 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 164.1, 161.8, 160.2, 145.2, 132.4, 132.3, 130.5, 129.7, 129.5, 129.3, 129.0, 128.3, 126.4, 115.4, 113.9, 92.7, 56.2; ESI-HRMS, m/z: Calcd for C25H19N2O2 [M + H]+: 379.1441, Found: 379.1442.
- (31) (E)-2-((4-Fluorobenzylidene)amino)-4,5-diphenylfuran-3-carbonitrile (4d): Yellow solid (77.0 mg, yield 70%), m.p. 173.4–175.3 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 9.13 (s, 1H, CH), 8.19–8.09 (m, 2H, ArH), 7.53–7.43 (m, 10H, ArH), 7.42–7.33 (m, 2H, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 165.5 (d, J = 251.4 Hz), 161.1, 159.4, 145.8, 132.7 (d, J = 9.5 Hz), 132.2, 130.3, 129.7, 129.5, 129.3, 128.8, 126.5, 124.0, 117.0 (d, J = 22.1 Hz), 113.5, 94.2; 19F NMR (564 MHz, DMSO-d6), δ, ppm: −105.189; ESI-HRMS, m/z: Calcd for C24H16FN2O [M + H]+: 367.1241, Found: 367.1242.
- (32) (E)-4,5-Diphenyl-2-((4-trifluoromethylbenzylidene)amino)furan-3-carbonitrile (4e): Yellow solid (90.0 mg, yield 72%), m.p. 176.7–178.4 °C; 1H NMR (600 MHz, DMSO-d6), δ, ppm: 9.22 (s, 1H, CH), 8.25 (d, J = 8.4 Hz, 2H, ArH), 7.93 (d, J = 8.4 Hz, 2H, ArH), 7.55- 7.51 (m, 5H, ArH), 7.49–7.47 (m, 2H, ArH), 7.41–7.37 (m, 3H, ArH); 13C NMR (150 MHz, DMSO-d6), δ, ppm: 160.7, 158.8, 146.4, 139.0, 132.5 (q, J = 32.0 Hz), 130.6, 130.2, 129.7, 129.5, 129.3, 128.7, 126.6, 126.5 (q, J = 3.9 Hz), 125.3 (q, J = 271.2 Hz), 113.3, 95.8; 19F NMR (564 MHz, DMSO-d6), δ, ppm: −61.483; ESI-HRMS, m/z: Calcd for C25H16F3N2O [M + H]+: 417.1209, Found: 417.1205.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meng, Z.; Yan, J.; Ning, C.; Shi, M.; Wei, Y. Construction of pyrroles, furans and thiophenes via intramolecular cascade desulfonylative/dehydrogenative cyclization of vinylidenecyclopropanes induced by NXS (X = I or Br). Chem. Sci. 2023, 14, 7648–7655. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; He, M.; Zhou, T.S.; Wang, Q.; He, L.L.; Wang, S.J.; Hu, B.; Wei, B.; Wang, H.; Cui, Z.N. 2,5-Disubstituted furan derivatives containing 1,3,4-thiadiazolemoiety as potenta-glucosidase and E. coli b-glucuronidase inhibitors. Eur. J. Med. Chem. 2021, 216, 113322. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.A.; Stoltz, B.M. Polycyclic furanobutenolide-derived cembranoid and norcembranoid natural products: Biosynthetic connections and synthetic efforts. Chem. Rev. 2017, 117, 7878–7909. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sinclair, A.J.; Faiza, M.; Li, D.M.; Han, X.L.; Yin, H.Y.; Wang, Y.H. Furan fatty acids—Beneficial or harmful to health? Prog. Lipid Res. 2017, 68, 119–137. [Google Scholar] [CrossRef]
- Chen, V.Y.; Kwon, O. Unified approach to furan natural products via phosphine-palladium catalysis. Angew. Chem. Int. Ed. 2021, 60, 8874–8881. [Google Scholar] [CrossRef]
- Li, X.J.; Wen, Y.F.; Wang, Y.; Zhou, X.P.; Xie, X.L. Modifying poly(propylene carbonate) with furan-based non-isocyanate polyurethanes. Chin. J. Polym. Sci. 2023, 41, 1069–1077. [Google Scholar] [CrossRef]
- Larin, A.A.; Degtyarev, D.D.; Ananyev, I.V.; Pivkina, A.N.; Fershtat, L.L. Linear furoxan assemblies incorporating nitrobifuroxan scaffold: En route to new high-performance energetic materials. Chem. Eng. J. 2023, 470, 144144. [Google Scholar] [CrossRef]
- Kucherov, F.A.; Romashov, L.V.; Galkin, K.I.; Ananikov, V.P. Chemical transformations of biomass-derived C6-furanic platform chemicals for sustainable energy research, materials science, and synthetic building blocks. ACS Sustain. Chem. Eng. 2018, 6, 8064–8092. [Google Scholar] [CrossRef]
- Fan, C.C.; Lou, S.Y.; Shen, C.J.; Liao, J.L.; Ni, H.; Chen, S.Y.; Zhu, Z.H.; Hu, X.P.; Xie, W.; Zhao, H.J.; et al. Natural product-inspired discovery of naphthoquinone-furo-piperidine derivatives as novel STAT3 inhibitors for the treatment of triple-negative breast cancer. J. Med. Chem. 2024, 67, 15291–15310. [Google Scholar] [CrossRef]
- Mohapatra, S.; Panda, J.; Mohapatra, S.; Nayak, S. Synthesis of polysubstituted furans: An update since 2019. Asian J. Org. Chem. 2023, 12, e202300304. [Google Scholar] [CrossRef]
- Qiu, S.Q.; Yu, X.Y.; Guo, H.X.; Li, J.; Li, X.Y.; Xu, P. Photocatalytic radical bis(trifluoromethyl)carbinolation of alkenes and heteroarenes. Angew. Chem. Int. Ed. 2025; early view. [Google Scholar] [CrossRef]
- Kondoh, A.; Aita, K.; Terada, M. Synthesis of polysubstituted naphthofurans and indenofurans based on a regiodivergent intramolecular carbometalation strategy with 2-(2′-alkynylaryl)-3-iodofurans. Chem. Eur. J. 2023, 29, e202300132. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.H.; Gao, S.J.; Li, X.Y.; Ma, M.T.; Shen, Z.L.; Chu, X.Q. “On-water” defluorinative cyclization of trifluoromethyl enones with phosphine oxides: Synthesis of polysubstituted furans. Org. Chem. Front. 2024, 11, 3974–3981. [Google Scholar] [CrossRef]
- Vu, T.T.N.; Trinh, T.N.; Pham, T.T.; Ha, M.T.; Mac, D.H.; Retailleau, P.; Nguyen, T.B. I2-promoted oxidative annulation of deoxybenzoin-chalcone adduct: Temperature-controlled access to tetrasubstituted 2,3-trans-dihydrofurans and furans. Adv. Synth. Catal. 2024, 366, 2691–2695. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, N.; Ren, J.Y.; Huang, H.H.; Luan, X.J.; Zuo, Z.J. Highly selective 1,4-diacylation/cycloisomerization of 1,3-enynes: De novo synthetic strategy to polysubstituted furans. Org. Lett. 2024, 26, 35–40. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Kan, Y.M.; Jia, X.N.; Huang, B.B.; Li, T.; Zhao, X.X. Electrocatalytic [3+2] annulation for the synthesis of polysubstituted furans. Org. Lett. 2023, 25, 4540–4545. [Google Scholar] [CrossRef]
- Wang, Y.M.; Pritchard, G.J.; Kimber, M.C. A General convergent strategy for the synthesis of tetra-substituted furan fatty acids (FuFAs). Eur. J. Org. Chem. 2020, 2020, 2914–2922. [Google Scholar] [CrossRef]
- Jin, H.M.; Fürstner, A. Modular synthesis of furans with up to four different substituents by a trans-carboboration strategy. Angew. Chem. Int. Ed. 2020, 59, 13618–13622. [Google Scholar] [CrossRef]
- Nguyen, V.P.; Nguyen, N.N.H.; Lai, N.D.; Mac, D.H.; Retailleau, P.; Nguyen, T.B. Sulfur-promoted oxidative cyclization of pentan-1-ones: Direct access to tetrasubstituted furans from deoxybenzoins and chalcones. Org. Lett. 2023, 25, 6419–6423. [Google Scholar] [CrossRef]
- Zhang, P.; Ti, W.Q.; Gao, T.F.; Zhu, J.; Lin, A.J.; Gao, S.; Li, X.Y.; Yao, H.Q. Three-component approach to modular synthesis of tetra-substituted furans and pyrroles. Org. Chem. Front. 2024, 11, 2554–2560. [Google Scholar] [CrossRef]
- Gewnld, K. Uber die reaktion von α-hydroxy-ketonen mit malodinitril. Heterocycles 1966, 99, 1002–1007. [Google Scholar] [CrossRef]
- Coumar, M.S.; Tsai, M.T.; Chu, C.Y.; Uang, B.J.; Lin, W.H.; Chang, C.Y.; Chang, T.Y.; Leou, J.S.; Teng, C.H.; Wu, J.S.; et al. Identification, SAR studies, and X-ray co-crystallographic analysis of a novel furanopyrimidine aurora kinase A inhibitor. ChemMedChem 2010, 5, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Haleem, A.H.; Kassem, M.A.; Elnagar, M.R.; Abbas, S.E.S.; El Kerdawy, A.M.; Farouk, A.K.B.A.W. Furan-and furopyrimidine-based derivatives: Synthesis, VEGFR-2 inhibition, and in vitro cytotoxicity. ACS Med. Chem. Lett. 2024, 15, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.Y.; Shou, J.R.; Qin, W.Y.; Mo, J.Y.; Huang, H.W. Selective formation of 2,3,5-trisubstituted furans from 1,3-dicarbonyls and hydroxyketones. Adv. Synth. Catal. 2023, 365, 4014–4020. [Google Scholar] [CrossRef]
- Wang, Y.M.; Pritchard, G.J.; Kimber, M.C. Total synthesis of the tetrasubstituted furan fatty acid metabolite CeDFP via Au-catalyzed intermolecular alkyne hydroarylation. Org. Lett. 2019, 21, 4892–4895. [Google Scholar] [CrossRef]
- Fan, J.; Liang, X.C.; Yao, L.L.; Wang, Y.T.; Wang, K.; Yao, B.B.; Liu, Y.J.; Xu, S.W. Synthesis of tetrasubstituted furans through a Cu/base-mediated cascade reaction from terminal alkynes and 1,2-diketones. Org. Biomol. Chem. 2025, 23, 1084–1088. [Google Scholar] [CrossRef]
- Huang, X.-Y.; Gao, S.-J.; Ge, D.H.; Ma, M.T.; Shen, Z.-L.; Chu, X.-Q. Modular synthesis of furans with four nonidentical substituents by aqueous defluorinative reaction of trifluoromethyl enones with two nucleophiles. Org. Lett. 2024, 27, 462–469. [Google Scholar] [CrossRef]
- Baranska, I.; Osmialowski, B.; Rafinska, K.; Rafinski, Z. Construction of highly functionalized 2-styrylfurans by N-heterocyclic carbene/Brønsted acid catalysis. Org. Lett. 2024, 26, 3514–3518. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Hu, Y.; Jin, L.Q.; Gu, Y.W.; Xie, Y.J. Rapid and controlled assembly of polysubstituted furans and their oligoaryls from alkynes and aldehydes facilitated by sequential deprotonation. Org. Lett. 2025, 27, 692–697. [Google Scholar] [CrossRef]
- Miyazaki, D.; Kudo, K.; Fujiki, Y.; Watanabe, N.; Matsui, T.; Ichikawa, J.J.; Fuchibe, K. Gold(I)-catalyzed [2+2] and [3+2] cycloadditions of 1,1-difluoroallenes with aldehydes: Switchable syntheses of fluorinated oxetanes and furans. Org. Lett. 2025, 27, 3807–3812. [Google Scholar] [CrossRef]
- Lian, P.F.; Li, Z.H.; Qiu, X.Y.; Ding, T.M.; Zhang, S.Y. Organocatalytic asymmetric synthesis of sulfonyl-substituted furans via a cascade 1,6-addition/cyclization/enantio-selective protonation pathway. ACS Catal. 2024, 14, 12717–12724. [Google Scholar] [CrossRef]
- Xiao, W.L.; Wang, N.; Yang, L.L.; Feng, Y.M.; Chu, P.L.; Zhang, J.J.; Liu, S.S.; Shao, W.B.; Zhou, X.; Liu, L.W.; et al. Exploiting natural maltol for synthesis of novel hydroxypyridone derivatives as promising anti-virulence agents in bactericides discovery. J. Agric. Food Chem. 2023, 71, 6603–6616. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.H.; He, R.; Zhang, X.; Zhang, W.B. An Ir/Zn dual catalysis for enantio- and diastereodivergent α-allylation of α-hydroxyketones. J. Am. Chem. Soc. 2016, 138, 11093–11096. [Google Scholar] [CrossRef]
- Gao, Y.-Y.; Hua, Y.-Z.; Wang, M.-C. Asymmetric 1,6-conjugate addition of para-quinone methides for the synthesis of chiral β,β-diaryl-α-hydroxy ketones. Adv. Synth. Catal. 2018, 360, 80–85. [Google Scholar] [CrossRef]
- Chen, P.; Cao, W.; Li, X.Q.; Shi, D.Y. A unified approach for divergent synthesis of heterocycles via TMSOTf-catalyzed formal [3+2] cycloaddition of electron-rich alkynes. Adv. Synth. Catal. 2021, 363, 4789–4794. [Google Scholar] [CrossRef]
- Liu, H.J.; Qi, C.R.; Wang, L.; Guo, Y.H.; Li, D.; Jiang, H.F. Base-promoted three-component cascade reaction of α-hydroxy ketones, malonodinitrile, and alcohols: Direct access to tetrasubstituted NH-pyrroles. J. Org. Chem. 2021, 86, 9610–9620. [Google Scholar] [CrossRef]
- Yu, S.-W.; Li, Z.-H.; Li, M.-X.; Zeng, Y.; Ye, W.-X.; Xie, J.-Y.; Wang, Z.-Y. A novel 2-methylimidazole promoted oxyacyloxylation of α-hydroxy ketones and anhydrides: An easy access to α-acyloxy ketones. Catalysts 2024, 14, 811. [Google Scholar] [CrossRef]
- CCDC 2431696 (for 3g), CCDC 2431698 (for 3s), CCDC 2431700 (for 3v) Contain Thesupplementary Crystallographic Data for This Paper. Available online: https://www.ccdc.cam.ac.uk/structures (accessed on 16 April 2025).
- Yu, S.-W.; Chen, Z.-J.; Li, H.-Q.; Li, W.-X.; Li, Y.; Li, Z.; Wang, Z.-Y. Keto sulfones catalyzed by BF3·OEt2 oxysulfonylation of alkynes with sodium sulfinates to access β-keto sulfones catalyzed by BF3·OEt2. Molecules 2024, 29, 3559. [Google Scholar] [CrossRef]
- Seifi, M.; Sheibani, H. Studies on condensation of 1,3-dicarbonyls with malononitrile: Synthesis of 2-pyridinones. Arab. J. Chem. 2018, 10, S2453–S2456. [Google Scholar] [CrossRef]
- Balha, M.; Mondal, B.; Pan, S.C. Organocatalytic asymmetric synthesis of dihydrofuran-spirooxindoles from benzylidene malononitriles and dioxindoles. Org. Biomol. Chem. 2019, 17, 6557–6561. [Google Scholar] [CrossRef]
- He, J.J.; Li, Z. Synthesis of 3,5-diaryl-2,6-dicyanoanilines from tandem reactions of ynones with malononitrile. ChemistrySelect 2019, 4, 5732–5734. [Google Scholar] [CrossRef]
- Wang, S.W.; Zhang, J.Z.; Deng, Z.B.; Lv, Y.Y.; Huang, D.F.; Wang, K.H.; Wang, J.J.; Hu, Y.L. Synthesis of fully substituted difluoromethylpyrazoles by cyclization of difluoro-acetohydrazonoyl bromides with 2-acylacetonitriles or malononitrile. J. Org. Chem. 2024, 89, 10591–10602. [Google Scholar] [CrossRef] [PubMed]
- Bathula, C.; Ahmed, A.A.; Kadam, A.; Sekar, S.; Hwang, J.H.; Lee, S.H.; Kim, H.S. Multi-functional Co3O4 embedded carbon nanotube architecture for oxygen evolution reaction and benzoin oxidation. J. Mol. Liq. 2021, 343, 117616. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kumar, N.; Majumder, A.B.; Pandey, M.; Goverdhan, R.P.V.; Ranganath, K.V.S. Chiral modification of ferrite nanoparticles for oxidative kinetic resolution of benzoins. Asian J. Org. Chem. 2023, 12, e202300325. [Google Scholar] [CrossRef]
- Haut, F.L.; Habiger, C.; Wein, L.A.; Wurst, K.; Podewitz, M.; Magauer, T. Rapid assembly of tetrasubstituted furans via pummerer-type rearrangement. J. Am. Chem. Soc. 2021, 143, 1216–1223. [Google Scholar] [CrossRef]
- Zhu, H.F.; Yang, X.M.; Liu, Y.; Zhou, H.; Wang, Y. Chalcogen bonding catalysis enables ring-opening of cyclopropene and ring expansion of aryl ketones. Angew. Chem. Int. Ed. 2025, 64, e202423746. [Google Scholar] [CrossRef]
- Prousek, L.; Jurasek, A.; Kovac, L. Reaction and spectral properties of 2-amino-3-cyano-4,5-disubstituted furane derivatives. Chem. Commun. 1980, 45, 1581–1588. [Google Scholar] [CrossRef]
- El-Shahawi, M.M.; El-Ziaty, A.K. Enaminonitrile as building block in heterocyclic synthesis: Synthesis of novel 4H-furo[2,3-d][1,3]oxazin-4-one and furo[2,3-d]pyrimidin-4(3H)-one derivatives. J. Chem. 2017, 2017, 5610707. [Google Scholar] [CrossRef]
- Iwai, K.; Ono, M.; Nanjo, Y.; Ema, T. Minimization of amounts of catalyst and solvent in NHC-catalyzed benzoin reactions of solid aldehydes: Mechanistic consideration of solid-to-solid conversion and total synthesis of isodarparvinol B. ACS Omega 2020, 5, 10207–10216. [Google Scholar] [CrossRef]
- Yu, C.X.; Lu, J.; Li, T.J.; Wang, D.L.; Qin, B.B.; Zhang, H.H.; Yao, C.S. A NHC-involved, cascade, metal-free, and three-component synthesis of 2,3-diarylated fully substituted furans under solvent-free conditions. Synlett 2011, 16, 2420–2424. [Google Scholar] [CrossRef]
- Feng, X.; Lancelot, J.C.; Prunier, H.; Rault, S. First synthesis of 4H-furo[3,2-f]pyrrolo[1,2-a][1,4]diazepines. J. Heterocycl. Chem. 1996, 33, 2007–2011. [Google Scholar] [CrossRef]
- Khan, M.W.; Uddin, M.K.; Ali, M.; Rahman, M.S.; Rashid, M.A.; Chowdhury, R. A convenient synthesis of new annelated pyrimidines and their biological importance. J. Heterocycl. Chem. 2014, 51, E216–E221. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield (%) [b] |
| 1 [c] | CsF | EtOH | 3 | 47 |
| 2 | CsF | EtOH | 3 | 90 |
| 3 [d] | CsF | EtOH | 3 | 72 |
| 4 | Cs2CO3 | EtOH | 3 | Trace |
| 5 | NaOH | EtOH | 3 | 41 |
| 6 | Ba(OH)2 | EtOH | 3 | 45 |
| 7 | AcONa | EtOH | 3 | 40 |
| 8 | K2CO3 | EtOH | 3 | 37 |
| 9 | Na2CO3 | EtOH | 3 | 52 |
| 10 | NaHCO3 | EtOH | 3 | 43 |
| 11 | tBuOK | EtOH | 3 | 33 |
| 12 | tBuONa | EtOH | 3 | 37 |
| 13 | DBU | EtOH | 3 | 23 |
| 14 | TEA | EtOH | 3 | 53 |
| 15 | Imidazole | EtOH | 3 | 57 |
| 16 | 2-MI | EtOH | 3 | 61 |
| 17 | Pyridine | EtOH | 3 | 43 |
| 18 | CsF | Toluene (110) [e] | 3 | 65 |
| 19 | CsF | H2O (100) | 3 | 42 |
| 20 | CsF | MeOH (70) | 3 | 82 |
| 21 | CsF | DMF (110) | 3 | 87 |
| 22 | CsF | DMSO (110) | 3 | 89 |
| 23 | CsF | THF (70) | 3 | 67 |
| 24 | CsF | MeCN (80) | 3 | 79 |
| 25 | CsF | 1,4-Dioxane (100) | 3 | 47 |
| 26 | CsF | DCM (40) | 3 | 43 |
| 27 | CsF | DEM (90) | 3 | 52 |
| 28 | CsF | Xylene (110) | 3 | 68 |
| 29 | CsF | EtOH (70) | 3 | 85 |
| 30 | CsF | EtOH (60) | 3 | 79 |
| 31 | CsF | EtOH | 4 | 94 |
| 32 | CsF | EtOH | 5 | 90 |
| 33 [f] | CsF | EtOH | 4 | 73 |
| 34 [g] | CsF | EtOH | 4 | 76 |
| 35 [h] | CsF | EtOH | 4 | 77 |
| 36 [i] | CsF | EtOH | 4 | 91 |
 |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Y.; Yang, S.-H.; Guo, J.-L.; Li, Y.; Lin, T.; Wang, Z.-Y. Metal-Free Catalytic Synthesis of Tetrasubstituted Furans from α-Hydroxy Ketones and Cyano Compounds. Molecules 2025, 30, 1832. https://doi.org/10.3390/molecules30081832
Zeng Y, Yang S-H, Guo J-L, Li Y, Lin T, Wang Z-Y. Metal-Free Catalytic Synthesis of Tetrasubstituted Furans from α-Hydroxy Ketones and Cyano Compounds. Molecules. 2025; 30(8):1832. https://doi.org/10.3390/molecules30081832
Chicago/Turabian StyleZeng, Yu, Shi-Hang Yang, Ji-Lin Guo, Yun Li, Ting Lin, and Zhao-Yang Wang. 2025. "Metal-Free Catalytic Synthesis of Tetrasubstituted Furans from α-Hydroxy Ketones and Cyano Compounds" Molecules 30, no. 8: 1832. https://doi.org/10.3390/molecules30081832
APA StyleZeng, Y., Yang, S.-H., Guo, J.-L., Li, Y., Lin, T., & Wang, Z.-Y. (2025). Metal-Free Catalytic Synthesis of Tetrasubstituted Furans from α-Hydroxy Ketones and Cyano Compounds. Molecules, 30(8), 1832. https://doi.org/10.3390/molecules30081832