1. Introduction
Naphthalene, the smallest member among the polycyclic aromatic hydrocarbons (PAHs) [
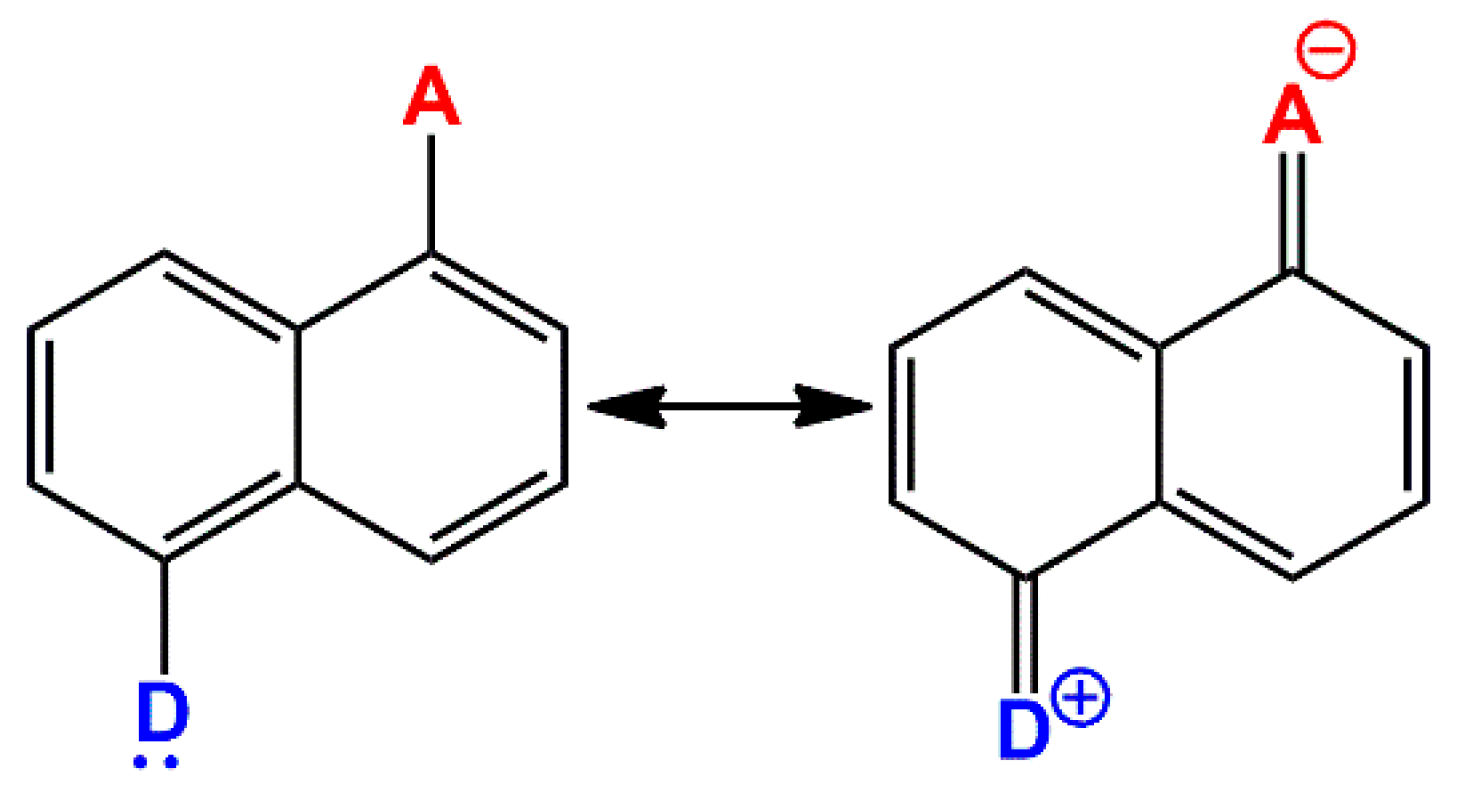
1], is characterized by two fused benzene rings that form a backbone structure resembling an extended π-conjugated “pipeline”. This molecular structure can be exploited to connect two or more substituents that control the properties and, thus, the reactivity of the naphthalene derivatives. The electron densities of the substituents, as well as their relative positions, play a crucial role, as they can function either as π-donors (D) or π-acceptors (A). This leads to several possible combinations, including D-π-D, A-π-A, and D-π-A, where electrons flow from the donor to the acceptor through the aromatic backbone (
through-conjugation) [
2]. In the case of the D-π-A configuration, this arrangement results in a push–pull (
captodative) system. The molecular structure of this system can be envisioned as a linear combination of the neutral and charge-separated resonance forms. The following
Scheme 1 reports, as an example, these two resonance structures in the case of 1,5-disubstituted-naphthalene.
Due to the significantly lower aromaticity of naphthalene compared with that of benzene, disubstituted-naphthalenes have attracted attention for designing various π-extended structures. For example, some naphthalene compounds with highly conjugated hydroxyl (OH) groups, such as naphthalene diols, have demonstrated promising applications in medical treatments due to their antioxidant, antibacterial, and anti-inflammatory properties [
3]. These beneficial effects have been linked to their ability to participate in reactions involving hydrogen atom transfer (HAT) with 2,2-diphenyl-1-picrylhydrazyl (DPPH•) radicals, which are commonly used to assess antioxidant activity. However, the biological properties of these compounds are not limited to naphthalene hydroxyl derivatives; amino-naphthol and amino-naphthalene compounds have also shown particularly effective antioxidant activity [
4].
The relative positions of hydroxy and amino substituents on the disubstituted-naphthalene aromatic backbone play a crucial role in determining the radical scavenging activity. This relationship is closely linked to the low bond dissociation energies (BDEs) of the N-H and O-H bonds found in these substituents. Compounds such as dihydroxy-naphthalene, amino-naphthol, and diamino-naphthalene exhibit these characteristics, making them effective hydrogen donors capable of neutralizing free radicals [
4]. This activity is correlated with the electron density of the overall molecule, and it can be further influenced by modifying the main functional groups (aniline and/or phenol) with substituents that enhance either the electro-donating or electro-withdrawing characteristics. In this context, Vullev and colleagues investigated how different amide derivatives affected the electronic properties of pyrene [
5]. They concluded that the significant electric dipoles and the ability to form ordered hydrogen-bonded networks in the amides, combined with the proper orientation of the amide groups, influence the π-conjugation of the chromophore. This, in turn, affects properties such as optical absorption and reduction potential. Additionally, the amide groups have permanent electric dipoles that influence the electrochemical state of the molecules. These dipoles impact the stabilization of any radical ions that may form, as well as their optical properties [
6].
One of the most studied disubstituted-naphthalenes is 1,5-diamino-naphthalene (DAN), a D-π-D type PAH characterized by a higher degree of molecular symmetry. The amino groups exhibit two opposing behaviors: on one hand, the nitrogen atom exerts an inductive electron-withdrawing effect; on the other hand, it displays a strong π-charge resonance, donating its lone pair to the aromatic ring. Typically, the resonance effect predominates, enhancing the negative potentials present in the π regions of the PAH [
7]. The symmetric arrangement of the two electro-donating amino groups in DAN appears to be primarily responsible for its strong antioxidant properties [
4]. Moreover, in our recent work, we evaluated also the pro-oxidant activity of a DAN derivative obtained by reacting it with the amino acid
l-glycine [
8]. This reaction produced carbon dot structures that exhibited biocidal properties. In that occasion, we observed that both amino groups of DAN reacted symmetrically with two molecules of
l-glycine, resulting in a new compound that retained the original molecular symmetry and most of the properties of DAN. For this reason, we deemed it of interest to understand how the pro-oxidant and antioxidant properties of DAN could be modulated by simultaneously breaking the molecular symmetry and altering the electro-donating features through a simple molecular functionalization.
The present study aimed to investigate the possibility of tuning the oxidant/antioxidant activities of a disubstituted-naphthalene by transforming its structure from a symmetric D-π-D type into an asymmetric captodative system, such as D-π-A. DAN was thus chosen as a model compound primarily because of its highly symmetrical disubstituted structure and the large literature support on its properties. A simple approach to induce this transformation, without altering the π-conjugated backbone of the naphthalene too much, can be achieved; for instance, by reducing the electro-donating mesomeric effect of a single aniline’s nitrogen through an amidation reaction. This target was achieved by breaking the molecular symmetry through the conversion of one of its aniline groups into a corresponding anilide of pyroglutamic acid (PyroGlu). The objective was to explore how the inclusion of a single amide functionality would influence the electronic properties of the π-conjugated moieties to which it is attached and its capacity to interact with radical species. In fact, the conversion of an aniline group into an anilide should alter the DAN capability of promoting either a radical formation or instead scavenging newly formed radicals. This conversion was successfully accomplished through a simple solventless thermal treatment of a 1:1 mixture of DAN and PyroGlu at 160 °C, followed by a simple purification process leading to the desired target, the mono-pyroglutanilide. The oxidant and antioxidant properties of this asymmetric derivative were characterized using two standard assays: indocyanine green (ICG) for the pro-oxidative and 2,2-diphenyl-1-picrylhydrazyl radical (DPPH•) for the antioxidative. The results were then compared with the symmetrical DAN. To the best of our knowledge, this is the first work that tries to understand how an asymmetric amidation of a disubstituted naphthalene could affect its pro-oxidant/antioxidant properties.
2. Results
To modify the symmetry-related properties of 1,5-diamino-napthalene, we needed to explore a synthetic pathway for derivatizing a single aniline moiety while minimizing the self-polymerization that often occurs in liquid media, which can often lead to macromolecular structures [
8]. Since we have recently investigated the flexibility of
l-glutamic acid (Glu) in the low-temperature copolymerization of high-melting amino acids [
9,
10], we found it worthwhile to apply this approach to create an anilide derivative of DAN. In fact, when treated with Glu at 160 °C for 4 h, Glu converts into pyroglutamic acid (PyroGlu), a cyclic amide that melts and acts as an effective dispersing agent, thus avoiding the use of other solvents.
Intrigued by the idea of thermally treating a mixture of Glu and DAN at low temperatures, we conducted a preliminary TG/DSC analysis of DAN, both in its pure form and in a mixture with Glu and PyroGlu, to determine the most suitable thermal conditions to achieve our target.
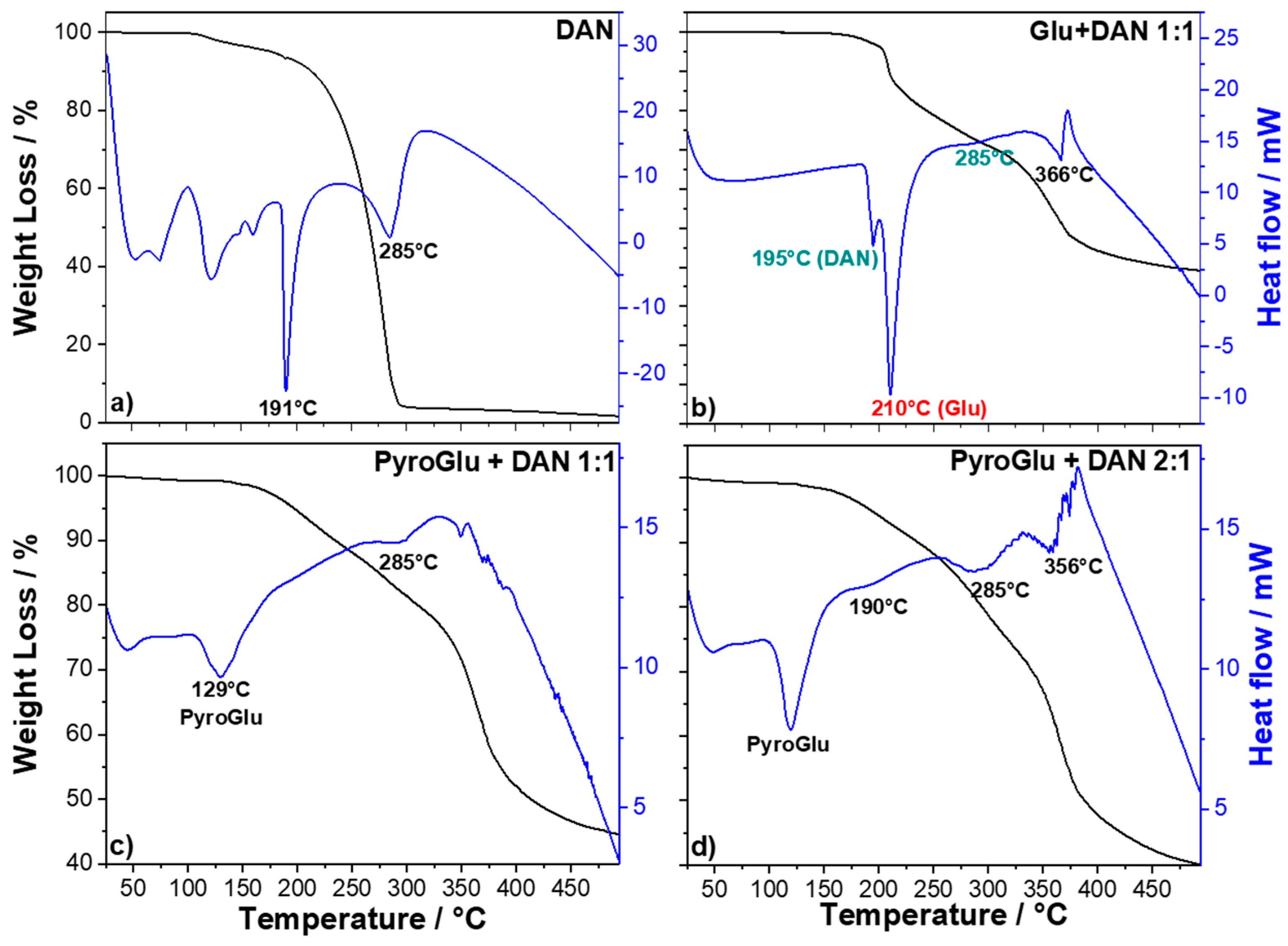
The TG-DSC profile of DAN (
Figure 1a) shows several endothermic events between 50 and 165 °C, associated with a 5% of weight loss, which can be assigned to the loss of adsorbed water and some phase transitions.
A sharp, intense endothermic event, peaking at 191 °C, marks the DAN melting point. After the melting process, the DAN molecules begin to degrade, as indicated by the subsequent endothermic event that peaks at 285 °C.
Figure 1b shows the thermal behavior of a 1:1 mixture of DAN and Glu (the thermal degradation of Glu was thoroughly discussed in our previous work [
9,
10]). The main endothermic phenomena are the melting of DAN at 195 °C, associated with a negligible weight loss (≅4%), and a more intense peak at 210 °C, related to the melting of
l-glutamic acid with a weight loss of about 12%. The latter event corresponds to a well-known dehydro-condensation process that leads to a cyclic amide of
l-glutamic acid named pyroglutamic acid (PyroGlu) [
10,
11]. This transformation is highly dependent on the heating/time conditions and a complete conversion into PyroGlu can be obtained by treating
l-glutamic acid at 160 °C for 4 h [
12]. The other two slightly endothermic events, peaking at 285 and 366 °C, can be related to the degradation of the DAN and the glutamic derivatives, respectively. Since our synthetic strategy is based on reacting PyroGlu with DAN, we have decided to perform the reaction starting directly from commercially available PyroGlu to minimize the possible side reactions occurring between Glu and DAN before the conversion into PyroGlu. We have, therefore, analyzed the TG/DSC profile of two reaction mixtures obtained by combining PyroGlu and DAN in 1:1 (
Figure 1c) and 2:1 (
Figure 1d) ratios. The TG-DSC profiles of the mixtures show a different thermal response with respect to the mixture including Glu. A thermal event related to the melting of PyroGlu can be observed at around 130 °C in both mixtures with no weight loss associated. Interestingly,
Figure 1c does not show any thermal event that can be correlated to DAN melting at 190 °C. This suggests that molten PyroGlu is acting as dispersing agent, thus promoting the reactivity of DAN, which shows a very little endothermic peak related to its thermal degradation (285 °C). The analysis of the 2:1 mixture (
Figure 1d) shows again the event associated with the PyroGlu melting, a little shoulder at 190 °C for the DAN melting and two evident peaks associated with the thermal degradation of unreacted DAN (285 °C) and PyroGlu derivatives (366 °C). The apparent reduced reactivity of DAN in the 2:1 mixture could be possibly attributed to a more relevant self-reaction of PyroGlu since the weight loss of the two samples looks very similar. Based on the TG/DSC analysis, we have decided to pursue a synthetic pathway using a 1:1 ratio of DAN and PyroGlu, which acts as a dispersing agent, enabling the two compounds to react under mild thermal conditions (4 h at 160 °C) (
Scheme 2).
After the reaction, the raw product was dispersed in milli-Q water, filtered, and the water dispersion was firstly purified by extracting the unreacted DAN with diethyl ether. Next, the residual water was then extracted with ethyl acetate, following the purification steps through TLC eluted with pure ethyl acetate. The final product was dried in an oven at 60 °C, resulting in a brownish-pink powder that was characterized using FTIR and NMR techniques.
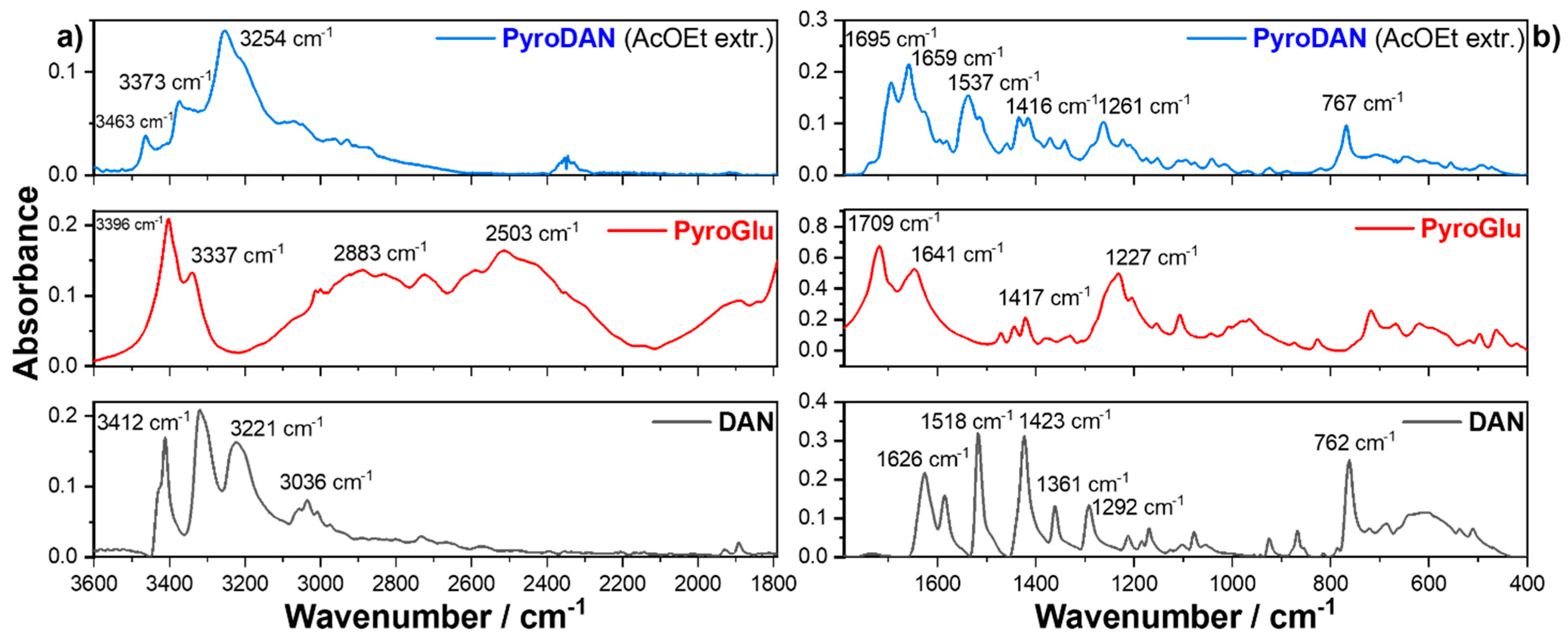
Figure 2 shows the FTIR absorption spectra of the ethyl acetate extract (top) compared with the spectra of pure PyroGlu (middle) and DAN (bottom).
The spectrum of DAN (
Figure 2a bottom) is characterized by the asymmetric and symmetric stretching vibrations of the free aniline (-NH
2), 3412 and 3319 cm
−1, respectively, and by the asymmetric and symmetric stretching of aromatic C-H, 3220 and 3033 cm
−1, respectively. The FT-IR spectrum of PyroGlu (
Figure 2a middle) is also characterized by the presence of two N-H stretching vibrations, asymmetric (3396 cm
−1) and symmetric (3333 cm
−1), related to the pyrrolidine nitrogen of the lactam. The broad signal peaking around 2900 cm
−1 can be related to O-H stretching of the free carboxylic acid whilst the broad signal peaking at 2500 cm
−1 can be associated with the C-H stretching of the aliphatic moiety. The spectrum of the ethyl acetate extract is characterized by a couple of signals, 3463 and 3373 cm
−1, which appear to be related, respectively, to the asymmetric and symmetric stretching of a free aniline group of a DAN moiety, though shifted to higher wavenumbers when compared with free DAN. The most prominent signal, peaking at 3254 cm
−1, can be associated with the N-H stretching of an anilide group, thus indicating the formation of an amide bond between the carboxylic acid of PyroGlu and one of the anilines of the DAN. This assumption is further confirmed by the disappearance of the signal related to the free OH stretching of the carboxylic acid. The spectra in
Figure 2b show the infrared profiles of the ethyl acetate extract in the region between 1790 and 400 cm
−1. The FT-IR spectrum of DAN (
Figure 2b bottom) is characterized by the presence of four peaks (1626, 1585, 1518, and 1423 cm
−1) corresponding to the symmetric and asymmetric C
ar=C
ar stretching of the aromatic backbone and N-H bending of the aniline moieties. The two signals at 1361 and 1292 cm
−1 are instead related to the C-N symmetric and asymmetric stretching. Finally, the signal peaking at 762 cm
−1 can be associated with the out-of-plane bending of three adjacent hydrogens [
13]. The infrared spectrum of PyroGlu (
Figure 2b middle) shows two infrared signals centered in 1709 and 1641 cm
−1, which can be associated with the stretching of the free carboxyl (−C=O) moiety and the amide group of the γ-lactam (−CONH−), respectively. The tern of weak but sharp signals peaking at 1468, 1443, and 1417 cm
−1 can be correlated with N-H, O-H, and C-H bending whilst the broad signal peaking at 1227 cm
−1 can be related to the C-O stretching [
14]. The analysis of the FTIR spectrum of the ethyl acetate extract (
Figure 2 top), when compared with the infrared spectra of the precursor, highlights some structural insights that allow us to hypothesize an attribution to a mono-pyroglutamide derivative of the DAN, which we have named PyroDAN. In fact, the infrared spectrum of PyroDAN shows a clear decrease in the C=O stretching of the free carboxylate (1709 cm
−1) in PyroGlu and the rise of C=O stretching in an Amide I bond (1643 cm
−1), suggesting a reaction between an aniline of DAN and the carboxyl group of PyroGlu. This assumption is further confirmed by the presence of a new Amide II bond (1537 cm
−1), the bending of PyroGlu (1416 cm
−1), and the slightly shifted out-of-plane bending of the DAN (767 cm
−1). Moreover, the C-O stretching of the PyroGlu free hydroxyl group (1227 cm
−1) disappeared and the C-N stretching of the primary amine of DAN at 1292 cm
−1 is replaced by the C-N stretching of a secondary C-N at 1261 cm
−1 [
13].
The comparison of the FTIR absorption spectra presented in
Figure 2 indicates that the ethyl acetate extract can reasonably be identified as a pyroglutamide derivative of DAN. To further clarify this identification, the ethyl acetate extract was structurally characterized using one-dimensional (1D) (
Figure 3 and
Figures S1 and S2) and two-dimensional (2D) (
1H-
1H COSY;
1H-
13C HSQC;
1H-
15N HSQC) (
Figure 4 and
Figures S5–S8) proton NMR spectroscopy in deuterated dimethyl sulfoxide (DMSO-
d6). The 1D proton NMR spectra of the starting materials, 1,5-DAN and PyroGlu, were also reported as a reference in the
Supplementary Information (Figures S3 and S4). Proton signals at 1.16, 1.99, and 4.02 ppm correspond to ethyl acetate residues resulting from the compound purification process [
15].
The
1H-NMR shows the presence of two distinct moieties: the first, aromatic (see also
Figure S1), related to DAN; and the second, aliphatic (see also
Figure S2), related to pyroglutamic acid. The two moieties can be unambiguously assigned by taking advantage of the previous work performed by our group on pyroglutamic [
9,
10] and 1,5-diamino-naphthalene derivatives [
8], as well as comparing the signals with those of the pure starting materials reported in the
Supplementary Information (Figures S3 and S4).
The aminoacidic proton, α-CH, at 4.40 ppm, integrating for one
1H, confirms the presence of the pyroglutamic moiety. This signal is downshifted with respect to the same proton in unreacted Glu (3.87 ppm) because of its involvement in the formation of the internal amide, which is characteristic of the pyroglutamic structure. The presence of the lactam is established by the signal of the secondary amidic proton at 7.98 ppm, integrating for one
1H and, as evidenced from the COSY-NMR (inset
Figure 4), clearly correlating with the α-CH proton. The formation of the five-membered ring is further proven by the presence of 2 γ-H at about 2.17 and 2.25 ppm, each integrating for one
1H.
Interestingly, the attribution of these two hydrogens is based upon their mutual coupling in the COSY-NMR, as well as their coupling with the two β-H around 2.10 and 2.40 ppm, which in turn are also clearly coupled with the α-CH (
Figure 4). The latter are two hydrogens that become diastereotopic as a result of the lactam formation.
The analysis of the aromatic moiety of DAN highlights the presence of only a free aniline group evidenced by the signal at 5.75 ppm, integrating for two
1H. The second aniline group has, in fact, reacted with the free carbonyl of pyroglutamic acid to give an anilide, an aromatic amide, whose presence is confirmed by the signal at 9.82 ppm, integrating for one
1H. This is crucial to confirm that the thermal reaction of DAN with pyroglutamic acid at 160 °C afforded the formation of an asymmetric species (PyroDAN) instead of a symmetric trimer, such as that previously obtained reacting the two aniline groups of DAN with two molecules of
l-glycine [
8]. The reaction pattern affording to the PyroDAN molecule is reported in
Scheme 2.
The aromatic region characteristic of diamino-naphthalene shows an asymmetric distribution of the benzenoid protons that suggests the presence of a partially reacted moiety. The signal at 6.70 ppm is attributed to the proton named
Hf (
Figure 3), which couples with the nearby proton named
He (7.23 ppm). The signal at 7.23 ppm integrates for 2H because there is also a second proton with very similar chemical shift, 7.24 ppm. The analysis of the
1H-NMR clearly shows a tern of signals, 7.92, 7.56, and 7.34 ppm, correlated by an evident roofing effect. In particular, from the multiplicity, the signal at 7.34 ppm can be assigned to the proton named
Hb, thus the signal at 7.92 can be attributed to the proton closer to the anilide group,
Hc, and that at 7.56 ppm should be attributed to the proton
Ha. The only remaining signal, at 7.24 ppm, should be then related to the proton named
Hd, which is very close to the signal at 7.23 ppm, explaining why together they integrate for 2H. The HSQC analysis (
Figures S5–S7), showing the one-bond heteronuclear correlation between
1H and
13C, also confirms the above attributions.
In particular,
Figure S5 shows the correlation of the α-CH proton of the pyroglutamic moiety, at 4.40 ppm, with the carbon at 56.6 ppm. The correlation of the two γ-H of the PyroGlu moiety at about 2.17 and 2.25 ppm, with their corresponding
13C at 29.7 ppm and the
1H-
13C correlation of the β-H with the carbon signal at 26 ppm, can be observed in
Figure S6.
In the aromatic region, as reported in
Figure S7, the assignment of the proton signal at 6.70 ppm to
Hf in
Figure 1 is supported by the correlation with the carbon signal at 108.16 ppm. The two overlapping proton signals
He and
Hd can be more clearly resolved by the
13C signals of the attached carbons at 110.31 and 127.07 ppm, respectively. The HSQC cross-peak at 7.57/122.35 ppm in
Figure S7 can be then assigned to the
Ha and the attached carbon, while the aromatic proton closer to the anilide group (
Hc in
Figure 1) correlates with the carbon resonating at 120.41 ppm.
The mono-amidation of 1,5-DAN is further confirmed by a one-bond heteronuclear correlation (HSQC) between
1H and
15N (
Figure S8), which shows the presence of three distinct nitrogen signals, 110.12, 121.53, and 123.41 ppm, correlated, respectively, to the free aniline (5.78 ppm), the lactam (7.99 ppm), and the anilide (9.83 ppm) of PyroDAN. An additional confirmation of the PyroDAN structure was obtained using HRMS-ESI+ spectra, which show a good matching between the calculated (270.12370) and experimental
m/
z (270.12354) for the molecular ion C
15H
16N
3O
2 [M+H]
+ (
Figure S9). A coherent fragmentation pattern of this molecular ion was obtained using higher-energy collisional dissociation (HCD) that highlights the breaking of the C-N and C-C bonds next to the hexocyclic carbonyl (
Figure S10). This HCD MS-MS, following a loss of carbon dioxide, leads to two major fragments at
m/
z equal to 84 and 159, corresponding, respectively, to fragments from the pyroglutamic and DAN moieties.
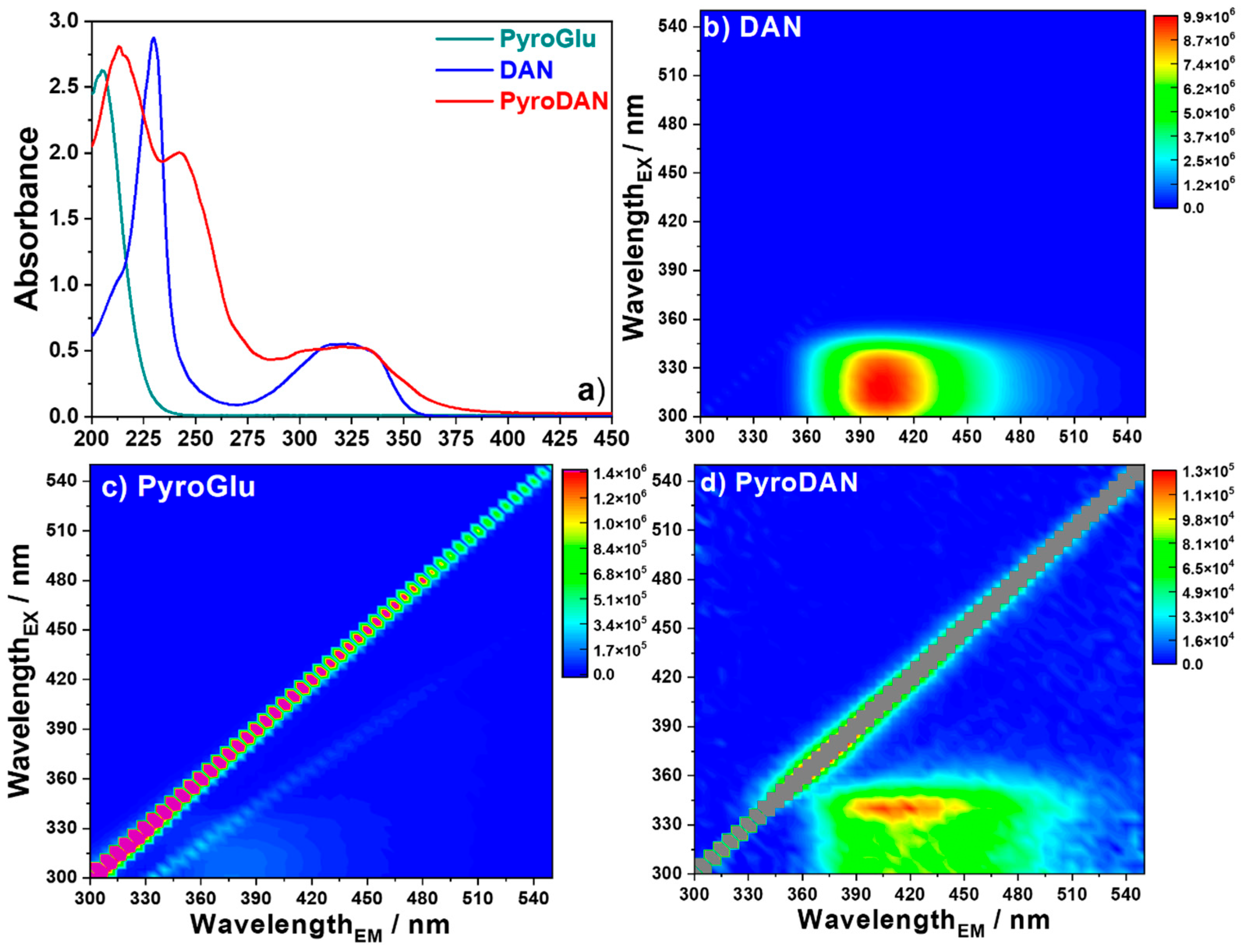
After the NMR analysis, we compared the optical properties of PyroDAN to those of the starting materials (
Figure 5).
Figure 5a shows the UV-Vis absorption spectra of PyroGlu, DAN, and PyroDAN. PyroGlu exhibits a strong absorption at around 202 nm (π→π* transition) due to the carbonyl moieties of the γ-lactam structure and the free carboxylic group. The absorption spectrum of DAN is characterized by a sharp strong band associated to a π→π* transition peaking at 230 nm with a small shoulder around 210 nm. A second broad band spanning between 275 and 360 nm and centered at 320 nm, is associated with the
n→π* electronic transition, which results from the electron transfer from the lone pair of the nitrogen to the π* orbital of the naphthalene moiety. This broad band is formed by the combination of three bands peaking at 315, 325, and 334 nm (
Figure S11). The poor resolution of the vibrational structure in the absorption spectrum can be correlated with the strong interaction between the amino group and a polar protic solvent, such as water. In fact, following Platt’s classification [
16], in polar protic solvents these bands can be correlated with
1A
g →
1L
b transitions since, in presence of a hydrogen-bonding medium, the polar state
1L
a is more destabilized than
1L
b [
17].
The absorption profile of PyroDAN is characterized by two strong bands peaking first at 213 nm and ascribed to the π→π* electronic transitions due to the carbonyl moieties of the γ-lactam structure and the new anilide moiety. A second absorption band, inherited from the DAN aromatic moiety, is slightly red-shifted at 242 nm with respect to the parent structure due to the introduction of a new amide bond. The characteristic combined broad band associated with the
n→π* electronic transition of the DAN, spanning from 290 to 340 nm, became wider and more structured when compared with DAN (
Figure S11). This broadening effect has been already observed for pyrenyl-amides by Vullev and colleagues, who discovered that amides, though breaking the pyrene symmetry, cause a small shift in both UV-Vis absorption and the fluorescence spectra [
5]. In fact, the peaks at 315 and 325 nm appeared both blue-shifted in PyroDAN, respectively, to 302 and 320 nm. In addition, two new signals, a shoulder peaking at 275 and a little band peaking 289 nm, can be associated most probably with the
n→π* electronic transition of the two carbonyl groups, the intra- and inter-molecular amides of the PyroGlu moiety. It is noteworthy that the changes in the fine structure of the long-wavelength band are ascribable to the alteration of the DAN symmetry due to the transformation of a single aniline to anilide operated by the PyroGlu moiety. In fact, the total dipole length of DAN is characterized by the bond moment of the individual amino groups that act in opposite directions to each other [
17]. The transformation of a single aniline into anilide breaks this symmetry, thus increasing the molecular dipole by replacing one of the anilines with a functional group, the pyroglutamide, characterized by a reduced electro-donating mesomeric effect when compared with the unreacted amino group.
The 3D fluorescence map (x-emission; y-excitation; z-false color intensity scale) of DAN (
Figure 5b) is characterized by a strong emission centered at 405 nm (λ
ex = 320 nm), in accordance with the previous findings [
17]. PyroDAN exhibits a broad fluorescent emission centered around 415 nm with an excitation maximum at 340 nm (
Figure 5d). In contrast, commercial PyroGlu does not display any significant emission (
Figure 5c). The fluorescence data suggest that the photoluminescence observed in PyroDAN is related to the conjugated bonds present in the aromatic moiety inherited from the DAN. However, the original fluorescence intensity of the DAN resulted in a dramatic reduction due to the electronic effect of the pyroglutamic anilide on the π-conjugation. Moreover, the small spectral broadening evidenced in the PyroDAN fluorescence confirms what was already found for the fluorescence spectra of pyrenyl-amides [
5]. To better understand how the structural modification introduced in PyroDAN affected its quantitative fluorescence emission in comparison with DAN, we have also acquired a fluorescence spectrum for DAN and PyroDAN at the same molar concentration equal to 63 µM in milli-Q water (
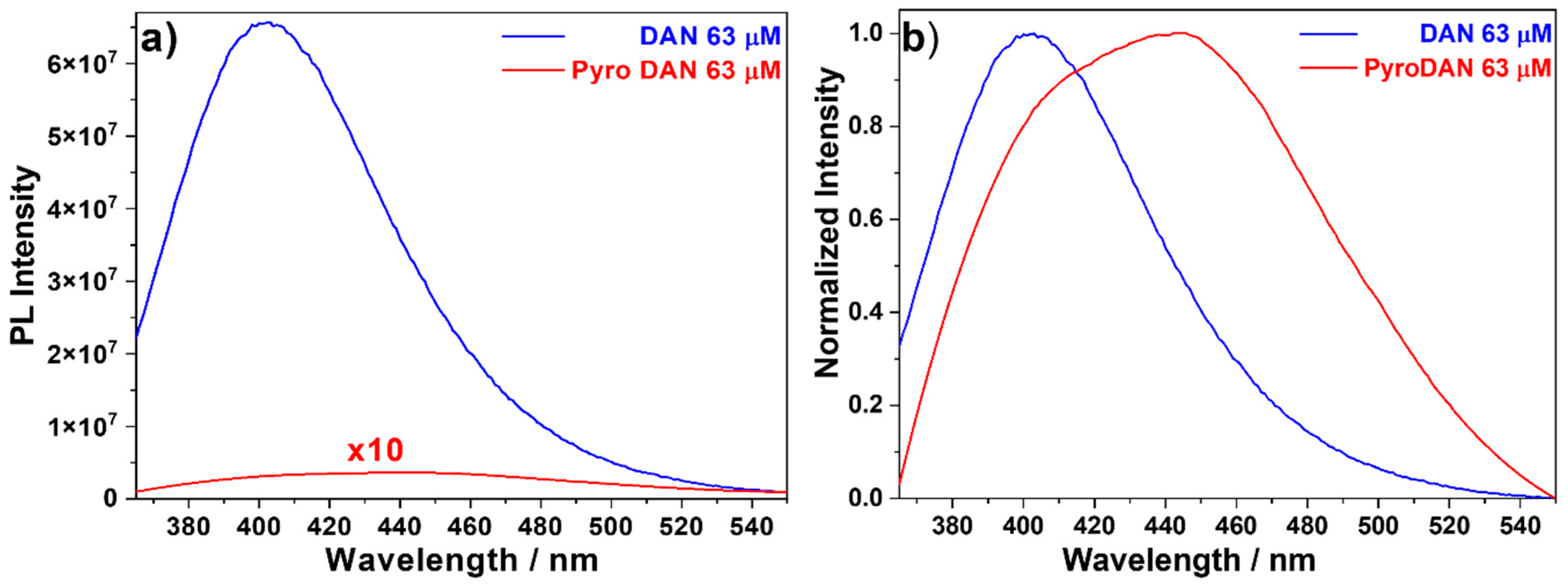
Figure 6), so as to compare directly the emissions in a more quantitative manner.
Figure 6a shows clearly that, at the same concentration, the intensity of DAN is about 200 times higher than PyroDAN. Given the high difference in intensity, the spectrum of PyroDAN needed to be multiplied by a factor of 10 to avoid being confused with the zero line. This phenomenon can be seen as a direct consequence of the electronic perturbation of the π-conjugation of the DAN backbone due to the functional transformation of a single aniline into a pyroglutamyl anilide, which decreases the electro-donating effect of its nitrogen lone pair. Moreover, after normalizing the two spectra of
Figure 6a to 1 (
Figure 6b), the differences in the fluorescence profile of the two molecular structures appear with more clarity. In fact, while the curve profile of DAN can be seen as a result of a single component with a maximum centered a 403 nm, the profile of PyroDAN is clearly resulting from a combination of at least two components: one closely related to the DAN moiety and a second centered at 445 nm. While the first component can be ascribed to a radiative decay internal to the π-conjugated system, the second band can be due to the presence of an additional moiety external to the π-conjugated system.
The fine structure of PyroDAN shown in
Figure 6b confirms what was already discussed above for the absorption profile of PyroDAN; in particular, the signal broadening of the long-wavelength band due to a desymmetrization of the original DAN with a charge transfer extended from the π-conjugated naphthalene backbone to the pyroglutamyl moiety.
2.1. Oxidant Activity
After analyzing the differences in the photoluminescence properties of PyroDAN in relationship with the parent structure of DAN, we have deemed it of interest to understand how the symmetry breaking due to the amidation could affect its activity. In a recent work, we have investigated the pro-oxidant activity of carbon dots obtained by reacting DAN with
l-glycine. This activity is promoted by the generation of singlet oxygen,
1O
2, a strong oxidant reactive oxygen species (ROS), and it can be monitored using the only near infra-red (NIR) probe approved by the Food and Drug Administration (FDA) of the USA, which is the indocyanine green (ICG) [
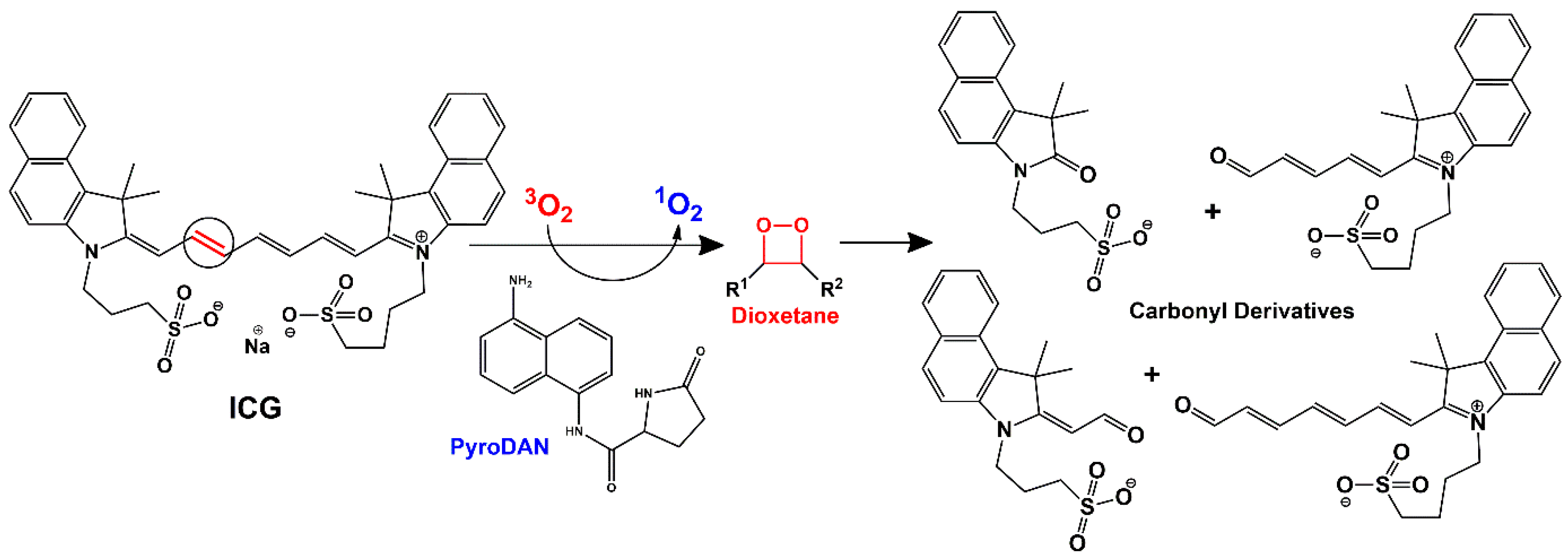
18]. ICG is a water-soluble tricarbocyanine dye (
Scheme 3) that displays a maximum absorption peak centered at 780 nm (
Figure S12). The irradiation of the photosensitizer generates a singlet oxygen, which induces the molecular degradation of the ICG conjugated polyolefin structure via dioxetane formation with subsequent oxidative fragmentation into several carbonyl derivatives [
18]. The fragmentation can be monitored by following the decrease in ICG absorption during light irradiation, thus monitoring the singlet oxygen effect produced by a specific photosensitizer.
Indeed, DAN is responsible for energy transfer (type II reaction) towards molecular oxygen in its triplet ground state,
3O
2. This process is followed by the spin pairing of the electrons in the
π* antibonding orbitals with the generation of highly reactive singlet oxygen, with irrelevant contributions of other ROS [
8,
18,
19].
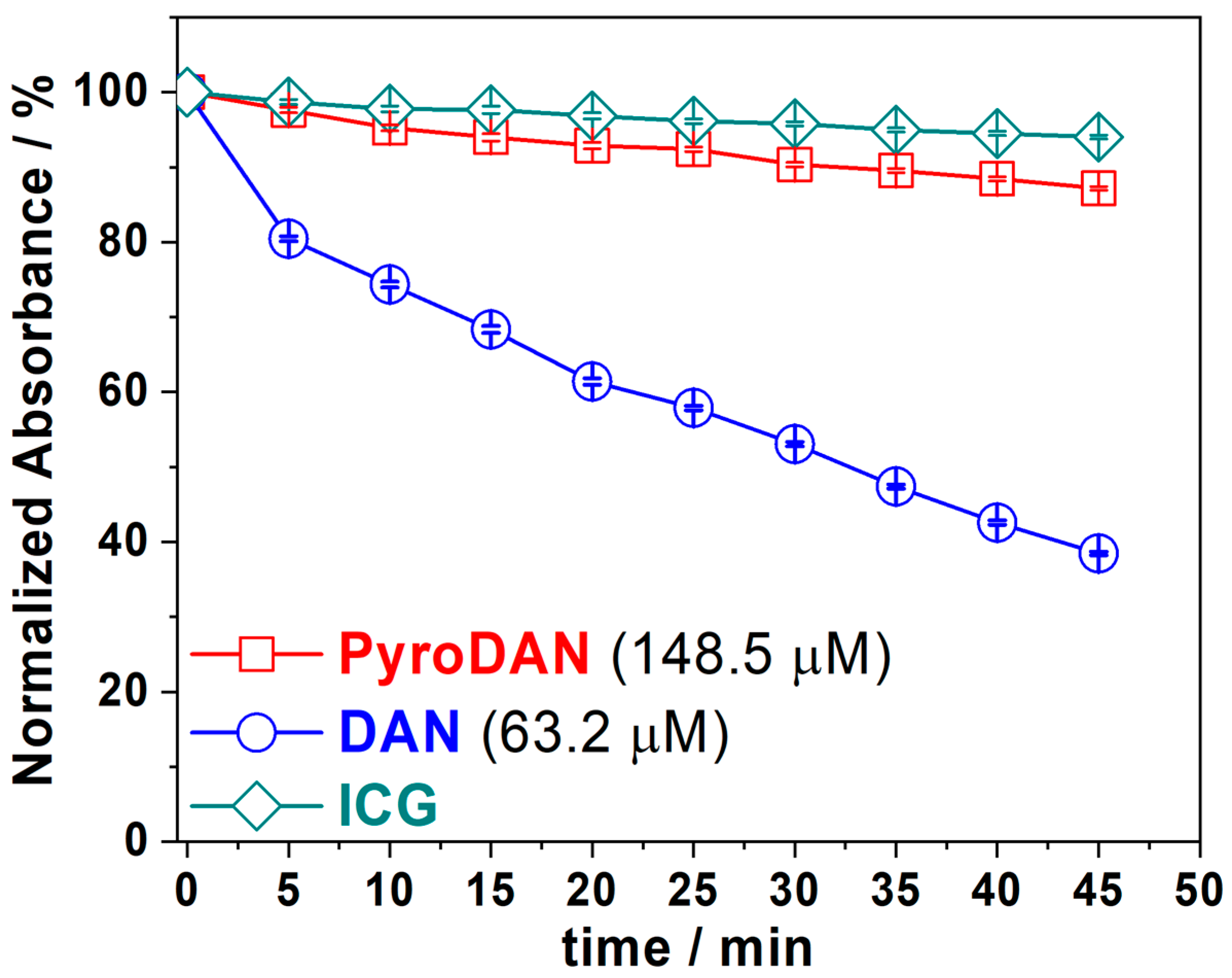
The oxidant activity of PyroDAN was also studied using the ICG photodegradation protocol and the results were compared with those of DAN (
Figure 7). This assay was performed by irradiating the photosensitizers (DAN or PyroDAN) with a fixed wavelength of 320 nm. The oxidant activity of the photosensitizer is evaluated as percentage of the ICG decomposition against time, measuring the absorption of residual ICG at 780 nm. In order to evaluate the background decomposition of ICG, a control solution of ICG was monitored at the same time intervals as the samples, and the error associated was estimated as a standard deviation of a triplicate set of measurements.
Figure 7 shows the ICG degradation curves for the ICG, ICG + DAN, and ICG + Pyro DAN, as a function of time.
The graph shows that derivatizing DAN as a mono-pyroglutanilide affects its capability of generating singlet oxygen and thus its oxidant activity. The PyroDAN profile closely resembles that of bare ICG, which serves as a control and shows only a 4% degradation of ICG within 45 min. In contrast, DAN is capable of oxidizing nearly 60% of ICG in the same time frame. Additionally, achieving this lower level of activity requires a concentration of PyroDAN that is over twice that of DAN. In conclusion, the derivatization of DAN into mono-pyroglutanilide almost completely suppresses its oxidizing capabilities. This likely occurs due to the symmetry breaking, with consequent electron density perturbation, resulting from the amidation of only one of the aniline moieties, which hampers the possibility of generating singlet oxygen.
2.2. Antioxidant Activity
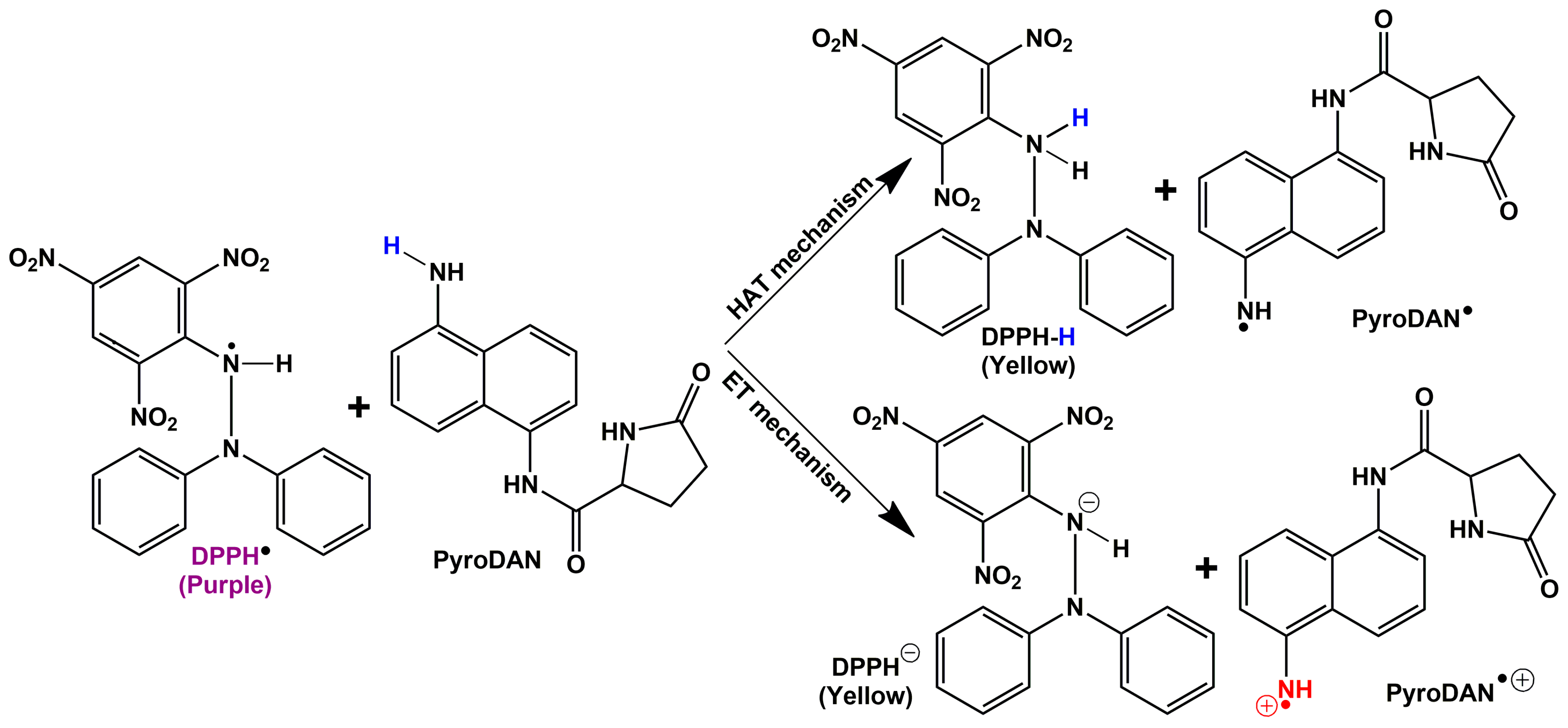
The antioxidant activity of PyroDAN was assessed using a standard DPPH• assay to determine the scavenging activity at various concentrations and compare the result with that of pure DAN. The assay is based on the capability of the sample to quench the stable radical DPPH• (2,2-diphenyl-1-picrylhydrazyl) by releasing either an atom of hydrogen (H•) through a mechanism known as hydrogen atom transfer (HAT) or through an electron transfer (ET). The latter can be of two kinds: single electron transfer followed by proton transfer (SET-PT) or sequential proton loss electron transfer (SPLET) [
4]. The assay is performed by monitoring the decrease of the UV-Vis absorption of the DPPH at 524 nm with a corresponding color shift from purple to pale yellow (
Figures S13 and S14).
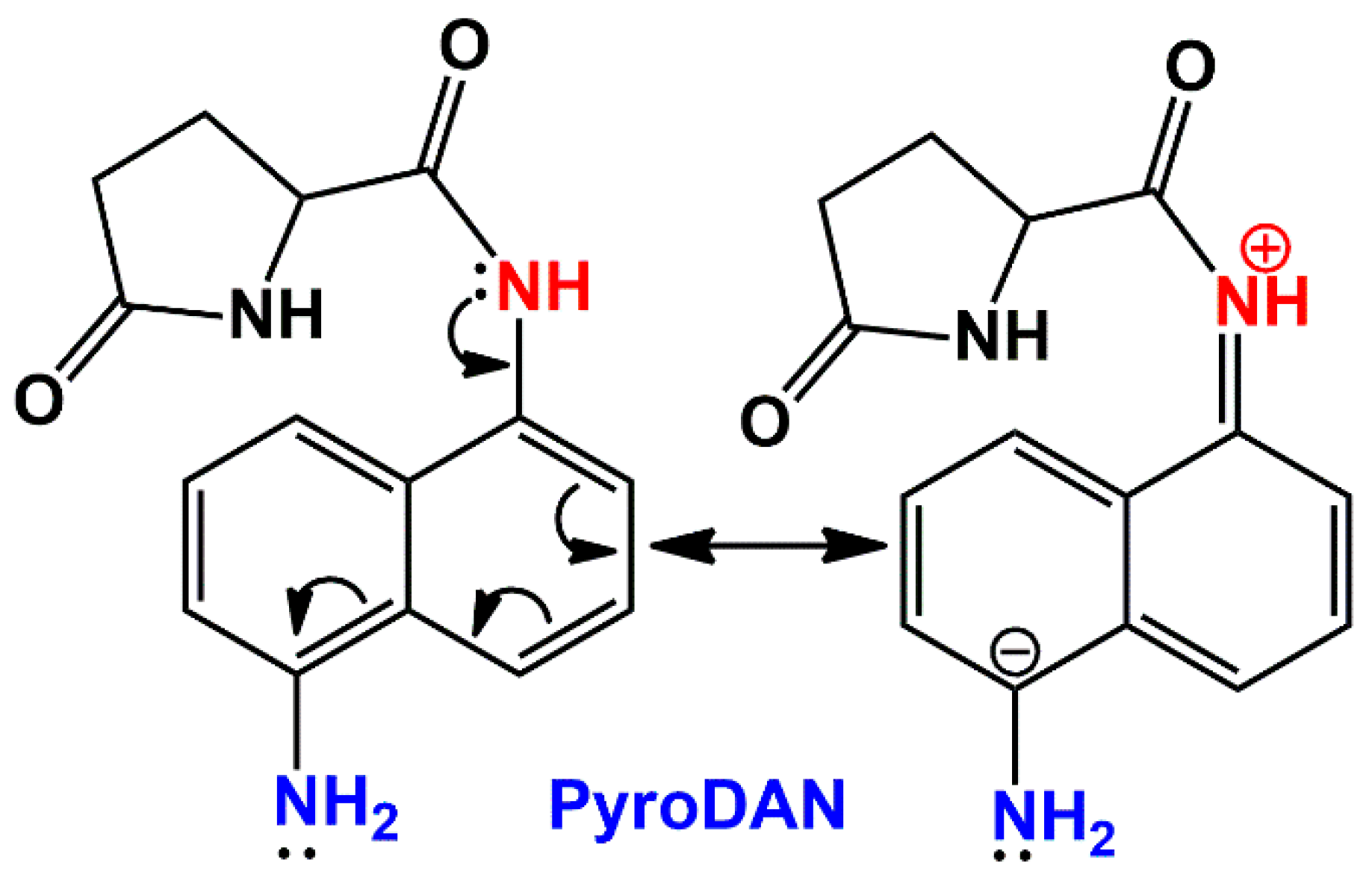
Scheme 4 illustrates these mechanisms applied to PyroDAN.
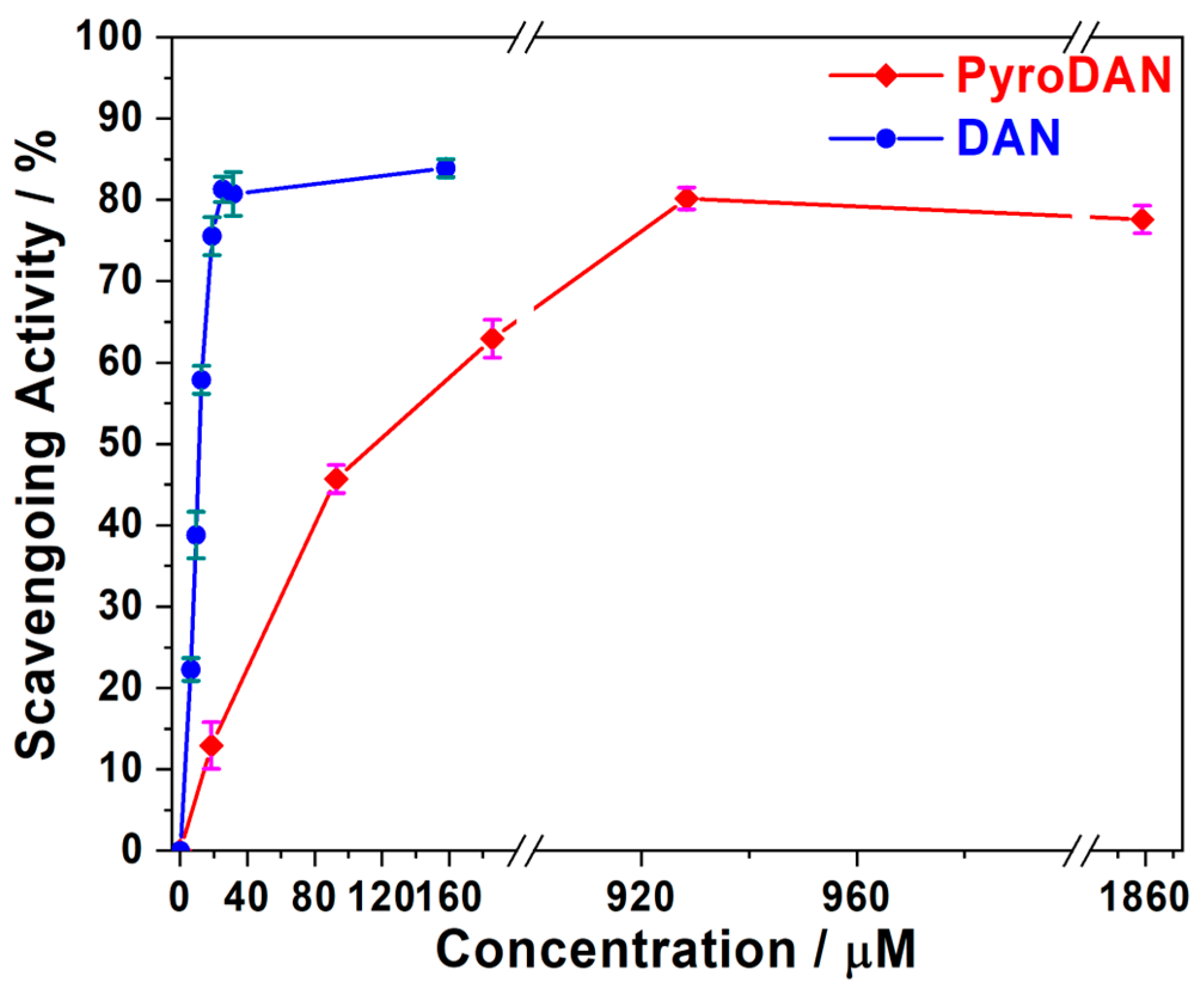
Figure 8 illustrates the antioxidant activity of DAN and PyroDAN resulting from the DPPH• assay.
The percentage of scavenging activity is shown as a function of the increasing concentrations of antioxidants (DAN or PyroDAN). At concentrations as low as 20 µM, DAN exhibits a significantly higher antioxidant activity, neutralizing about 80% of the DPPH• radical. In contrast, PyroDAN only achieves this level of neutralization at a much higher concentration of 930 µM, indicating that DAN is approximately 45 times more efficient than its mono-pyroglutanilide counterpart. Interestingly, although the PyroDAN antioxidant activity is considerably weaker, some activity is still retained. The transformation of aniline into anilide, with the subsequent alteration of the electronic environment of the aromatic structures, greatly reduces the PyroDAN capability of hydrogen donation. An explanation can be found in the change of resonance structures characterizing the asymmetric PyroDAN with respect to the highly symmetric and planar DAN. The lone pair of the amidic nitrogen, in fact, can be delocalized either on the aromatic ring, increasing its electron density, or on the amidic carbonyl, generating a positively charged nitrogen that could drain out electron density from the π-conjugate system.
The mesomeric electro-donating effect (
Scheme 5) is predominant and thus the increased electron density on the π-conjugated system, paired with the symmetry breaking, makes the aniline nitrogen more basic and the hydrogen less acidic and less prone to homolitically dissociate and join the free DPPH• radical. Moreover, although the previous literature reported the bland antioxidant activity of some acetanilides [
19], in our case, the already poor acidic hydrogen of the anilide group (pK
a ≅ 18) is less disposed to dissociate homolitically.
2.3. Conclusions
This study demonstrates the successful synthesis and characterization of a novel mono-pyroglutanilide derivative of 1,5-diamino-naphthalene (DAN), PyroDAN, via a solventless thermal coupling with l-pyroglutamic acid. The derivatization process effectively modified the electronic and reactive properties of DAN, as evidenced by the extensive spectroscopic and analytical evaluations.
The antioxidant properties, while significantly reduced when compared with DAN, are still retained at a mild level, as shown by the DPPH• assay. Importantly, the oxidant activity of PyroDAN was almost entirely suppressed, as confirmed by the ICG photodegradation experiment. These results indicate that the symmetry-breaking modification of the DAN structure has a profound impact on its redox properties.
To the best of our knowledge, this is the first work that tries to understand how an asymmetric amidation of a diamino-naphthalene could affect its pro-oxidant/antioxidant properties. The tuneable fluorescent properties of PyroDAN—the mild antioxidant activity and the inhibition of the cytologically harmful pro-oxidant properties—suggest promising applications in bioimaging and other biological fields. Future research will focus on further exploring its biological compatibility and performance in targeted applications.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}