
Plasticity and Co-Factor-Dependent Structural Changes in the RecA Nucleoprotein Filament Studied by Small-Angle X-Ray Scattering (SAXS) Measurements and Molecular Modeling

 , , , , ,
, , , , ,  , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Estimation of the Filament Length Based on Experimental SAXS Signals
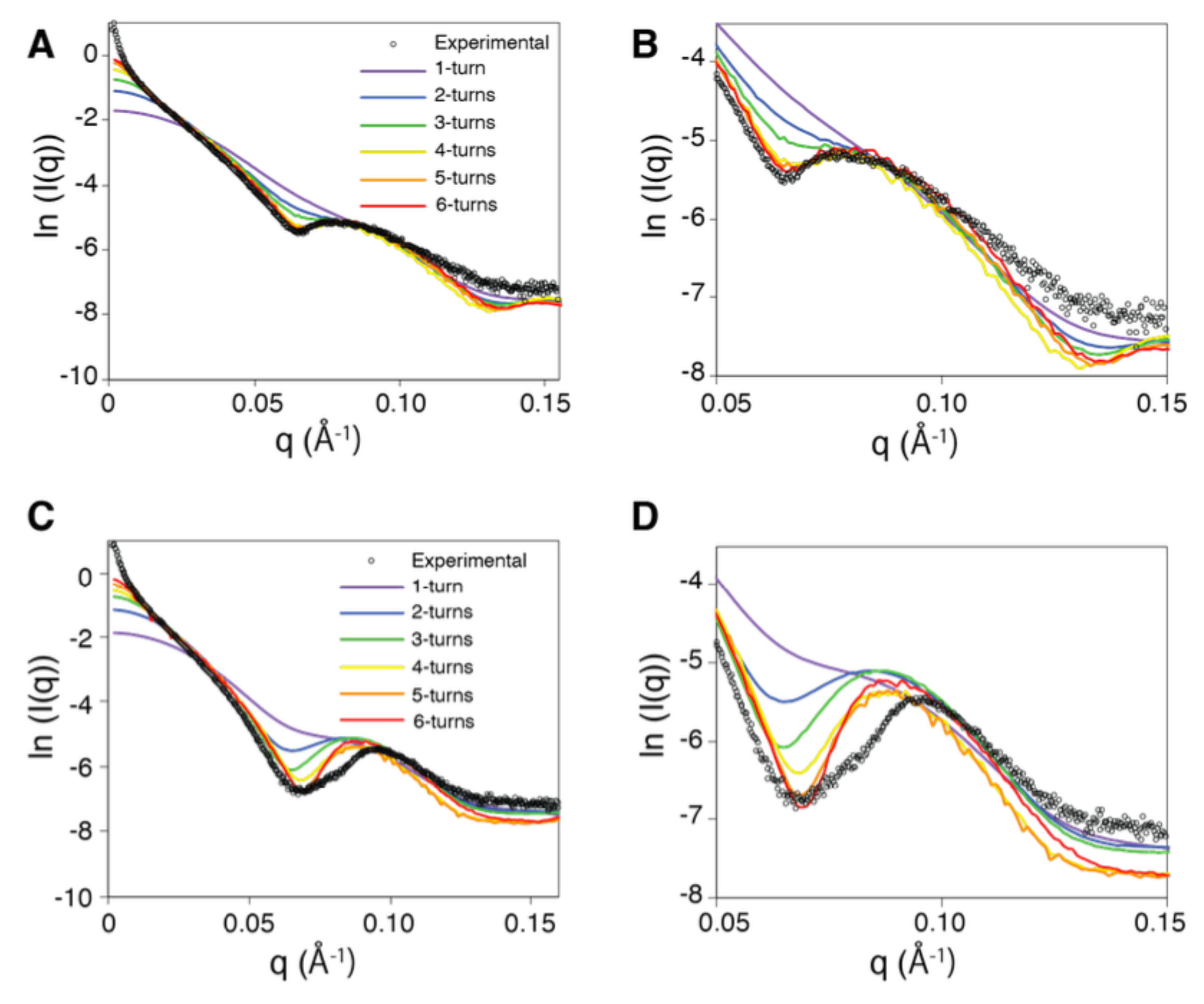
2.2. The Solution Structure of the Extended Form of the RecA Filament Matches the Crystallographic Structure
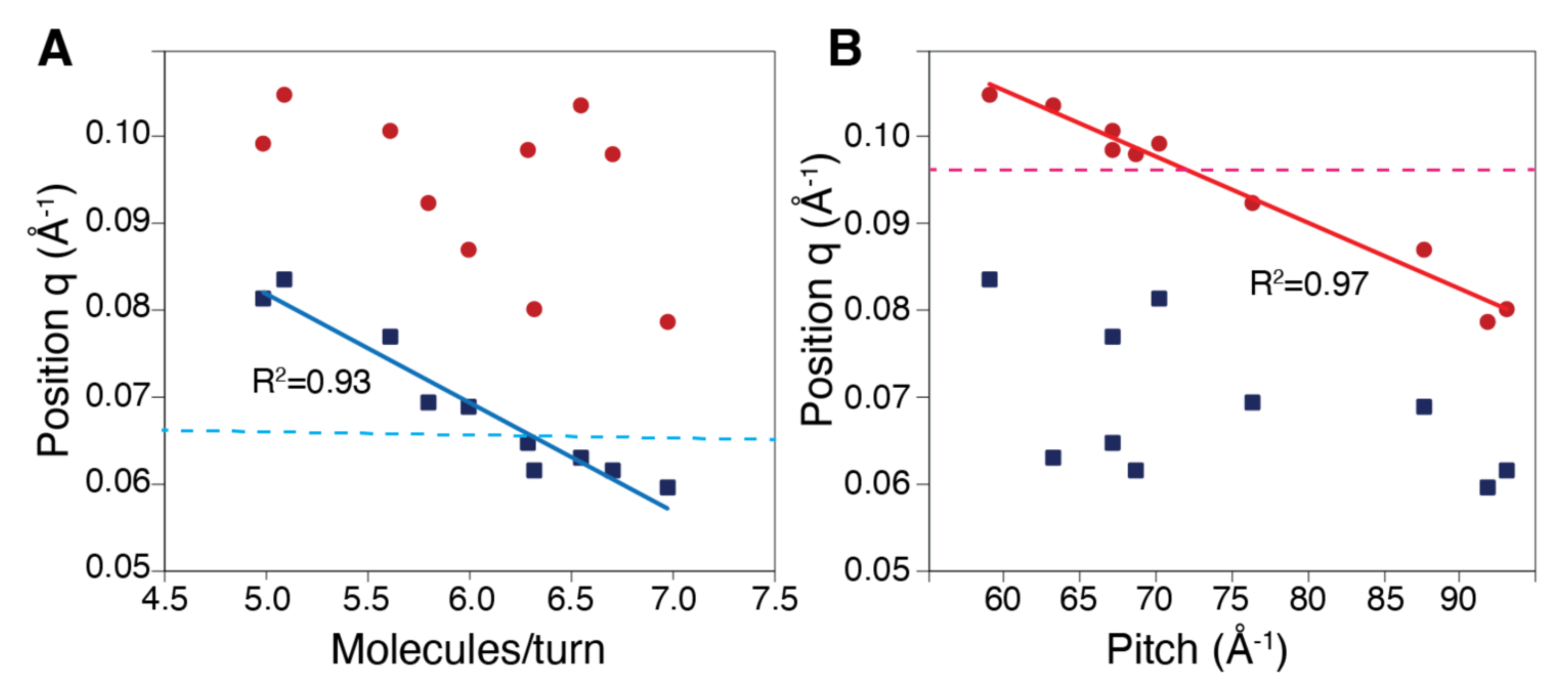
2.3. Solution Structure of the Compressed Form of the RecA Filament
2.4. Experimental Observation of the Mg-Driven Transition Between the RecA–ATP and RecA–ADP Filament Forms
3. Materials and Methods
3.1. Sample Preparation of RecA Protein and RecA Protein Filaments
3.2. Small-Angle X-Ray Scattering (SAXS)
3.3. Modeling Filament Forms Based on Selected Interfaces
3.4. Interface Sampling and Selection
3.5. Interface Adjustment
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Goodsell, D.S.; Olson, A.J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Abeta and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid beta-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, D.; Löwe, J. Collaborative protein filaments. EMBO J. 2015, 34, 2312–2320. [Google Scholar] [CrossRef]
- Chiesa, G.; Kiriakov, S.; Khalil, A.S. Protein assembly systems in natural and synthetic biology. BMC Biol. 2020, 18, 35. [Google Scholar] [CrossRef]
- Assaiya, A.; Burada, A.P.; Dhingra, S.; Kumar, J. An overview of the recent advances in cryo-electron microscopy for life sciences. Emerg. Top. Life Sci. 2021, 5, 151–168. [Google Scholar]
- Zhang, Q.; Lam, W.H.; Zhai, Y. Assembly and activation of replicative helicases at origin DNA for replication initiation. Curr. Opin. Struct. Biol. 2024, 88, 102876. [Google Scholar] [CrossRef]
- Story, R.M.; Weber, I.T.; Steitz, T.A. The structure of the E. coli recA protein monomer and polymer. Nature 1992, 355, 318–325. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, H.; Pavletich, N.P. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 2008, 453, 489–494. [Google Scholar] [CrossRef]
- Murakami, K.; Yasunaga, T.; Noguchi, T.Q.P.; Gomibuchi, Y.; Ngo, K.X.; Uyeda, T.Q.P.; Wakabayashi, T. Structural basis for actin assembly, activation of ATP hydrolysis, and delayed phosphate release. Cell 2010, 143, 275–287. [Google Scholar] [CrossRef]
- Nogales, E. An electron microscopy journey in the study of microtubule structure and dynamics. Protein Sci. 2015, 24, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Egelman, E.H. Structural data suggest that the active and inactive forms of the RecA filament are not simply interconvertible. J. Mol. Biol. 1992, 227, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jacobs, S.A.; West, S.C.; Ogawa, T.; Egelman, E.H. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc. Natl. Acad. Sci. USA 2001, 98, 8419–8424. [Google Scholar] [CrossRef]
- Jegou, A.; Romet-Lemonne, G. The many implications of actin filament helicity. Semin. Cell Dev. Biol. 2020, 102, 65–72. [Google Scholar] [CrossRef]
- Gong, R.; Jiang, F.; Moreland, Z.G.; Reynolds, M.J.; de Los Reyes, S.E.; Gurel, P.; Shams, A.; Heidings, J.B.; Bowl, M.R.; Bird, J.E.; et al. Structural basis for tunable control of actin dynamics by myosin-15 in mechanosensory stereocilia. Sci. Adv. 2022, 8, eabl4733. [Google Scholar] [CrossRef]
- Thomsen, N.D.; Berger, J.M. Running in reverse: The structural basis for translocation polarity in hexameric helicases. Cell 2009, 139, 523–534. [Google Scholar] [CrossRef]
- Ma, W.; Schulten, K. Mechanism of substrate translocation by a ring-shaped ATPase motor at millisecond resolution. J. Am. Chem. Soc. 2015, 137, 3031–3040. [Google Scholar] [CrossRef]
- Boyer, B.; Ezelin, J.; Poulain, P.; Saladin, A.; Zacharias, M.; Robert, C.H.; Prévost, C. An integrative approach to the study of filamentous oligomeric assemblies, with application to RecA. PLoS ONE 2015, 10, e0116414. [Google Scholar] [CrossRef]
- Egelman, E.H. Reconstruction of helical filaments and tubes. Methods Enzymol. 2010, 482, 167–183. [Google Scholar]
- Prévost, C.; Sacquin-Mora, S. Moving pictures: Reassessing docking experiments with a dynamic view of protein interfaces. Proteins 2021, 89, 1315–1323. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef] [PubMed]
- Delalande, O.; Molza, A.E.; Dos Santos Morais, R.; Chéron, A.; Pollet, É.; Raguenes-Nicol, C.; Tascon, C.; Giudice, E.; Guilbaud, M.; Nicolas, A.; et al. Dystrophin’s central domain forms a complex filament that becomes disorganized by in-frame deletions. J. Biol. Chem. 2018, 293, 6637–6646. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M. Modeling structure and dynamics of protein complexes with SAXS profiles. Methods Mol. Biol. 2018, 1764, 449–473. [Google Scholar] [PubMed]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef] [PubMed]
- DiCapua, E.; Schnarr, M.; Ruigrok, R.W.; Lindner, P.; Timmins, P.A. Complexes of RecA protein in solution. A study by small angle neutron scattering. J. Mol. Biol. 1990, 214, 557–570. [Google Scholar] [CrossRef]
- Timmins, P.A.; Ruigrok, R.W.; DiCapua, E. The solution structure of recA filaments by small angle neutron scattering. Biochimie 1991, 73, 227–230. [Google Scholar] [CrossRef]
- Nordén, B.; Elvingson, C.; Kubista, M.; Sjöberg, B.; Ryberg, H.; Ryberg, M.; Mortensen, K.; Takahashi, M. Structure of RecA-DNA complexes studied by combination of linear dichroism and small-angle neutron scattering measurements on flow-oriented samples. J. Mol. Biol. 1992, 226, 1175–1191. [Google Scholar] [CrossRef]
- Lebedev, D.V.; Baitin, D.M.; Islamov, A.K.; Kuklin, A.I.; Shalguev, V.K.; Lanzov, V.A.; Isaev-Ivanov, V.V. Analytical model for determination of parameters of helical structures in solution by small angle scattering: Comparison of RecA structures by SANS. FEBS Lett. 2003, 537, 182–186. [Google Scholar] [CrossRef]
- Burian, M.; Amenitsch, H. Dummy-atom modelling of stacked and helical nanostructures from solution scattering data. IUCrJ 2018, 5, 390–401. [Google Scholar] [CrossRef]
- Sabei, A.; Prentiss, M.; Prévost, C. Modeling the Homologous Recombination Process: Methods, Successes and Challenges. Int. J. Mol. Sci. 2023, 24, 14896. [Google Scholar] [CrossRef]
- Trewhella, J.; Duff, A.P.; Durand, D.; Gabel, F.; Guss, J.M.; Hendrickson, W.A.; Hura, G.L.; Jacques, D.A.; Kirby, N.M.; Kwan, A.H.; et al. 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. D Struct. Biol. 2017, 73, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Santuz, H.; Laurent, B.; Robert, C.H.; Prévost, C. Heligeom: A web resource to generate, analyze, and visualize filament architectures based on pairwise association geometries of biological macromolecules. J. Mol. Biol. 2025, 169019. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016, 44, W424–W429. [Google Scholar] [CrossRef]
- Kim, R.; Kanamaru, S.; Mikawa, T.; Prévost, C.; Ishii, K.; Ito, K.; Uchiyama, S.; Oda, M.; Iwasaki, H.; Kim, S.K.; et al. RecA requires two molecules of Mg2+ ions for its optimal strand exchange activity in vitro. Nucleic Acids Res. 2018, 46, 2548–2559. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Bohrmann, B.; Hewat, E.; Engel, A.; Kellenberger, E.; DiCapua, E. The inactive form of recA protein: The ’compact’ structure. EMBO J. 1993, 12, 9–16. [Google Scholar] [CrossRef]
- Lusetti, S.L.; Shaw, J.J.; Cox, M.M. Magnesium ion-dependent activation of the RecA protein involves the C terminus. J. Biol. Chem. 2003, 278, 16381–16388. [Google Scholar] [CrossRef]
- Cazenave, C.; Toulmé, J.J.; Hélène, C. Binding of RecA protein to single-stranded nucleic acids: Spectroscopic studies using fluorescent polynucleotides. EMBO J. 1983, 2, 2247–2251. [Google Scholar] [CrossRef]
- Stasiak, A.; Di Capua, E. The helicity of DNA in complexes with recA protein. Nature 1982, 299, 185–186. [Google Scholar] [CrossRef]
- Jeffries, C.M.; Ilavsky, J.; Martel, A.; Hinrichs, S.; Meyer, A.; Pedersen, J.S.; Sokolova, A.V.; Svergun, D.I. Small-angle X-ray and neutron scattering. Nat. Rev. Methods Prim. 2021, 1, 1–39. [Google Scholar] [CrossRef]
- Mendez, R.; Leplae, R.; Maria, L.D.; Wodak, S.J. Assessment of blind predictions of protein-protein interactions: Current status of docking methods. Proteins 2003, 52, 51–67. [Google Scholar] [CrossRef]
- Boyer, B.; Danilowicz, C.; Prentiss, M.; Prévost, C. Weaving DNA strands: Structural insight on ATP hydrolysis in RecA-induced homologous recombination. Nucleic Acids Res. 2019, 47, 7798–7808. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, T.; Ikawa, S.; Iwasaki, W.; Hiraki, T.; Hikima, T.; Mikawa, T.; Arai, N.; Kamiya, N.; Shibata, T. Loop L1 governs the DNA-binding specificity and order for RecA-catalyzed reactions in homologous recombination and DNA repair. Nucleic Acids Res. 2015, 43, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, A.; Morozova, N.; Vedyaykin, A.; Yakimov, A.; Khodorkovskii, M.; Pobegalov, G. Single-molecule characterization of compressed RecA nucleoprotein filaments. Biochem. Biophys. Res. Commun. 2022, 614, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Conover, A.J.; Danilowicz, C.; Gunaratne, R.; Coljee, V.W.; Kleckner, N.; Prentiss, M. Changes in the tension in dsDNA alter the conformation of RecA bound to dsDNA-RecA filaments. Nucleic Acids Res. 2011, 39, 8833–8843. [Google Scholar] [CrossRef]
- Alekseev, A.; Serdakov, M.; Pobegalov, G.; Yakimov, A.; Bakhlanova, I.; Baitin, D.; Khodorkovskii, M. Single-molecule analysis reveals two distinct states of the compressed RecA filament on single-stranded DNA. FEBS Lett. 2020, 594, 3464–3476. [Google Scholar] [CrossRef]
- DiCapua, E.; Ruigrok, R.W.; Timmins, P.A. Activation of recA protein: The salt-induced structural transition. J. Struct. Biol. 1990, 104, 91–96. [Google Scholar] [CrossRef]
- Petukhov, M.; Lebedev, D.; Shalguev, V.; Islamov, A.; Kuklin, A.; Lanzov, V.; Isaev-Ivanov, V. Conformational flexibility of RecA protein filament: Transitions between compressed and stretched states. Proteins 2006, 65, 296–304. [Google Scholar] [CrossRef]
- Shinohara, T.; Arai, N.; Iikura, Y.; Kasagi, M.; Masuda-Ozawa, T.; Yamaguchi, Y.; Suzuki-Nagata, K.; Shibata, T.; Mikawa, T. Nonfilament-forming RecA dimer catalyzes homologous joint formation. Nucleic Acids Res. 2018, 46, 10855–10869. [Google Scholar] [CrossRef]
- Boyer, B.; Laurent, B.; Robert, C.H.; Prévost, C. Modeling Perturbations in Protein Filaments at the Micro and Meso Scale Using NAMD and PTools/Heligeom. Bio Protoc. 2021, 11, e4097. [Google Scholar] [CrossRef] [PubMed]
- Saladin, A.; Fiorucci, S.; Poulain, P.; Prévost, C.; Zacharias, M. PTools: An opensource molecular docking library. BMC Struct. Biol. 2009, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.F.; Chen, L.T.; Wang, A.H.J. Right or left turn? RecA family protein filaments promote homologous recombination through clockwise axial rotation. Bioessays 2008, 30, 48–56. [Google Scholar] [CrossRef]
- Zacharias, M. Protein-protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003, 12, 1271–1282. [Google Scholar] [CrossRef]
- Garmay, Y.; Shvetsov, A.; Karelov, D.; Lebedev, D.; Radulescu, A.; Petukhov, M.; Isaev-Ivanov, V. Correlated motion of protein subdomains and large-scale conformational flexibility of RecA protein filament. J. Phys. Conf. Ser. 2012, 340, 012094. [Google Scholar] [CrossRef]
- Chang, C.F.; Rankert, D.A.; Jeng, T.W.; Morgan, D.G.; Schmid, M.F.; Chiu, W. Cryo electron microscopy of unstained, unfixed RecA-cssDNA complexes. J. Ultrastruct. Mol. Struct. Res. 1988, 100, 166–172. [Google Scholar] [CrossRef]
- VanLoock, M.S.; Yu, X.; Yang, S.; Galkin, V.E.; Huang, H.; Rajan, S.S.; Anderson, W.F.; Stohl, E.A.; Seifert, H.S.; Egelman, E.H. Complexes of RecA with LexA and RecX differentiate between active and inactive RecA nucleoprotein filaments. J. Mol. Biol. 2003, 333, 345–354. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Yang, D.; Boyer, B.; Prévost, C.; Danilowicz, C.; Prentiss, M. Integrating multi-scale data on homologous recombination into a new recognition mechanism based on simulations of the recassdna/ dsdna structure. Nucleic Acids Res. 2015, 43, 10251–10263. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inaba-Inoue, S.; Sabei, A.; Molza, A.-E.; Prentiss, M.; Mikawa, T.; Sekiguchi, H.; Prévost, C.; Takahashi, M. Plasticity and Co-Factor-Dependent Structural Changes in the RecA Nucleoprotein Filament Studied by Small-Angle X-Ray Scattering (SAXS) Measurements and Molecular Modeling. Molecules 2025, 30, 1793. https://doi.org/10.3390/molecules30081793
Inaba-Inoue S, Sabei A, Molza A-E, Prentiss M, Mikawa T, Sekiguchi H, Prévost C, Takahashi M. Plasticity and Co-Factor-Dependent Structural Changes in the RecA Nucleoprotein Filament Studied by Small-Angle X-Ray Scattering (SAXS) Measurements and Molecular Modeling. Molecules. 2025; 30(8):1793. https://doi.org/10.3390/molecules30081793
Chicago/Turabian StyleInaba-Inoue, Satomi, Afra Sabei, Anne-Elisabeth Molza, Mara Prentiss, Tsutomu Mikawa, Hiroshi Sekiguchi, Chantal Prévost, and Masayuki Takahashi. 2025. "Plasticity and Co-Factor-Dependent Structural Changes in the RecA Nucleoprotein Filament Studied by Small-Angle X-Ray Scattering (SAXS) Measurements and Molecular Modeling" Molecules 30, no. 8: 1793. https://doi.org/10.3390/molecules30081793
APA StyleInaba-Inoue, S., Sabei, A., Molza, A.-E., Prentiss, M., Mikawa, T., Sekiguchi, H., Prévost, C., & Takahashi, M. (2025). Plasticity and Co-Factor-Dependent Structural Changes in the RecA Nucleoprotein Filament Studied by Small-Angle X-Ray Scattering (SAXS) Measurements and Molecular Modeling. Molecules, 30(8), 1793. https://doi.org/10.3390/molecules30081793