

Influence of Partial Disentanglement of Macromolecules on the Rheological, Thermal, and Mechanical Properties of Polypropylene–Polyethylene Blends




Abstract

1. Introduction
2. Results and Discussion
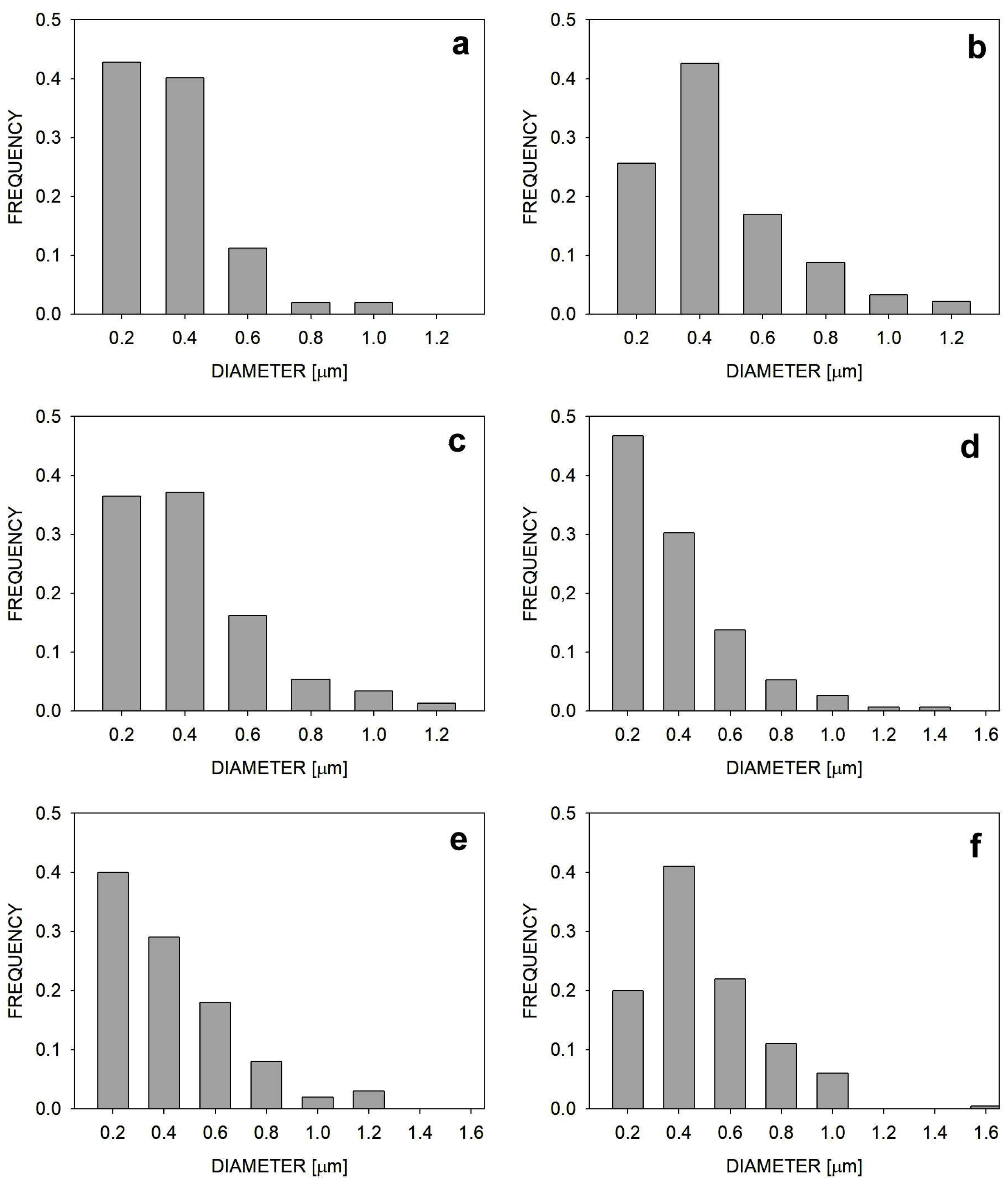
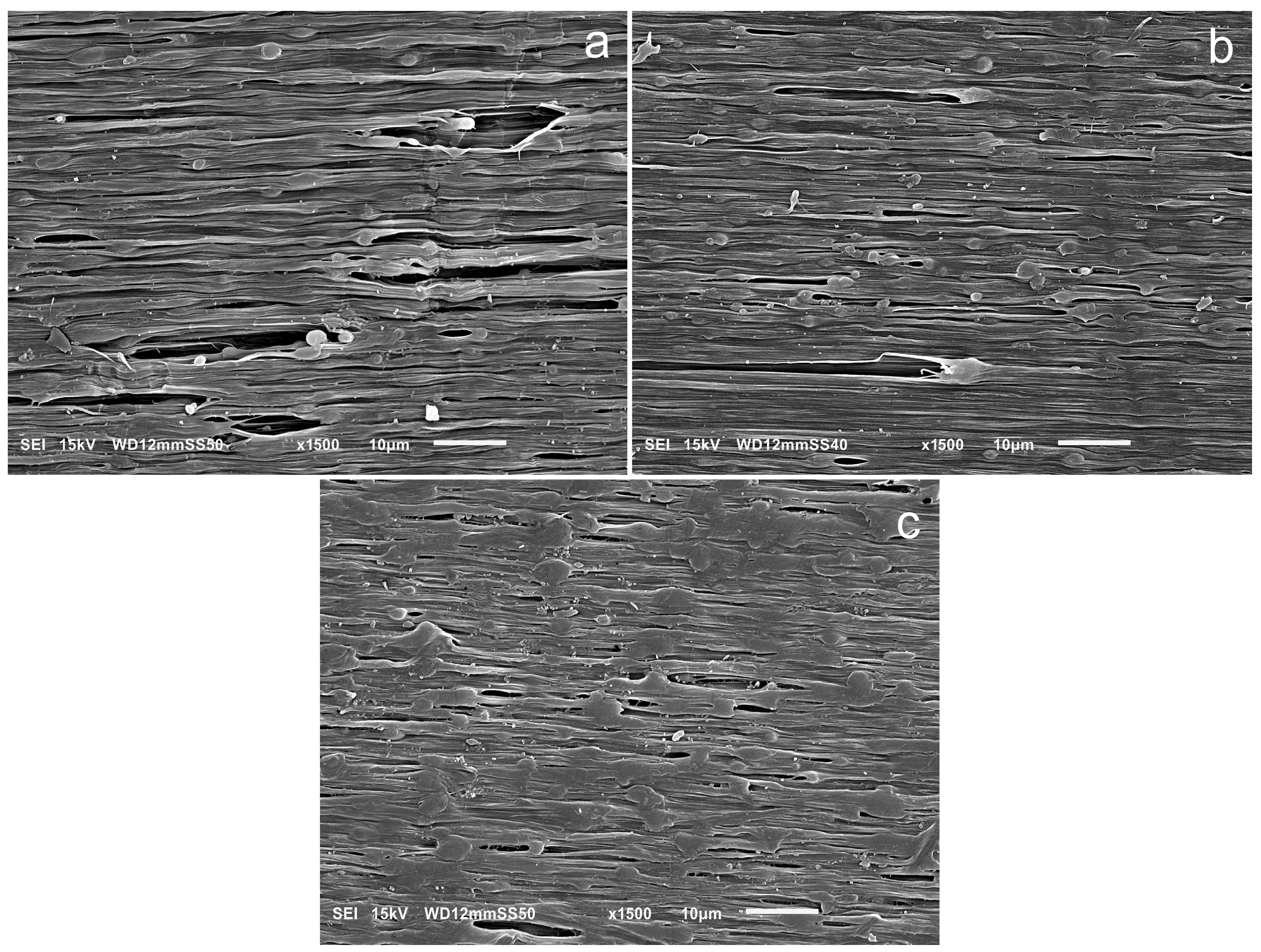
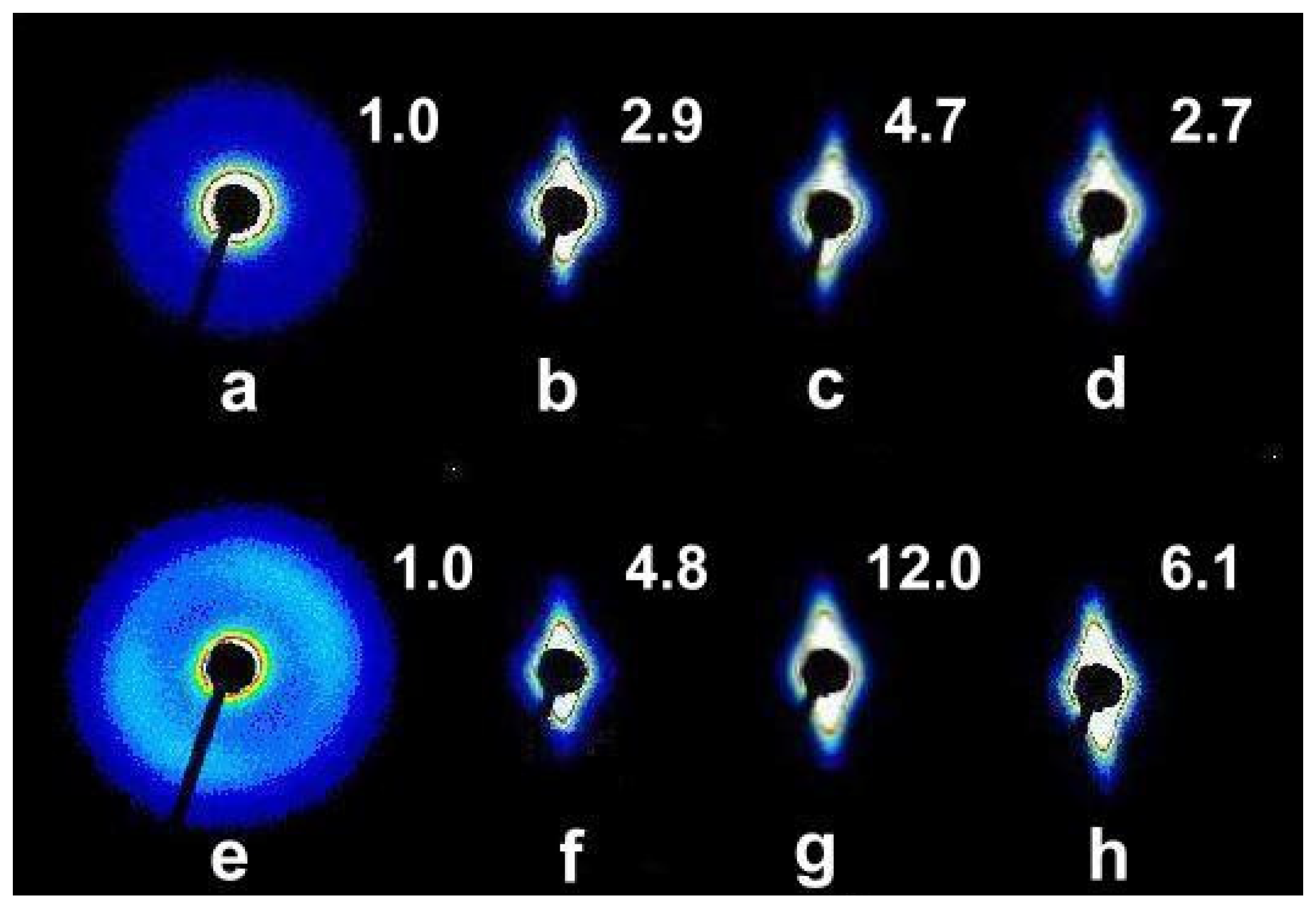
2.1. Morphologies of Blends
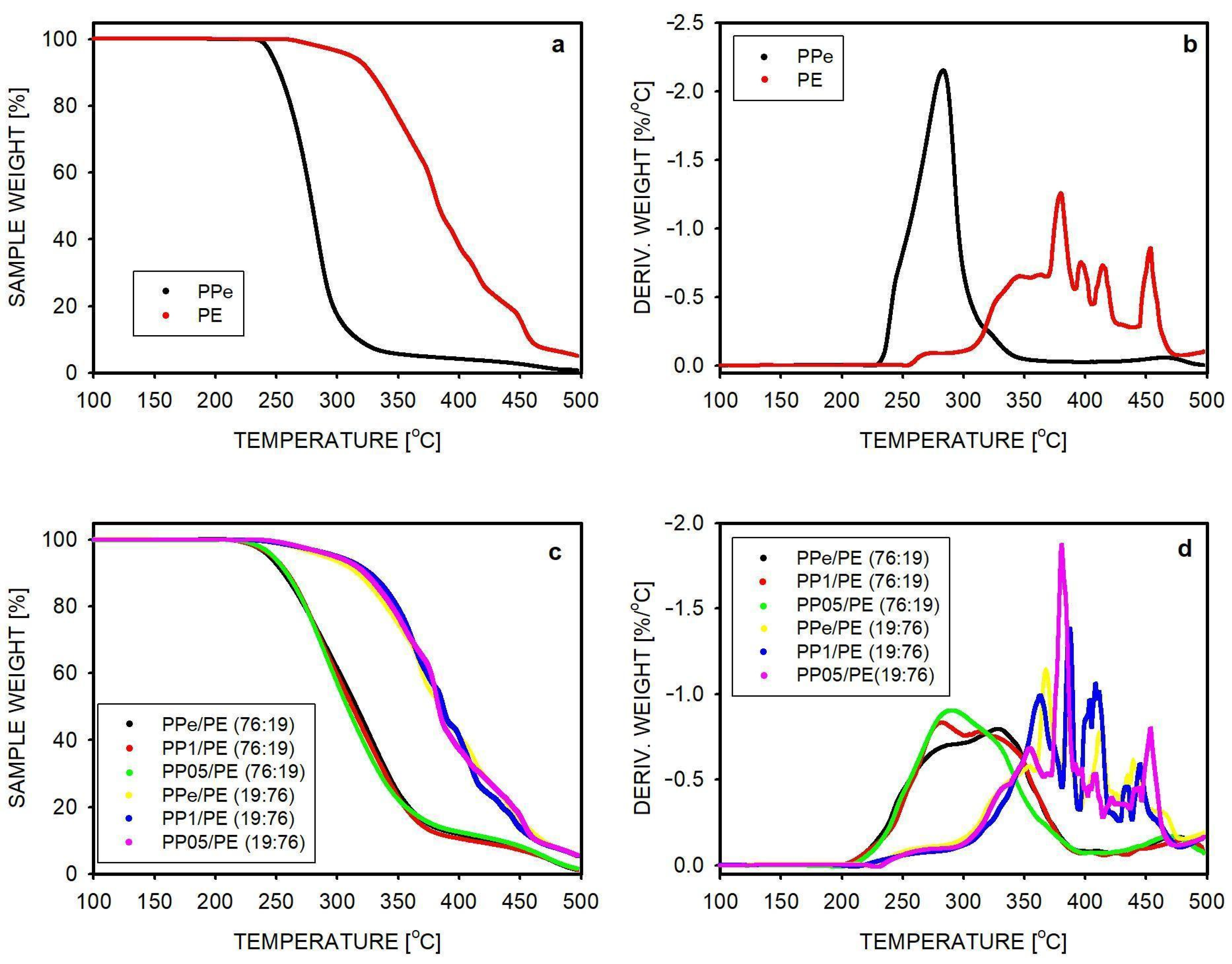
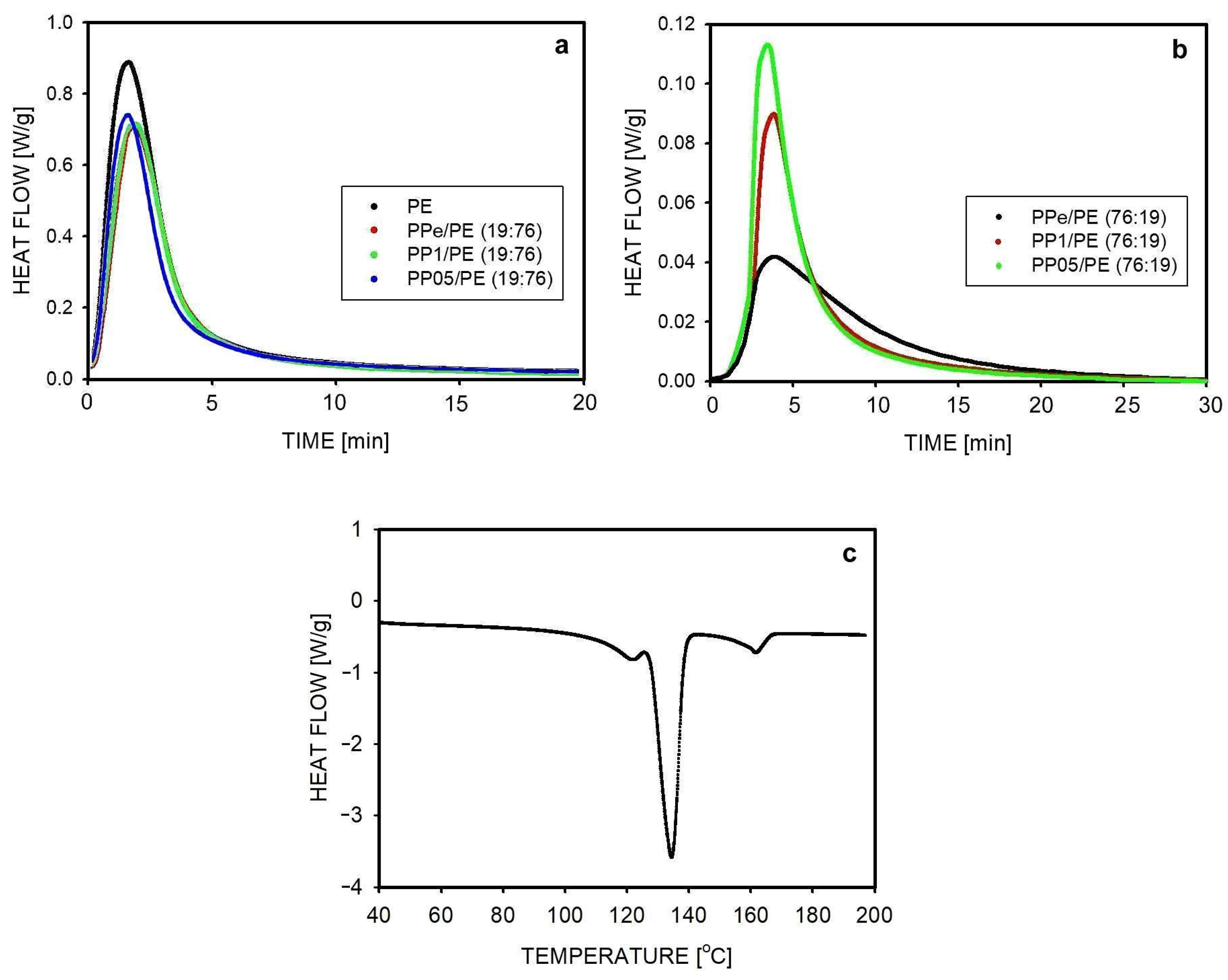
2.2. Thermal Stability
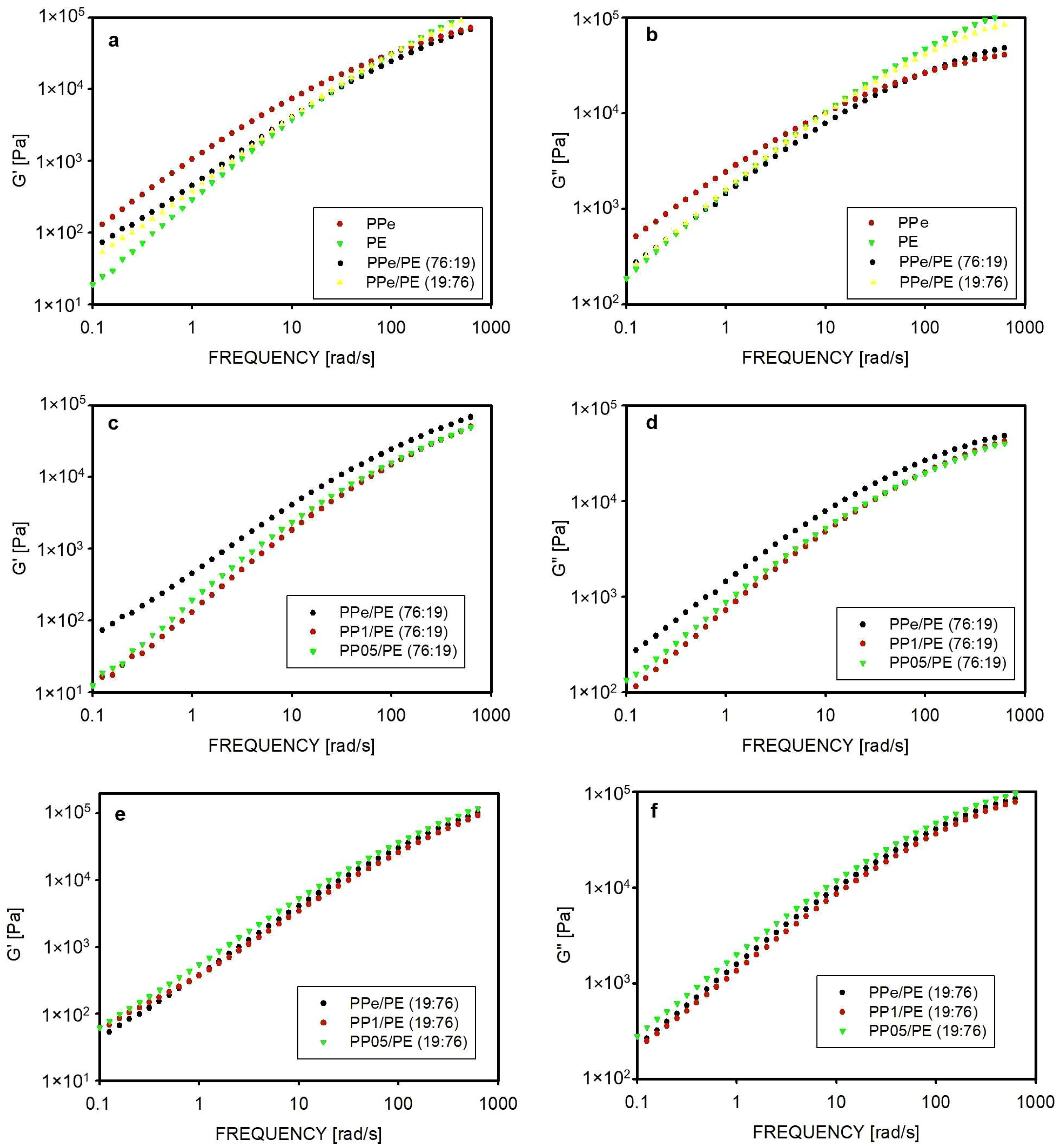
2.3. Rheology
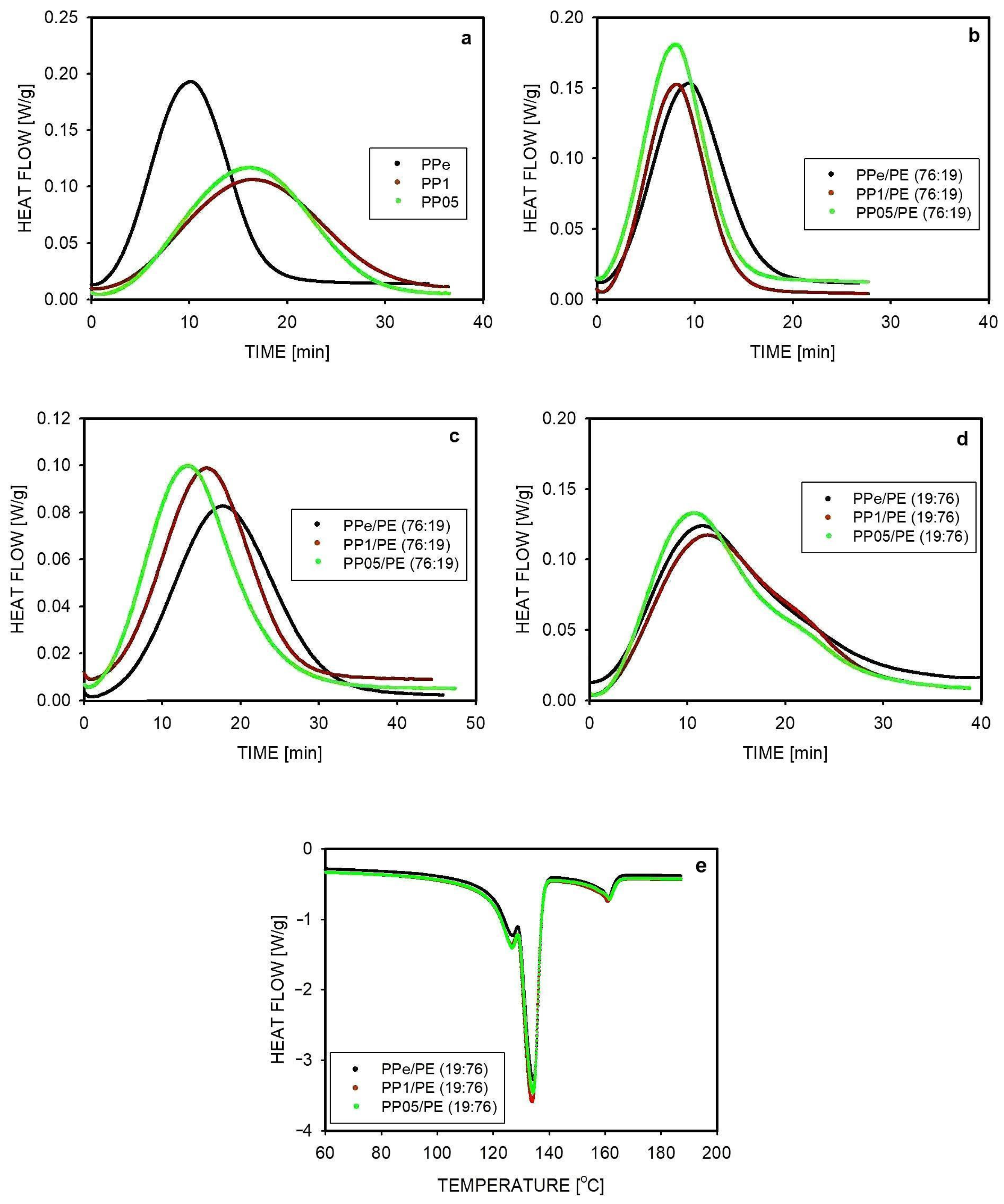
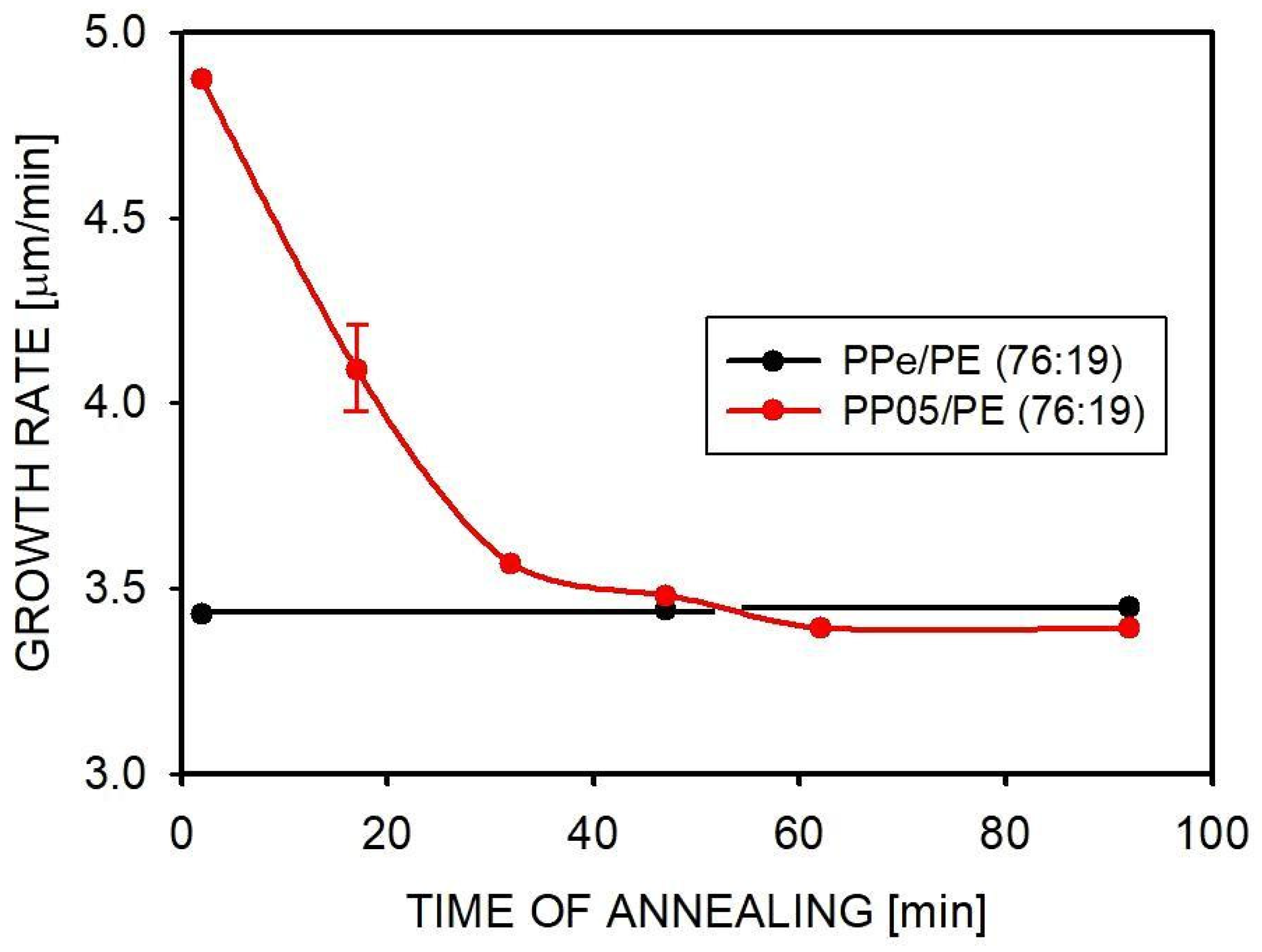
2.4. Isothermal Crystallization
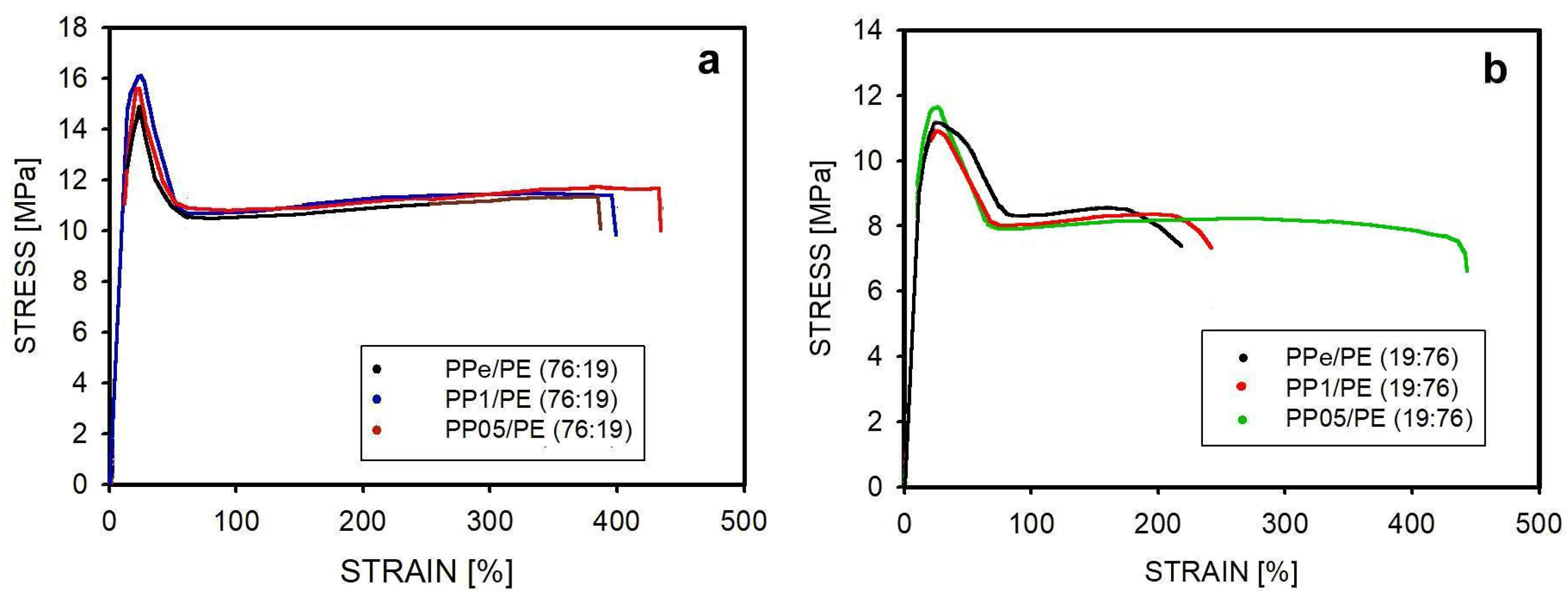
2.5. Mechanical Properties
3. Materials and Methods
3.1. Materials
3.2. Methods of Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berry, G.C.; Fox, T.G. The viscosity of polymers and their concentrated solutions. Adv. Polym. Sci. 1968, 5, 261–357. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. The connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]
- Pawlak, A. The Entanglements of Macromolecules and Their Influence on the Properties of Polymers. Macromol. Chem. Phys. 2019, 220, 1900043. [Google Scholar] [CrossRef]
- Wang, X.H.; Liu, R.; Wu, M.; Wang, Z.; Huang, Y. Effect of chain disentanglement on melt crystallization behavior of isotactic polypropylene. Polymer 2009, 50, 5824–5827. [Google Scholar] [CrossRef]
- Eckstein, A.; Suhm, J.; Friedrich, C.; Maier, R.; Sassmannshausen, J.; Bochmann, M.; Mulhaupt, R. Determination of Plateau Moduli and Entanglement Molecular Weights of Isotactic, Syndiotactic, and Atactic Polypropylenes Synthesized with Metallocene Catalysts. Macromolecules 1998, 31, 1335–1340. [Google Scholar] [CrossRef]
- Bu, H.S.; Gu, F.M.; Bao, L.; Chen, M. Influence of entanglements on crystallization of macromolecules. Macromolecules 1998, 31, 7108–7110. [Google Scholar] [CrossRef]
- Ji, G.; Ni, H.; Wang, C.; Xue, G.; Liao, Y.-T. Concentration Dependence of Crystalline Poly(ethylene terephthalate) Prepared by Freeze-Extracting Solutions. Macromolecules 1996, 29, 2691–2693. [Google Scholar] [CrossRef]
- Bastiaansen, C.W.M.; Meyer, H.E.H.; Lemstra, P.J. Memory effects in polyethylenes: Influence of processing and crystallization history. Polymer 1990, 31, 1435–1440. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J. Progress in Studies of Disentangled Polymers and Composites. J. Compos. Sci. 2023, 7, 521. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Chen, J.; Luo, J.; Min, J.; Fu, Q.; Zhang, J. Efficient disentanglement of polycarbonate melts under complex shear field. Polymer 2020, 201, 122610. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Y.; Shen, K.; Fu, Q.; Zhang, J. Insight into Shear-Induced Modification for Improving Processability of Polymers: Effect of Shear Rate on the Evolution of Entanglement State. J. Polym. Sci. B Polym. Phys. 2019, 57, 598–606. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, S.; Yu, X.; Xin, Z.; Ye, C.; Li, Z.; Xia, J. Nascent Particle Sizes and Degrees of Entanglement Are Responsible for the Significant Differences in Impact Strength of Ultrahigh Molecular Weight Polyethylene. J. Polym. Sci. B Polym. Phys. 2019, 57, 632–641. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. Cavitation phenomenon and mechanical properties of partially disentangled polypropylene. Polymer 2018, 151, 15–26. [Google Scholar] [CrossRef]
- Romano, D.; Tops, N.; Andablo-Reyes, E.; Ronca, S.; Rastogi, S. Influence of Polymerization Conditions on Melting Kinetics of Low Entangled UHMWPE and Its Implications on Mechanical Properties. Macromolecules 2014, 47, 4750–4760. [Google Scholar] [CrossRef]
- Hu, H.; Chen, J.; Yang, T.; Wang, P.; Min, J.; Fu, Q.; Zhang, J. Regulation of Entanglement Networks under Different Shear Fields and Its Effect on the Properties of Poly(L-lactide). Ind. Eng. Chem. Res. 2023, 62, 7434–7446. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. The crystallization of polypropylene with reduced density of entanglements. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 748–756. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Peng, F.; Cao, R.; Liu, Z.; Xu, T.; Li, L. Influence of Entanglement Density on Polymer Glass Transition Temperature. Macromolecules 2025, 58, 1512–1520. [Google Scholar] [CrossRef]
- Krajenta, J.; Safandowska, M.; Pawlak, A. The re-entangling of macromolecules in polypropylene. Polymer 2019, 175, 215–226. [Google Scholar] [CrossRef]
- Barangizi, H.; Krajenta, J.; Pawlak, A. The influence of entanglements of macromolecules on the mechanical and thermal properties of polylactide composites with carbon nanotube. Express Polym. Lett. 2023, 17, 738–758. [Google Scholar] [CrossRef]
- Barangizi, H.; Pawlak, A. Crystallization of partially disentangled polypropylene in nanocomposites with aluminum oxide. Polymer 2022, 254, 125049. [Google Scholar] [CrossRef]
- Chai, S.-C.; Xu, T.-Y.; Cao, X.; Wang, G.; Chen, Q.; Li, H.-L. Ultrasmall Nanoparticles Diluted Chain Entanglement in Polymer Nanocomposites. Chin. J. Polym. Sci. 2019, 37, 797–805. [Google Scholar] [CrossRef]
- Luo, J.; Chen, J.; Liu, M.; Min, J.; Fu, Q.; Zhang, J. Investigating the Influence of Incorporation of Boron Nitride on the Kinetics of Isotactic Polypropylene Entanglement Recovery. Ind. Eng. Chem. Res. 2021, 60, 12901–12910. [Google Scholar] [CrossRef]
- Xie, M.; Li, M. Viscosity reduction and disentanglement in ultrahigh molecular weight polyethylene melt: Effect of blending with polypropylene and poly(ethylene glycol). Eur. Polym. J. 2007, 43, 3480–3487. [Google Scholar] [CrossRef]
- Schirmeister, C.G.; Hees, T.; Dolynchuk, O.; Licht, E.H.; Thurn-Albrecht, T.; Muelhaupt, R. Digitally Tuned Multidirectional All-Polyethylene Composites via Controlled 1D Nanostructure Formation during Extrusion-Based 3D Printing. ACS Appl. Polym. Mater. 2021, 3, 1675–1686. [Google Scholar] [CrossRef]
- Tao, G.; Chen, Y.; Mu, J.; Zhang, L.; Ye, C.; Li, W. Exploring the entangled state and molecular weight of UHMWPE on the microstructure and mechanical properties of HDPE/UHMWPE blends. J. Appl. Polym. Sci. 2021, 138, 50741. [Google Scholar] [CrossRef]
- Tang, X.; Xing, J.; Yan, X.; Ye, C.; Zhang, L.; Zhang, Y.; Shu, B.; Mu, J.; Wei, L.; Wang, J.; et al. Metallocene Polyolefins Reinforced by Low-Entanglement UHMWPE through Interfacial Entanglements. Adv. Polym. Technol. 2022, 2022, 9344096. [Google Scholar] [CrossRef]
- Chen, Y.; Li, W.; Zhang, L.; Ye, C.; Tao, G.; Ren, C.; Jiang, B.; Wang, J.; Yang, Y. In Situ Synthesized Self-Reinforced HDPE/UHMWPE Composites with High Content of Less Entangled UHMWPE and High Gradient-Distributed Oriented Structures. ACS Appl. Polym. Mater. 2023, 5, 88–98. [Google Scholar] [CrossRef]
- Paul, D.R.; Bucknall, C.B. Introduction. In Polymer Blends. Volume 1: Formulation; Paul, D.R., Bucknall, C.B., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2000; pp. 1–14. [Google Scholar]
- Bartlett, D.W.; Barlow, J.W.; Paul, D.R. Mechanical Properties of Blends Containing HDPE and PP. J. Appl. Polym. Sci. 1982, 27, 2351–2360. [Google Scholar] [CrossRef]
- D’Orazio, L.; Greco, R.; Mancarella, C.; Martuscelli, E.; Ragosta, G.; Silvestre, C. Effect of the addition of ethylene-propylene random copolymers on the properties of high-density polyethylene/isotactic polypropylene blends: Part 1—Morphology and impact behavior of molded samples. Polym. Eng. Sci. 1982, 22, 536–544. [Google Scholar] [CrossRef]
- Khabbaz, H.S.; Demets, R.; Gahleitner, M.; Duscher, B.; Stam, R.; Dimitrova, A.; Fiorio, R.; Gijsman, P.; Ragaert, K.; Gooneie, A. Rheological insights into the degradation behavior of PP/HDPE blends. Polym. Degrad. Stabil. 2024, 225, 110819. [Google Scholar] [CrossRef]
- Lovinger, A.J.; Williams, M.L. Tensile Properties and Morphology of Blends of Polyethylene and Polypropylene. J. Appl. Polym. Sci. 1980, 25, 1703–1713. [Google Scholar] [CrossRef]
- Li, J.; Shanks, R.A.; Long, Y. Mechanical Properties and Morphology of Polyethylene–Polypropylene Blends with Controlled Thermal History. J. Appl. Polym. Sci. 2000, 76, 1151–1164. [Google Scholar] [CrossRef]
- Elmendorp, J.J.; Maalcke, R.J. A study on polymer blending microrheology: Part 1. Polym. Eng. Sci. 1985, 25, 1041–1047. [Google Scholar] [CrossRef]
- Lin, J.H.; Pan, Y.J.; Liu, C.F.; Huang, C.L.; Hsieh, C.T.; Chen, C.K.; Lin, Z.I.; Lou, C.W. Preparation and Compatibility Evaluation of Polypropylene/High Density Polyethylene Polyblends. Materials 2015, 8, 8850–8859. [Google Scholar] [CrossRef]
- Kock, C.; Gahleitner, M.; Schausberger, A.; Ingolic, E. Polypropylene/Polyethylene Blends as Models for High-Impact Propylene–Ethylene Copolymers, Part 1: Interaction Between Rheology and Morphology. J. Appl. Polym. Sci. 2013, 128, 1484–1496. [Google Scholar] [CrossRef]
- Valenza, A.; La Mantia, F.P.; Acierno, D. Rheological characteristic of blends of isotactic polypropylene with high density polyethylene. Eur. Polym. J. 1984, 20, 727–731. [Google Scholar] [CrossRef]
- Al-Mulla, A.; Shaban, H. Study of compatibility of recycled polypropylene/high density polyethylene blends using rheology. Polym. Bull. 2014, 71, 2335–2352. [Google Scholar] [CrossRef]
- Wenig, W.; Meyer, K. Investigation of the crystallization behavior of polypropylene-polyethylene blends by optical microscopy. Coll. Polym. Sci. 1980, 258, 1009–1014. [Google Scholar] [CrossRef]
- Noel, O.F.; Carley, J.F. properties of polypropylene-polyethylene blends. Polym. Eng. Sci. 1975, 15, 117–126. [Google Scholar] [CrossRef]
- Sherman, E.S. Reinforcement of polyethylene with propylene by a blending and deformation process. J. Mater. Sci. 1984, 19, 4014–4020. [Google Scholar] [CrossRef]
- Varin, R.A.; Djokovic, D. The effect of annealing at 135o on the mechanical properties of injection molded high density polyethylene-polypropylene blends. Polym. Eng. Sci. 1988, 28, 1477–1483. [Google Scholar] [CrossRef]
- Jose, S.; Aprem, A.S.; Francis, B.; Chandy, M.C.; Werner, P.; Alstaedt, V.; Thomas, S. Phase morphology, crystallization behaviour and mechanical properties of isotactic polypropylene/high density polyethylene blends. Eur. Polym. J. 2004, 40, 2105–2115. [Google Scholar] [CrossRef]
- Jones, H.; McClements, J.; Ray, D.; Hindle, C.S.; Kalloudis, M.; Koutsos, V. Thermomechanical Properties of Virgin and Recycled Polypropylene -High-Density Polyethylene Blends. Polymers 2023, 15, 4200. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.W. Structure and Properties of Polyethylene-Polypropylene Blend. J. Appl. Polym. Sci. 1983, 28, 605–618. [Google Scholar] [CrossRef]
- Martuscelli, E.; Pracella, M.; Avella, M.; Greco, R.; Ragosta, G. Properties of polyethylene-polypropylene blends: Crystallization behavior. Makromol. Chem. 1980, 181, 957–967. [Google Scholar] [CrossRef]
- Galeski, A.; Pracella, M.; Martuscelli, E. Polypropylene spherulite morphology and growth rate changes in blends with low-density polyethylene. J. Polym. Sci. Polym. Phys. Ed. 1984, 22, 739–747. [Google Scholar] [CrossRef]
- Martuscelli, E.; Pracella, M.; Della Volpe, G.; Greco, P. Morphology, crystallization, and thermal behavior of isotactic polypropylene/low density polyethylene blends. Makromol. Chem. 1984, 185, 1041–1061. [Google Scholar] [CrossRef]
- Galeski, A.; Bartczak, Z.; Pracella, M. Spherulite nucleation in polypropylene blends with low density polyethylene. Polymer 1984, 25, 1323–1326. [Google Scholar] [CrossRef]
- Bartczak, Z.; Galeski, A.; Pracella, M. Spherulite nucleation in blends of isotactic polypropylene with high-density polyethylene. Polymer 1986, 27, 537–543. [Google Scholar] [CrossRef]
- Eder, M.; Włochowicz, A. Analysis of the kinetics of crystallization of polyethylene-polypropylene blends. Acta Polym. 1984, 35, 548–553. [Google Scholar] [CrossRef]
- Shanks, R.A.; Li, J.; Yu, L. Polypropylene-polyethylene blend morphology controlled by time-temperature-miscibility. Polymer 2000, 41, 2133–2139. [Google Scholar] [CrossRef]
- Rybnikar, F. Crystallization and morphology in blends of isotactic polypropylene and linear polyethylene. J. Macrom. Sci. Part B Phys. 1988, 27, 125–144. [Google Scholar] [CrossRef]
- Mehrabi-Mazidi, M.; Sharifi, H. Post-consumer recycled high-density polyethylene/polypropylene blend with improved overall performance through modification by impact polypropylene copolymer: Morphology, properties and fracture resistance. Polym. Int. 2021, 70, 1701–1716. [Google Scholar] [CrossRef]
- Teh, J.W.; Rudin, A.; Keung, J.C. A review of polyethylene-polypropylene blends and their compatibilization. Adv. Polym. Technol. 1994, 13, 1–23. [Google Scholar] [CrossRef]
- Chukov, N.A.; Ligidov, M.K.; Pakhomov, S.I.; Mikitaev, A.K. Polypropylene polymer blends. Russ. J. Gen. Chem. 2017, 87, 2238–2249. [Google Scholar] [CrossRef]
- de Ballesteros, O.R.; Rispo, A.; Femina, G.; Davide, S.; Nocella, F.; Romano, R.; Cipullo, R.; Auriemma, F. Compatibilization of isotactic polypropylene (iPP) and polyethylene (PE) with PP-based olefin block copolymers. Polymer 2025, 319, 128040. [Google Scholar] [CrossRef]
- Graziano, A.; Jaffer, S.; Sain, M. Review on modification strategies of polyethylene/polypropylene immiscible thermoplastic blends for enhancing their mechanical behavior. J. Elastom. Plast. 2018, 51, 291–336. [Google Scholar] [CrossRef]
- Vervoort, S.; den Doedler, J.; Tocha, E.; Genoyer, J.; Walton, K.L.; Hu, Y.; Munro, J.; Jeltsch, K. Compatibilization of Polypropylene-Polyethylene Blends. Polym. Eng. Sci. 2018, 58, 460–465. [Google Scholar] [CrossRef]
- Kazemi, Y.; Kakroodi, A.R.; Rodrigue, D. Compatibilization Efficiency in Post-Consumer Recycled Polyethylene/Polypropylene Blends: Effect of Contamination. Polym. Eng. Sci. 2015, 55, 2368–2376. [Google Scholar] [CrossRef]
- Tselios, C.; Bikaris, D.; Maslis, V.; Panayiotou, C. In situ compatibilization of polypropylene-polyethylene blends: A thermomechanical and spectroscopic study. Polymer 1998, 39, 6807–6817. [Google Scholar] [CrossRef]
- Lin, Y.; Yakovleva, V.; Chen, H.; Hiltner, A.; Baer, E. Comparison of olefin copolymers as compatibilizers for polypropylene and high-density polyethylene. J. Appl. Polym. Sci. 2009, 113, 1945–1952. [Google Scholar] [CrossRef]
- Kruszynski, J.; Nowicka, W.; Rozanski, A.; Liu, Y.; Parisi, D.; Yang, L.; Pasha, F.A.; Bouyahyi, M.; Jasinska-Walc, L.; Duchateau, R. iPP/HDPE blends compatibilized by a polyester: An unconventional concept to valuable products. Sci. Adv. 2024, 10, eado1944. [Google Scholar] [CrossRef] [PubMed]
- Klimovica, K.; Pan, S.; Lin, Y.; Peng, X.; Ellison, C.J.; LaPointe, A.M.; Bates, F.S.; Coates, G.W. Compatibilization of iPP/HDPE Blends with PE-h-iPP Graft Copolymers. ACS Macro Lett. 2020, 9, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-W.; Padilla-Velez, O.; Kaewdeewong, P.; LaPointe, A.M.; Coates, G.W.; Eagan, J.M. Advances in Nonreactive Polymer Compatibilizers for Commodity Polyolefin Blends. Chem. Rev. 2024, 124, 9609–9632. [Google Scholar] [CrossRef]
- Liao, H.; Gao, J.; Liu, C.; Tao, G. Rheological Investigation on a Polypropylene/Low Density Polyethylene Blending Melt. J. Polym. Mater. 2024, 41, 45–54. [Google Scholar] [CrossRef]
- Sangroniz, L.; Carmeli, E.; Vulusic, L.; Hristov, V.; Galatini, M.; Tranchida, D.; Cavallo, D. Lamellar thickness of the polypropylene matrix determines surface induced nucleation of polyethylene droplets in immiscible blends. Polymer 2025, 321, 128144. [Google Scholar] [CrossRef]
- Namnidi, S.D.; van Breemen, L.C.A.; Loojmans, S. Structure-property relations in PP/HDPE blends: From processing to performance. Polymer 2025, 323, 128150. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y. Compatibility evaluation and mechanical properties of isotactic polypropylene/high density polyethylene (iPP/HDPE) blends. J. Polym. Res. 2024, 31, 161. [Google Scholar] [CrossRef]
- Sun, Q.; Fu, Q.; Xue, G.; Chen, W. Crystallization Behavior of Syndiotactic Poly(propylene) Freeze-Dried from Toluene at Very Dilute Concentration. Macromol. Rapid Comm. 2001, 22, 1182–1185. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R. Test of the reptation concept: Crystal growth rate as a function of molecular weight in polyethylene crystallized from the melt. Macromolecules 1988, 21, 3038–3051. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: Theory and experiment. Polymer 1997, 38, 3151–3212. [Google Scholar] [CrossRef]
- Yamazaki, S.; Hikosaka, M.; Toda, A.; Wataoka, I.; Gu, F. Role of entanglement in nucleation and ‘melt relaxation’ of polyethylene. Polymer 2002, 43, 6585–6593. [Google Scholar] [CrossRef]
- Haward, R.N. Strain hardening of thermoplastics. Macromolecules 1993, 26, 5860–5869. [Google Scholar] [CrossRef]
- ISO 1133; Plastics. Determination of the Melt Mass-Flow Rate (MFR) and Melt Volume-Flow Rate (MVR) of Thermoplastics Part 1: Standard Method. International Organization for Standardization: Vernier (Geneva), Switzerland, 2022.
- Krajenta, J.; Pawlak, A.; Galeski, A. Deformation of Disentangled Polypropylene Crystalline Grains into Nanofibers. J. Polym. Sci. B Polym. Phys. 2016, 54, 1983–1994. [Google Scholar] [CrossRef]
- Krigbaum, W.R.; Uematsu, I. Heat and entropy of fusion of isotactic polypropylene. J. Polym. Sci.-Part A Gen. Pap. 1965, 3, 767–776. [Google Scholar] [CrossRef]
- Wunderlich, B.; Czornyj, G. A Study of Equilibrium Melting of Polyethylene. Macromolecules 1977, 10, 906–913. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Material | Composition [wt.%] |
|---|---|---|
| PPe | PP, initial, equilibrium entangled | - |
| PP1 | PP, partially disentangled, obtained from 1 wt.% xylene solution | - |
| PP05 | PP, partially disentangled, obtained from 0.5 wt.% xylene solution | - |
| PE | HDPE, equilibrium entangled | - |
| PPe/PE (76:19) | Blend of PPe, PE, and EOC | 76:19:5 |
| PPe/PE (19:76) | Blend of PPe, PE, and EOC | 19:76:5 |
| PP1/PE (76:19) | Blend of PP1, PE, and EOC | 76:19:5 |
| PP1/PE (19:76) | Blend of PP1, PE, and EOC | 19:76:5 |
| PP05/PE (76:19) | Blend of PP05, PE, and EOC | 76:19:5 |
| PP05/PE (19:76) | Blend of PP05, PE, and EOC | 19:76:5 |
| Sample | Polypropylene | Polyethylene | ||||||
|---|---|---|---|---|---|---|---|---|
| T = 137 °C | T = 135 °C | T = 125 °C | T = 123 °C | |||||
| Hc [J/g] | Cr [%] | Hc [J/g] | Cr [%] | Hc [J/g] | Cr [%] | Hc [J/g] | Cr [%] | |
| PE | - | - | 187 | 64 | ||||
| PPe | 100 | 48 | - | - | ||||
| PP1 | 100 | 48 | - | - | ||||
| PP05 | 104 | 50 | - | - | ||||
| PPe/PE (76:19) | 94 | 45 | 96 | 46 | 115 | 36 | ||
| PP1/PE (76:19) | 102 | 49 | 97 | 46 | 84 | 39 | ||
| PP05/PE(76:19) | 99 | 47 | 97 | 46 | 63 | 43 | ||
| PPe/PE (19:76) | It did not crystallize | It did not crystallize | 206 | 70 | 210 | 72 | ||
| PP1/PE (19:76) | It did not crystallize | It did not crystallize | 209 | 71 | 209 | 72 | ||
| PP05/PE (19:76) | It did not crystallize | It did not crystallize | 214 | 73 | 208 | 71 | ||
| Material | Growth Rate [µm/min] | Density of Nucleation [Nuclei/m3] |
|---|---|---|
| PPe | 3.90 | 2.01 × 1013 |
| PP1 | 4.83 | 1.85 × 1013 |
| PP0.5 | 5.40 | 1.63 × 1013 |
| PPe/PE (76:19) | 3.43 | 3.24 × 1013 |
| PP1/PE (76:19) | 4.41 | 2.75 × 1013 |
| PP05/PE (76:19) | 4.91 | 2.09 × 1013 |
| Material | σy [MPa] | εy [%] | εb [%] |
|---|---|---|---|
| PPe/PE (76:19) | 14.8 ± 0.3 | 19 ± 1 | 217 ± 91 |
| PP1/PE (76:19) | 16.2 ± 0.4 | 18 ± 1 | 128 ± 66 |
| PP05/PE (76:19) | 15.8 ± 0.3 | 21 ± 1 | 173 ± 74 |
| PPe/PE (19:76) | 10.6 ± 0.4 | 25 ± 1 | 108 ± 62 |
| PP1/PE (19:76) | 11.3 ± 0.4 | 26 ± 1 | 130 ± 36 |
| PP05/PE (19:76) | 11.4 ± 0.3 | 27 ± 1 | 259 ± 101 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krajenta, J.; Lipinska, M.; Pawlak, A. Influence of Partial Disentanglement of Macromolecules on the Rheological, Thermal, and Mechanical Properties of Polypropylene–Polyethylene Blends. Molecules 2025, 30, 1786. https://doi.org/10.3390/molecules30081786
Krajenta J, Lipinska M, Pawlak A. Influence of Partial Disentanglement of Macromolecules on the Rheological, Thermal, and Mechanical Properties of Polypropylene–Polyethylene Blends. Molecules. 2025; 30(8):1786. https://doi.org/10.3390/molecules30081786
Chicago/Turabian StyleKrajenta, Justyna, Magdalena Lipinska, and Andrzej Pawlak. 2025. "Influence of Partial Disentanglement of Macromolecules on the Rheological, Thermal, and Mechanical Properties of Polypropylene–Polyethylene Blends" Molecules 30, no. 8: 1786. https://doi.org/10.3390/molecules30081786
APA StyleKrajenta, J., Lipinska, M., & Pawlak, A. (2025). Influence of Partial Disentanglement of Macromolecules on the Rheological, Thermal, and Mechanical Properties of Polypropylene–Polyethylene Blends. Molecules, 30(8), 1786. https://doi.org/10.3390/molecules30081786