
Synthesis and Electrochemiluminescence of a Di-Boron Thermally Activated Delayed Fluorescence Emitter


Abstract

1. Introduction
2. Results
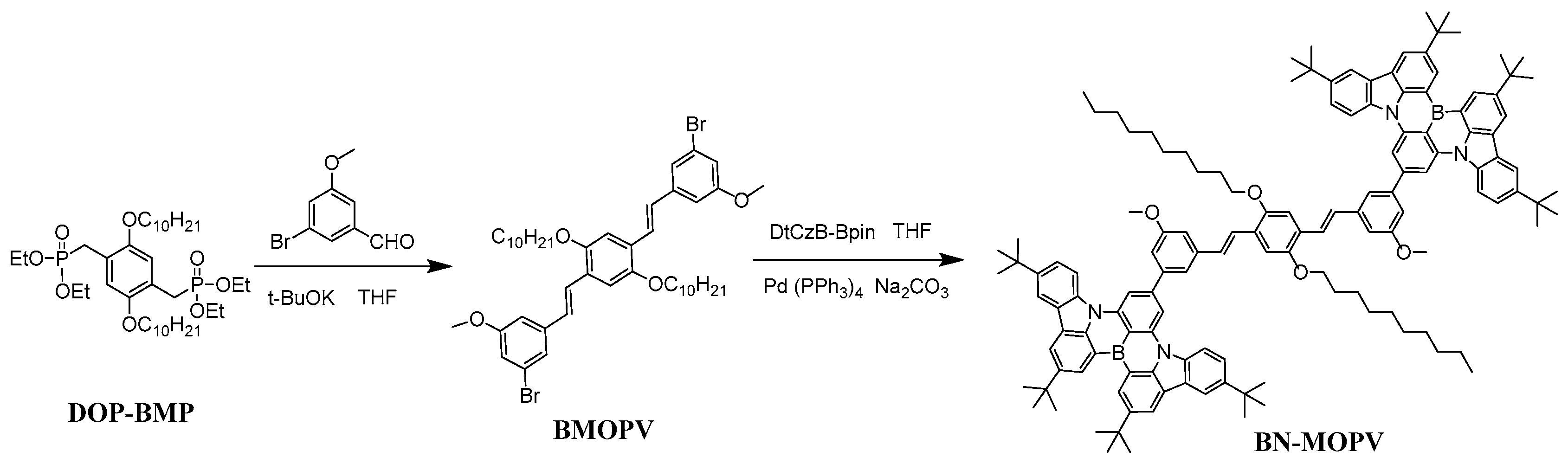
2.1. Design and Synthesis of a Diboron-Embedded Emitter
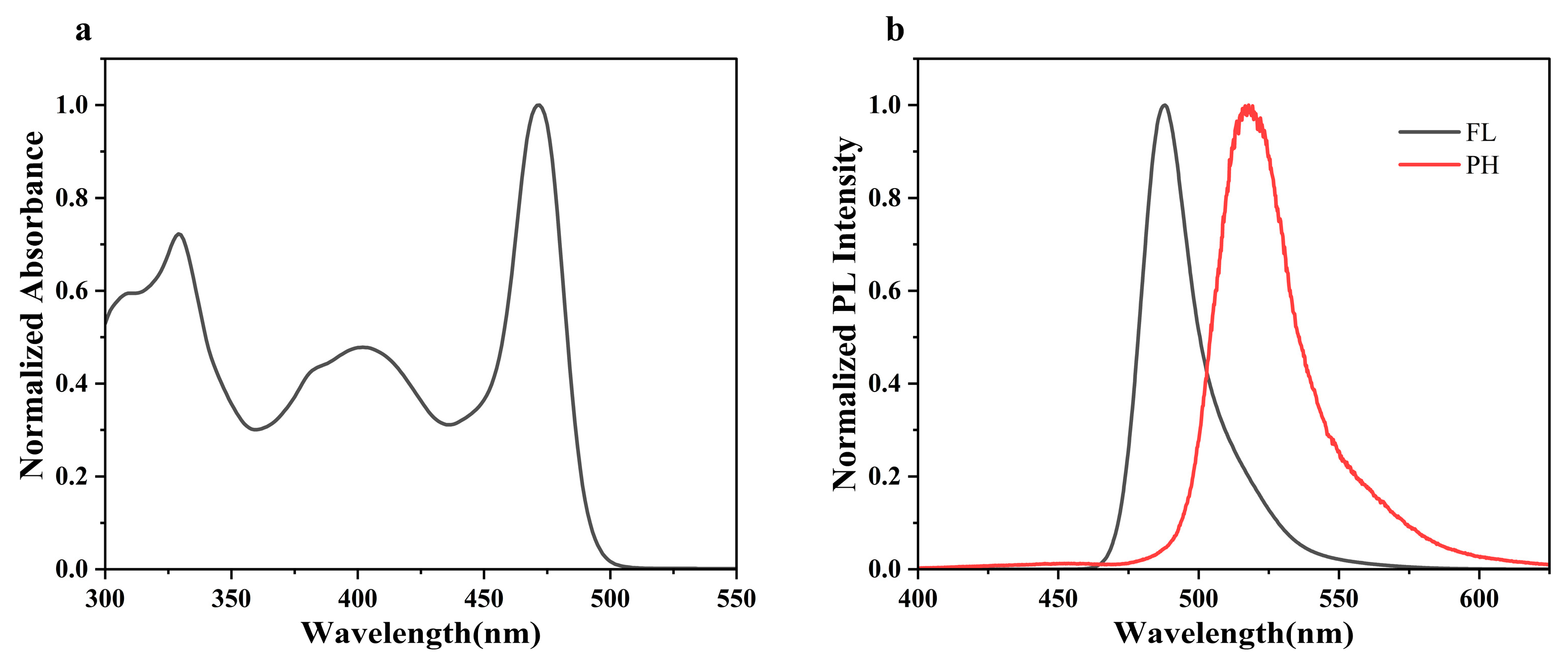
2.2. Photophysical Properties
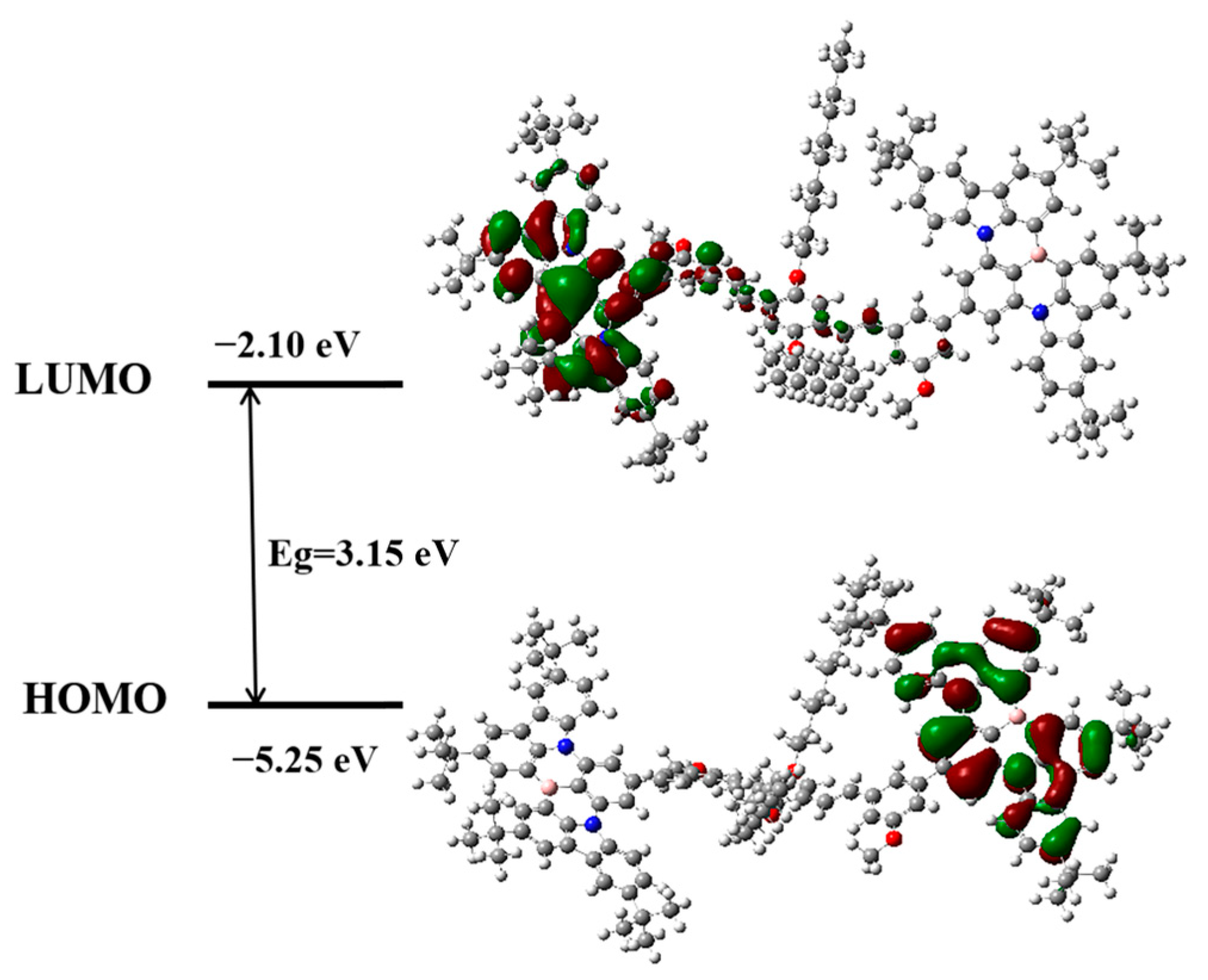
2.3. Theoretical Calculation
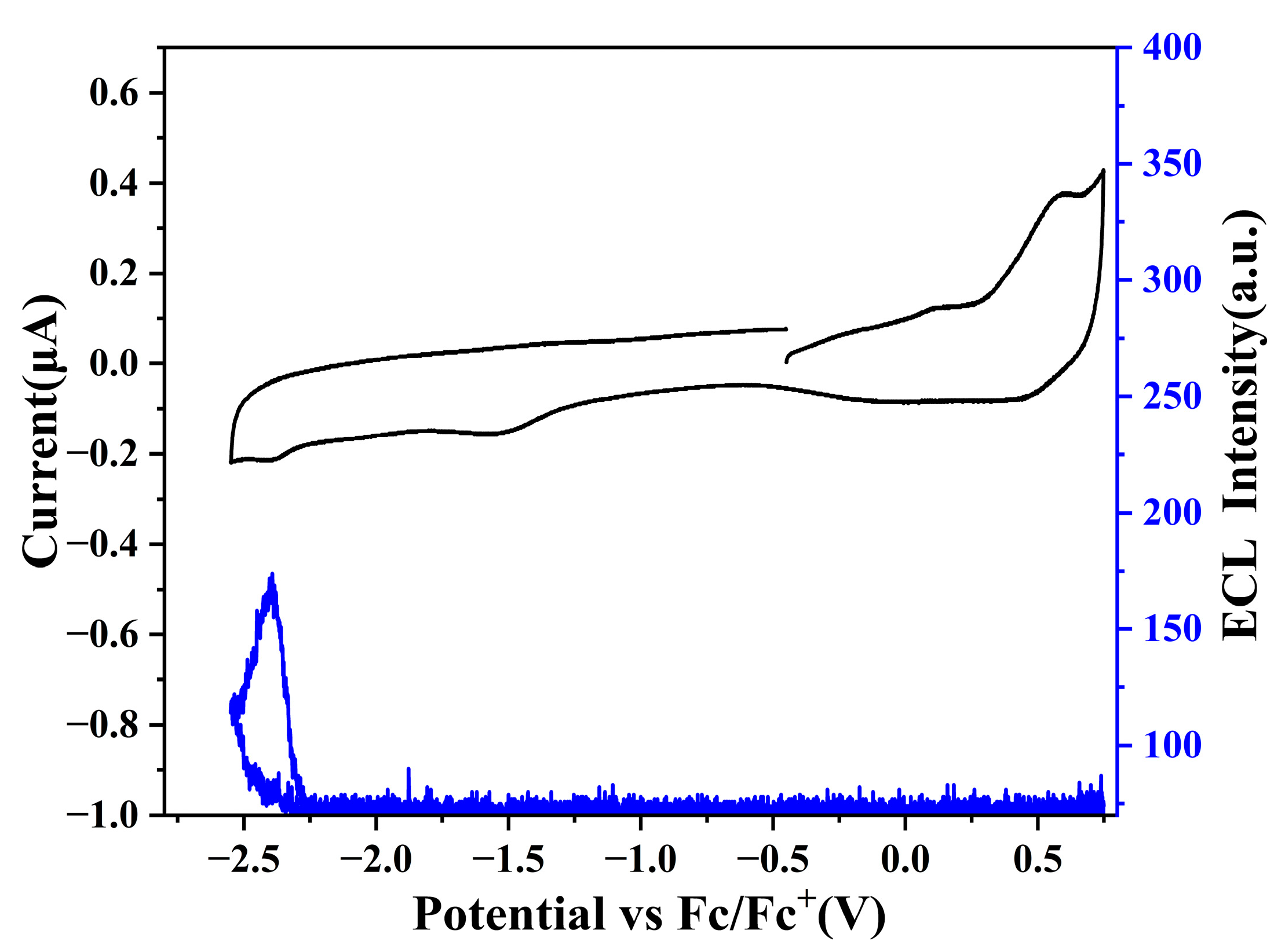
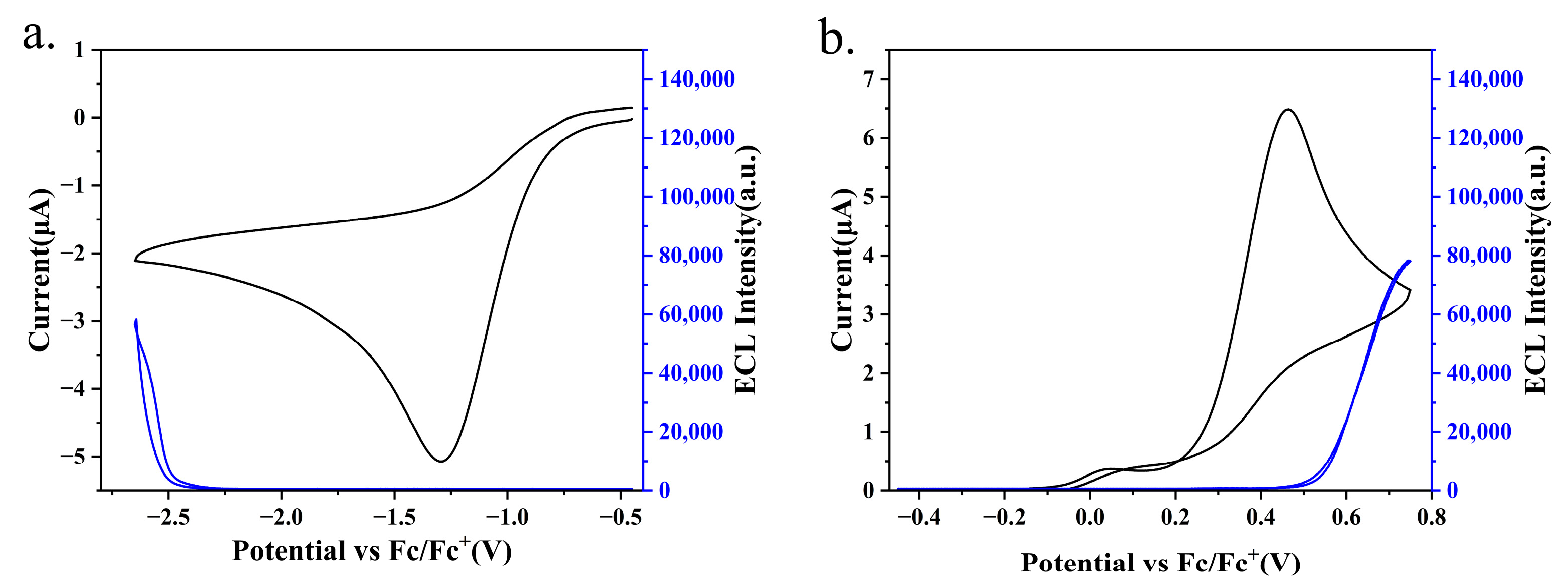
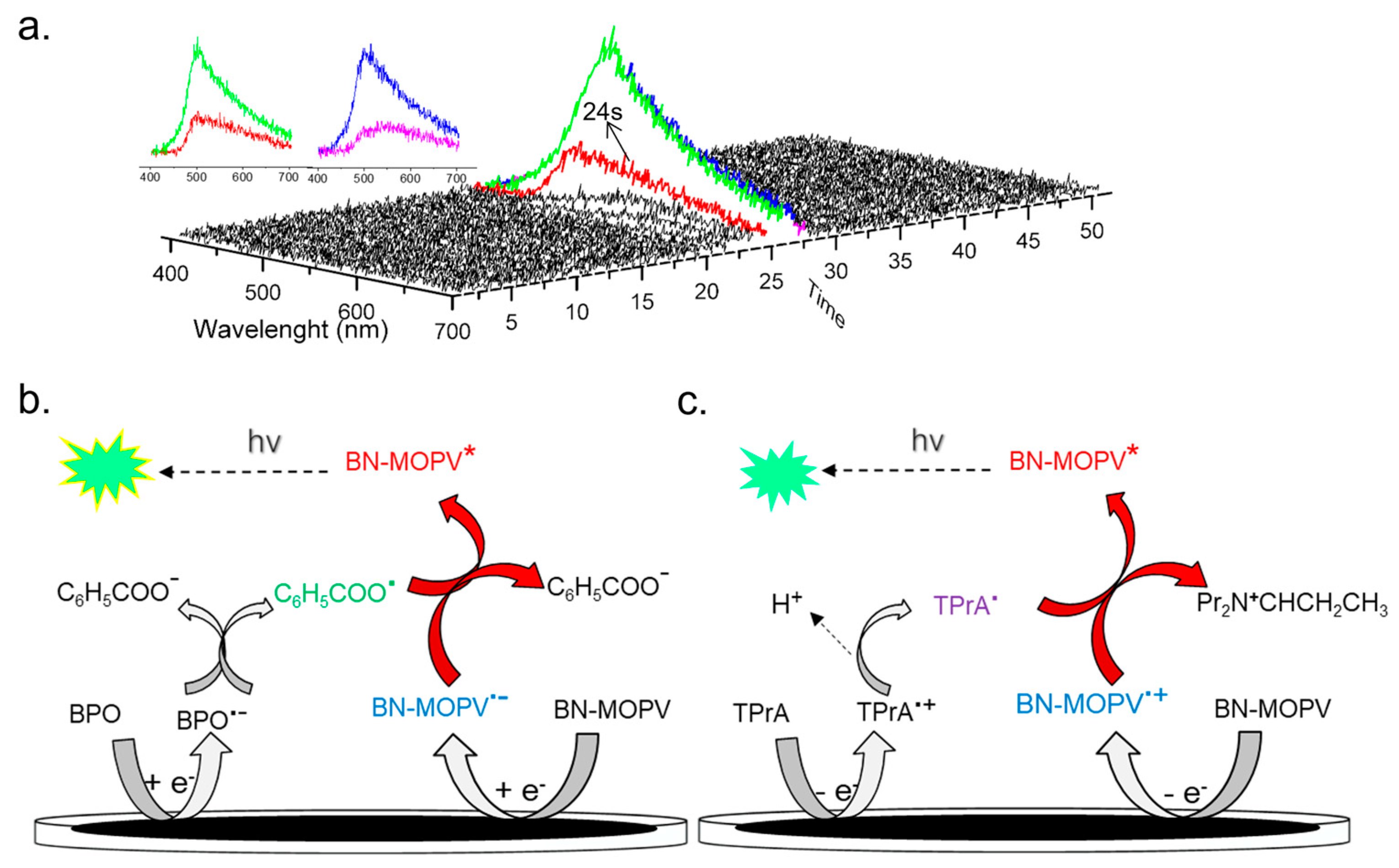
2.4. Electrochemistry and Annihilation ECL
2.5. ECL of Co-Reactant System
3. Materials and Methods
3.1. Synthesis of a Diboron-Embedded Emitter
- Synthesis of compound BMOPV
- Synthesis of compound BN-MOPV
3.2. Determination of Photophysical Properties
3.3. DFT Calculations
3.4. Electrochemistry and ECL Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.; Lei, P.; Feng, Y.; Fu, S.; Liu, X.; Zhang, S.; Tu, B.; Chen, C.; Li, Y.; Wang, L.; et al. Topologically engineering of π-conjugated macrocycles: Tunable emission and photochemical reaction toward multi-cyclic polymers. Chin. Chem. Lett. 2024, 35. [Google Scholar] [CrossRef]
- Fu, H.; Xu, Z.; Hou, H.; Luo, R.; Ju, H.; Lei, J. Framework-Enhanced Electrochemiluminescence in Biosensing. Chemosensors 2023, 11, 422. [Google Scholar] [CrossRef]
- Qin, X.; Zhan, Z.; Ding, Z. Progress in electrochemiluminescence biosensors based on organic framework emitters. Curr. Opin. Electrochem. 2023, 39, 101283. [Google Scholar] [CrossRef]
- Yang, L.; Li, J. Recent Advances in Electrochemiluminescence Emitters for Biosensing and Imaging of Protein Biomarkers. Chemosensors 2023, 11, 432. [Google Scholar] [CrossRef]
- Meier, S.B.; Tordera, D.; Pertegás, A.; Roldán-Carmona, C.; Ortí, E.; Bolink, H.J. Light-emitting electrochemical cells: Recent progress and future prospects. Mater. Today 2014, 17, 217–223. [Google Scholar] [CrossRef]
- Yang, Z.; Su, H. Recent Advances in Optical Engineering of Light-Emitting Electrochemical Cells. Adv. Funct. Mater. 2020, 30, 1906788. [Google Scholar] [CrossRef]
- Wang, C.; Wu, J.; Huang, H.; Xu, Q.; Ju, H. Electrochemiluminescence of Polymer Dots Featuring Thermally Activated Delayed Fluorescence for Sensitive DNA Methylation Detection. Anal. Chem. 2022, 94, 15695–15702. [Google Scholar] [CrossRef]
- Fang, J.; Dai, L.; Ren, X.; Wu, D.; Cao, W.; Wei, Q.; Ma, H. Protein-driven interaction enhanced electrochemiluminescence biosensor of hydrogen-bonded biohybrid organic frameworks for sensitive immunoassay of disease markers. Biosens. Bioelectron. 2024, 266, 116726. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Lu, Y.; Xu, Y.; Chen, F.; Yang, J.; Chen, Y.; Feng, J. Direct imaging of single-molecule electrochemical reactions in solution. Natur 2021, 596, 244–249. [Google Scholar] [CrossRef]
- Wu, Y.; Qin, D.; Yang, J.; Hu, S.; Xu, L.; Deng, B. Emitter-embedded metal–organic framework combined with molecularly-imprinted electrochemiluminescence microscopy for drug imaging detection. Microchem. J. 2024, 207. [Google Scholar] [CrossRef]
- Gao, H.; Shi, S.; Wang, S.; Tao, Q.; Ma, H.; Hu, J.; Chen, H.; Xu, J. Aggregation-induced delayed electrochemiluminescence of organic dots in aqueous media. Aggregate 2023, 5, e394. [Google Scholar] [CrossRef]
- Chu, K.; Ding, Z.; Zysman-Colman, E. Materials for Electrochemiluminescence: TADF, Hydrogen-Bonding, and Aggregation- and Crystallization-Induced Emission Luminophores. Chem. A Eur. J. 2023, 29, e202301504. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Niu, Y.; Yang, Y.; Peng, H.; Zhang, J.; Yao, C. Towards efficient blue aggregation-induced emission and delayed fluorescence molecules by locking the skeleton of indolocarbazole derivatives for non-doped OLEDs. Mater. Today Chem. 2024, 40. [Google Scholar] [CrossRef]
- Wong, J.M.; Zhang, R.; Xie, P.; Yang, L.; Zhang, M.; Zhou, R.; Wang, R.; Shen, Y.; Yang, B.; Wang, H.; et al. Revealing Crystallization-Induced Blue-Shift Emission of a Di-Boron Complex by Enhanced Photoluminescence and Electrochemiluminescence. Angew. Chem. Int. Ed. Engl. 2020, 59, 17461–17466. [Google Scholar] [CrossRef]
- Alberoni, C.; Pavan, G.; Scattolin, T.; Aliprandi, A. Critical Aspects and Challenges in the Design of Small Molecules for Electrochemiluminescence (ECL) Application. Chempluschem 2024, 89, e202400142. [Google Scholar] [CrossRef]
- Wang, X.; Wan, R.; Tang, Y.; Sun, S.; Chen, H.; Li, L.; Chen, J.; Wei, J.; Chi, Z.; Li, H. Aggregation-induced emission materials-based Electrochemiluminescence emitters for sensing applications: Progress, challenges and perspectives. Co-Ord. Chem. Rev. 2025, 531, 216520. [Google Scholar] [CrossRef]
- Huang, P.; Zou, X.; Xu, Z.; Lan, Y.; Chen, L.; Zhang, B.; Niu, L. Studies on Annihilation and Coreactant Electrochemiluminescence of Thermally Activated Delayed Fluorescent Molecules in Organic Medium. Molecules 2022, 27, 7457. [Google Scholar] [CrossRef]
- Fiorani, A.; Difonzo, M.; Rizzo, F.; Valenti, G. Versatile electrochemiluminescent organic emitters. Curr. Opin. Electrochem. 2022, 34, 100998. [Google Scholar] [CrossRef]
- Adachi, C. Third-generation organic electroluminescence materials. Jpn. J. Appl. Phys. 2014, 53, 060101. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Ren, Z.; Yan, S.; Bryce, M.R. All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 2018, 3, 18020. [Google Scholar] [CrossRef]
- Wei, J.; Yang, N.; Li, F.; Cai, S.; Zhang, B.; Cai, Z. Direct Comparative Studies Revealing the Contribution of TADF Activity of Organic Emitters Towards Efficient Electrochemiluminescence. Chem. A Eur. J. 2024, 30, e202401036. [Google Scholar] [CrossRef]
- Liu, X.; Cai, Z.; Wang, Q. Thermally Activated Delayed Fluorescence Emitters for Efficient Electrochemiluminescence. ChemPhotoChem 2024, 8, e202400221. [Google Scholar] [CrossRef]
- Zou, X.; Zeng, Z.; Zhao, B.; Yue, Y.; Xu, Z.; Zhang, Y.; Fu, Y.; Xie, Z.; Niu, L.; Zhang, B. Thermally Activated Delayed Fluorescent Polymer Dots for Electrochemiluminescent Sensing of Cu2+ Ions. ACS Appl. Polym. Mater. 2023, 5, 10116–10126. [Google Scholar] [CrossRef]
- Ishimatsu, R.; Matsunami, S.; Kasahara, T.; Mizuno, J.; Edura, T.; Adachi, C.; Nakano, K.; Imato, T. Electrogenerated Chemiluminescence of Donor–Acceptor Molecules with Thermally Activated Delayed Fluorescence. Angew. Chem. Int. Ed. Engl. 2014, 53, 6993–6996. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhang, B.; Hu, Q.; Zhao, B.; Zhu, Y.; Zhang, Y.; Kong, Y.; Zeng, Z.; Bao, Y.; Wang, W.; et al. Polymer Electrochemiluminescence Featuring Thermally Activated Delayed Fluorescence. Chemphyschem 2021, 22, 726–732. [Google Scholar] [CrossRef]
- Yang, M.; Park, I.S.; Yasuda, T. Full-Color, Narrowband, and High-Efficiency Electroluminescence from Boron and Carbazole Embedded Polycyclic Heteroaromatics. J. Am. Chem. Soc. 2020, 142, 19468–19472. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, J.; Zhang, D.; Yin, C.; Li, G.; Liu, Z.; Jia, X.; Qiao, J.; Duan, L. Sterically Wrapped Multiple Resonance Fluorophors for Suppression of Concentration Quenching and Spectrum Broadening. Angew. Chem. Int. Ed. Engl. 2021, 61, e202113206. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.H.; Urban, V.S.; Littrell, K.C.; Thiyagarajan, P.; Yu, L. Syntheses of Amphiphilic Diblock Copolymers Containing a Conjugated Block and Their Self-Assembling Properties. J. Am. Chem. Soc. 2000, 122, 6855–6861. [Google Scholar] [CrossRef]
- Fang, L.; Hu, Y.; Li, Q.; Xu, S.; Dhinakarank, M.K.; Gong, W.; Ning, G. Fluorescent cross-linked supramolecular polymers constructed from a novel self-complementary AABB-type heteromultitopic monomer. Org. Biomol. Chem. 2016, 14, 4039–4045. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Shiren, K.; Nakajima, K.; Nomura, S.; Nakatsuka, S.; Kinoshita, K.; Ni, J.; Ono, Y.; Ikuta, T. Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO–LUMO Separation by the Multiple Resonance Effect. Adv. Mater. 2016, 28, 2777–2781. [Google Scholar] [CrossRef]
- Qiu, Y.; Xia, H.; Miao, J.; Huang, Z.; Li, N.; Cao, X.; Han, J.; Zhou, C.; Zhong, C.; Yang, C. Narrowing the Electroluminescence Spectra of Multiresonance Emitters for High-Performance Blue OLEDs by a Peripheral Decoration Strategy. ACS Appl. Mater. Interfaces 2021, 13, 59035–59042. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, I.L.K.; Bouffier, L.; Sojic, N.; Kumar, S.S. Enhanced Electrochemiluminescence by Knocking Out Gold Active Sites. Angew. Chem. Int. Ed. Engl. 2024, 64, e202421185. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Abs a [nm] | λFL b [nm] | λPH c [nm] | FWHM d [nm] | ΦPL e [%] | Eg f [eV] | ES1 g [eV] | ET1 h [eV] | ΔEST [eV] |
|---|---|---|---|---|---|---|---|---|---|
| BN-MOPV | 328,400,472 | 493 | 517 | 22 | 67(88) | 2.77 | 2.58 | 2.38 | 0.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Cheng, J.; Wang, H. Synthesis and Electrochemiluminescence of a Di-Boron Thermally Activated Delayed Fluorescence Emitter. Molecules 2025, 30, 1718. https://doi.org/10.3390/molecules30081718
Zhou X, Cheng J, Wang H. Synthesis and Electrochemiluminescence of a Di-Boron Thermally Activated Delayed Fluorescence Emitter. Molecules. 2025; 30(8):1718. https://doi.org/10.3390/molecules30081718
Chicago/Turabian StyleZhou, Xiaojie, Jun Cheng, and Hongbo Wang. 2025. "Synthesis and Electrochemiluminescence of a Di-Boron Thermally Activated Delayed Fluorescence Emitter" Molecules 30, no. 8: 1718. https://doi.org/10.3390/molecules30081718
APA StyleZhou, X., Cheng, J., & Wang, H. (2025). Synthesis and Electrochemiluminescence of a Di-Boron Thermally Activated Delayed Fluorescence Emitter. Molecules, 30(8), 1718. https://doi.org/10.3390/molecules30081718