



General Synthesis of 2-Substituted Benzoxazoles Based on Tf2O-Promoted Electrophilic Activation of Tertiary Amides


Abstract
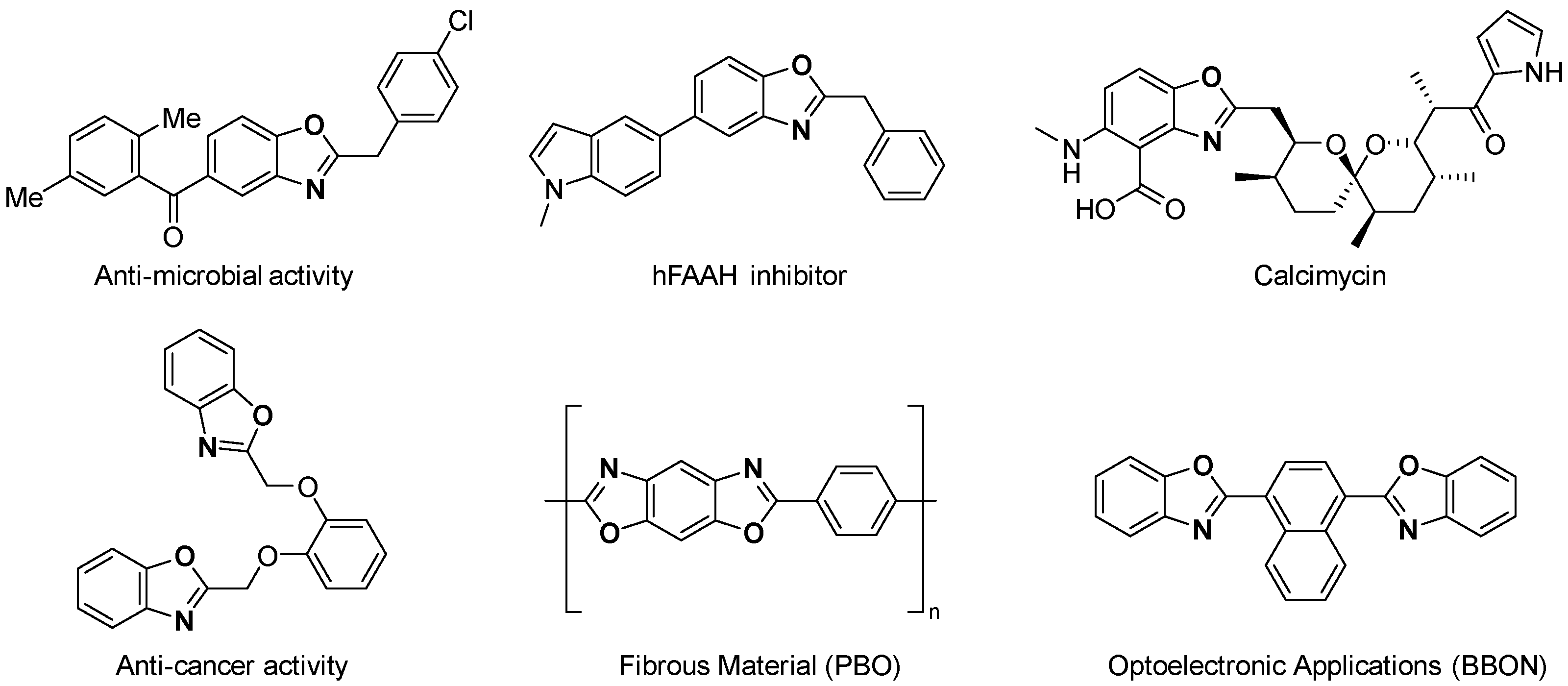
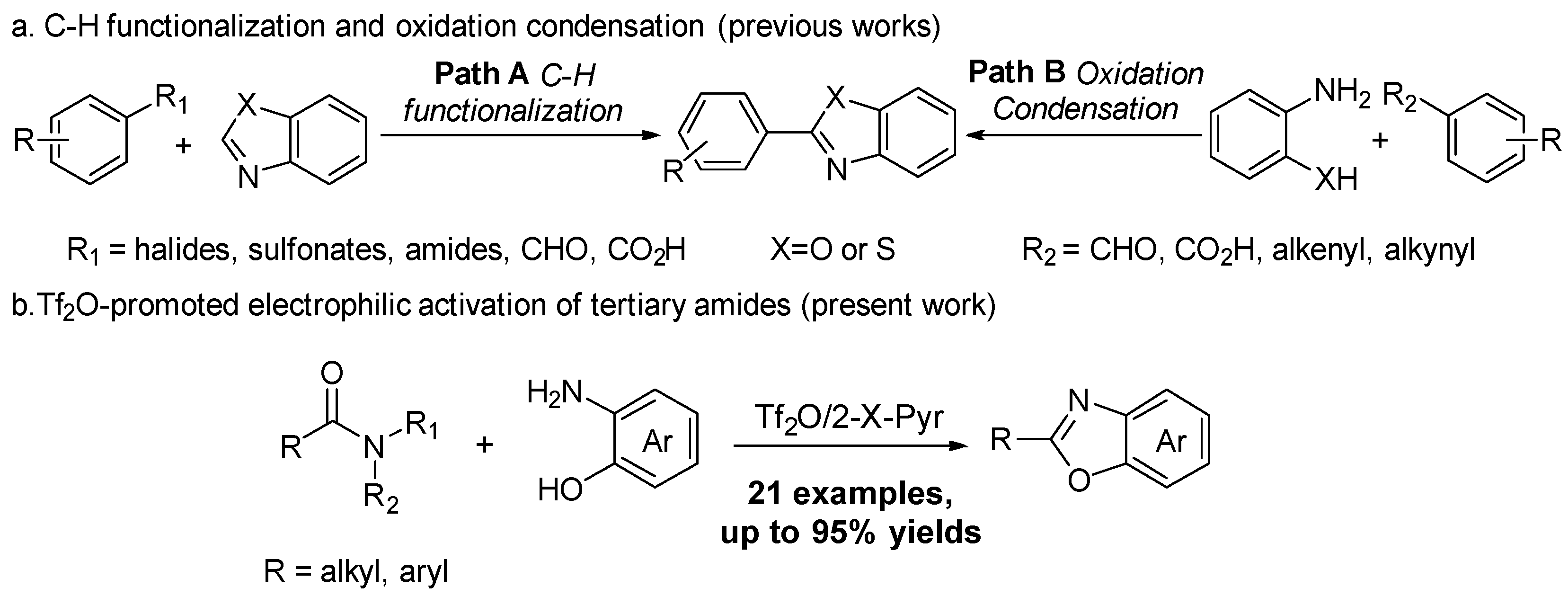
:1. Introduction
2. Results and Discussion
2.1. Synthesis
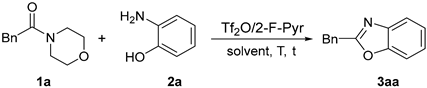
2.1.1. Optimization of Reaction Conditions
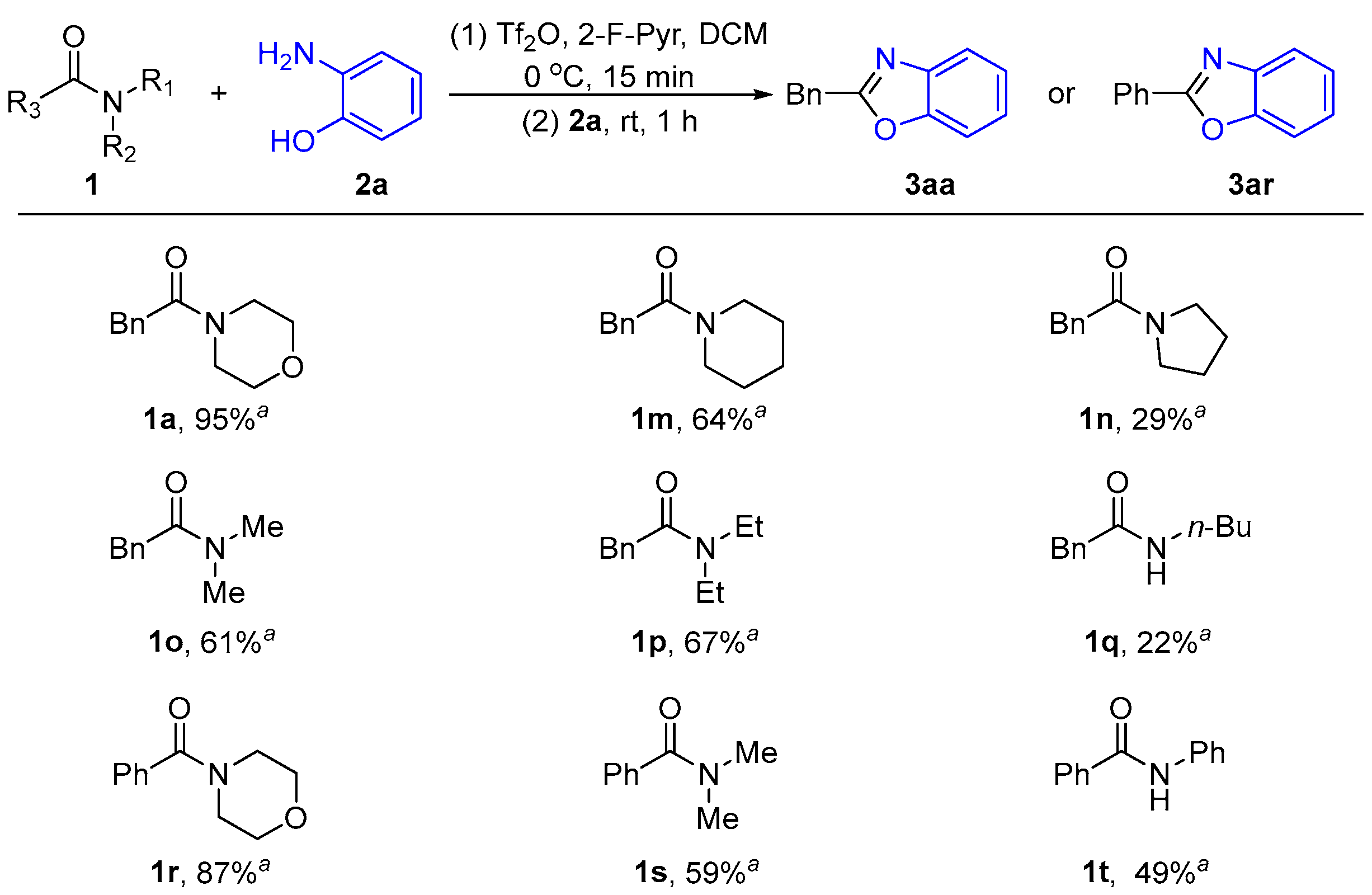
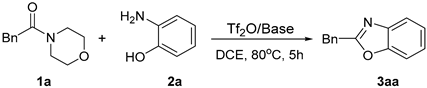
2.1.2. The Effect of Different Amide N-Leaving Groups on the Reaction
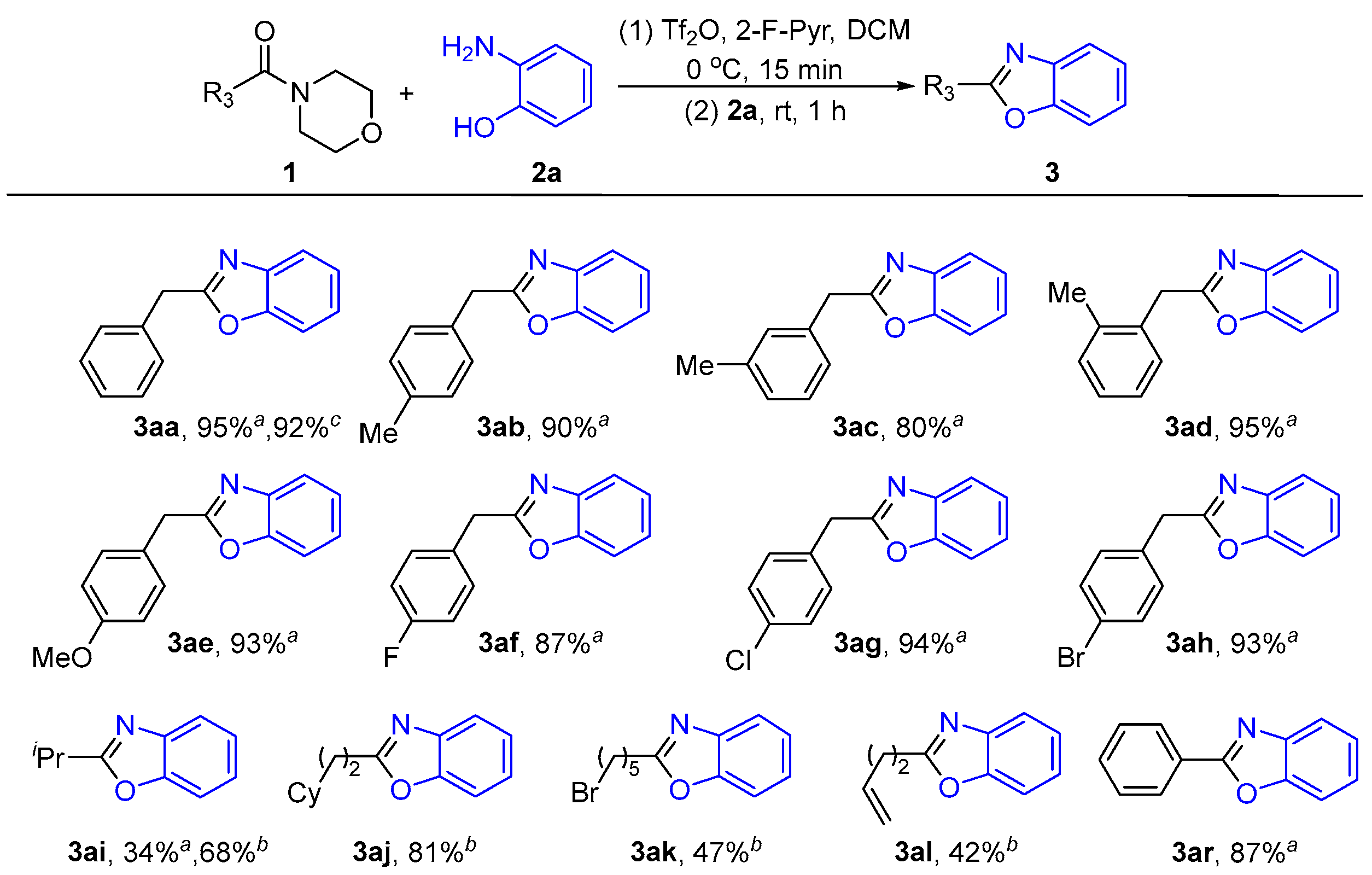
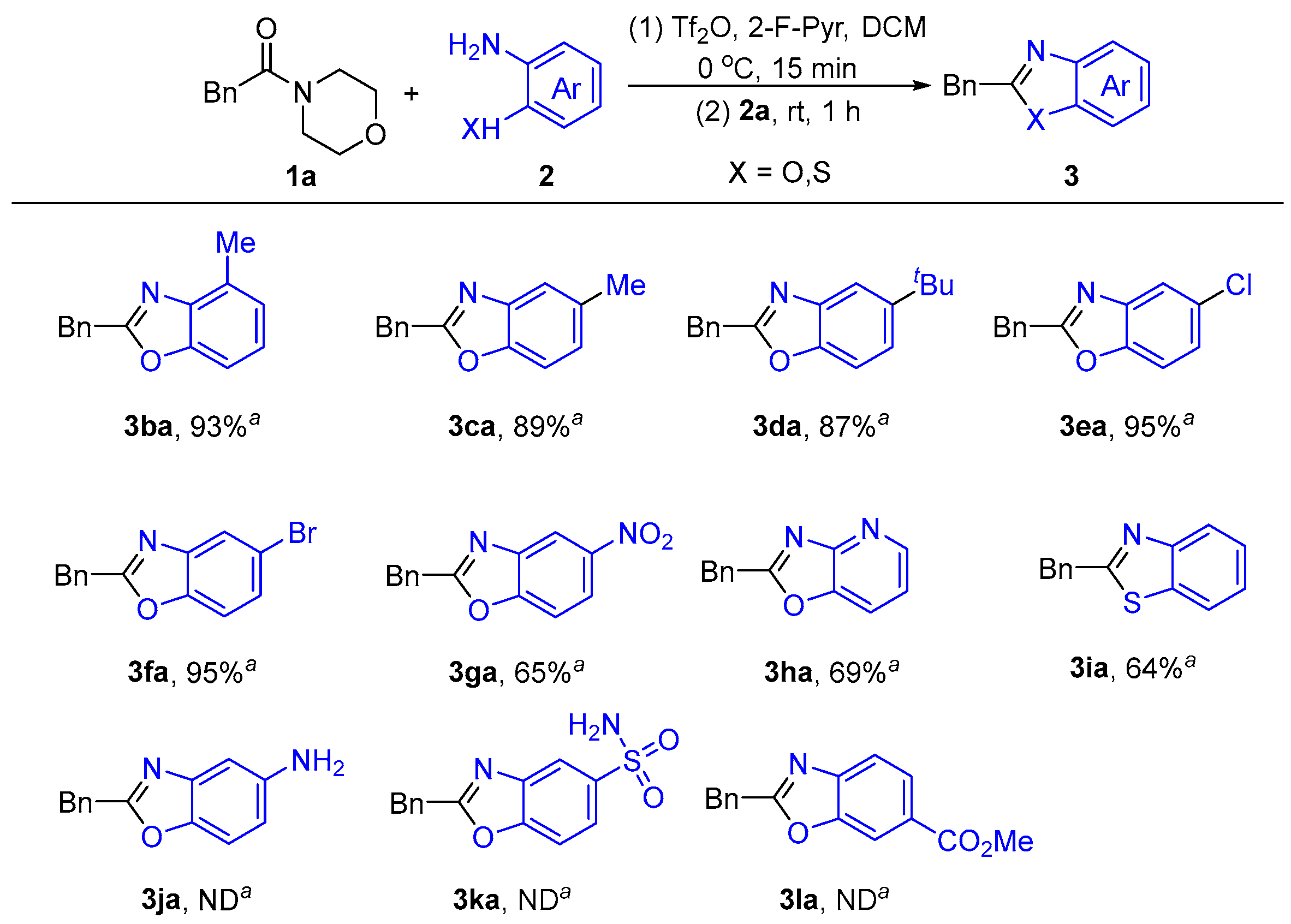
2.1.3. Substrates’ Scope Under Optimized Conditions
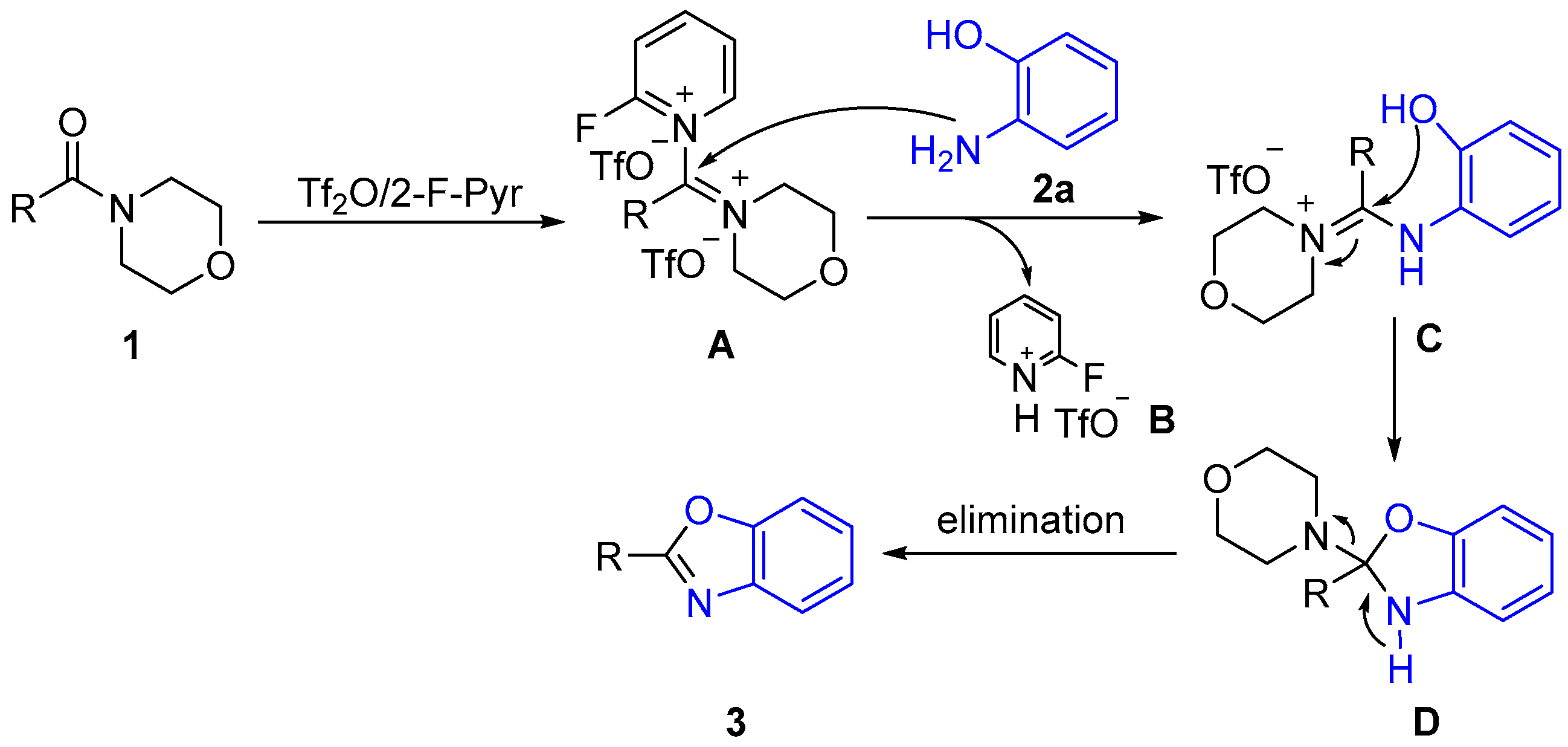
2.2. Proposed Mechanism
3. Materials and Methods
3.1. Materials and General Methods
3.2. General Procedure for the Preparation of 2-Substituted Benzoxazoles 3
3.3. Analytical Data of Compound 3aa–3ar and 3ba–ia
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, S.; Veeraswamy, G.; Bhattarai, D.; Goo, J.-I.; Lee, K.; Choi, Y. Recent Advances in the Development of Pharmacologically Active Compounds that Contain a Benzoxazole Scaffold. Asian J. Org. Chem. 2015, 4, 1338–1361. [Google Scholar] [CrossRef]
- Arulmurugan, S.; Kavitha, H.P.; Vennila, J.P. Review on the Synthetic Methods of Biologically Potent Benzoxazole Derivatives. MINI-Rev. Org. Chem. 2021, 18, 769–785. [Google Scholar] [CrossRef]
- Wong, X.K.; Yeong, K.Y. A Patent Review on the Current Developments of Benzoxazoles in Drug Discovery. ChemMedChem 2021, 16, 3237–3262. [Google Scholar] [CrossRef]
- Di Martino, S.; De Rosa, M. The Benzoxazole Heterocycle: A Comprehensive Review of the Most Recent Medicinal Chemistry Developments of Antiproliferative, Brain-Penetrant, and Anti-inflammatory Agents. Top. Curr. Chem. 2024, 382, 33. [Google Scholar] [CrossRef]
- Arakawa, K.; Inamasu, M.; Matsumoto, M.; Okumura, K.; Yasuda, K.; Akatsuka, H.; Kawanami, S.; Watanabe, A.; Homma, K.; Saiga, Y.; et al. Novel Benzoxazole 2,4-Thiazolidinediones as Potent Hypoglycemic Agents. Synthesis and Structure-Activity Relationships. Chem. Pharm. Bull. 1997, 45, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Tekiner-Gulbas, B.; Temiz-Arpaci, O.; Yildiz, I.; Altanlar, N. Synthesis and in Vitro Antimicrobial Activity of New 2-[p-substituted-benzyl]-5-[substituted-carbonylamino]benzoxazoles. Eur. J. Med. Chem. 2007, 42, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Oksuzoglu, E.; Temiz-Arpaci, O.; Tekiner-Gulbas, B.; Eroglu, H.; Sen, G.; Alper, S.; Yildiz, I.; Diril, N.; Aki-Sener, E.; Yalcin, I. A Study on the Genotoxic Activities of Some New Benzoxazoles. Med. Chem. Res. 2008, 16, 1–14. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, X.; Dou, W.; Zhang, H.; Liu, W.; Wang, C.; Zheng, J. Synthesis and Characterization of the Ligand Based on Benzoxazole and its Transition Metal Complexes: DNA-binding and Antitumor Activity. J. Inorg. Biochem. 2010, 104, 583–591. [Google Scholar] [CrossRef]
- Chancellor, D.R.; Davies, K.E.; De Moor, O.; Dorgan, C.R.; Johnson, P.D.; Lambert, A.G.; Lawrence, D.; Lecci, C.; Maillol, C.; Middleton, P.J.; et al. Discovery of 2-Arylbenzoxazoles as Upregulators of Utrophin Production for the Treatment of Duchenne Muscular Dystrophy. Med. Chem. 2011, 54, 3241–3250. [Google Scholar] [CrossRef]
- Sattar, R.; Mukhtar, R.; Atif, M.; Hasnain, M.; Irfan, A. Synthetic Transformations and Biological Screening of Benzoxazole Derivatives: A Review. J. Heterocycl. Chem. 2020, 57, 2079–2107. [Google Scholar] [CrossRef]
- Demmer, C.S.; Bunch, L. Benzoxazoles and Oxazolopyridines in Medicinal Chemistry Studies. Eur. J. Med. Chem. 2015, 97, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Rubner, R. Innovation via Photosensitive Polyimide and Poly(benzoxazole) Precursors—A Review by Inventor. J. Sci. Technol. 2004, 17, 685–691. [Google Scholar] [CrossRef]
- Kuroyanagi, J.I.; Kanai, K.; Sugimoto, Y.; Horiuchi, T.; Achiwa, I.; Takeshita, H.; Kawakami, K. 1,3-Benzoxazole-4-Carbonitrile as a Novel Antifungal Scaffold of β-1,6-glucan Synthesis Inhibitors. Bioorg. Med. Chem. 2010, 18, 7593–7606. [Google Scholar] [CrossRef]
- Estiarte, M.A.; Johnson, R.J.; Kaub, C.J.; Gowlugari, S.; O’Mahony, D.J.R.; Nguyen, M.T.; Emerling, D.E.; Kelly, M.G.; Kincaid, J.; Vincent, F.; et al. 2-Amino-5-arylbenzoxazole Derivatives as Potent Inhibitors of Fatty Acid Amide Hydrolase (FAAH). MedChemComm 2012, 3, 611–619. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, W.; Zhang, X.; Yin, L.; Chen, B.; Song, J. Synthesis and Pharmacological Evaluation of Piperidine (piperazine)-substituted Benzoxazole Derivatives as Multi-target Antipsychotics. Bioorg. Med. Chem. Lett. 2015, 25, 5299–5305. [Google Scholar] [CrossRef]
- Sałaga, M.; Sobczak, M.; Fichna, J. Inhibition of Fatty Acid Amide Hydrolase (FAAH) as a Novel Therapeutic Strategy in the Treatment of Pain and Inflammatory Diseases in the Gastrointestinal Tract. Eur. J. Pharm. Sci. 2014, 52, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ugale, V.G.; Bari, S.B. Quinazolines: New horizons in anticonvulsant therapy. Eur. J. Med. Chem. 2014, 80, 447–501. [Google Scholar] [CrossRef]
- Holmes, G.A.; Rice, K.; Snyder, C.R. Ballistic Fibers: A Review of the Thermal, Ultraviolet and Hydrolytic Stability of the Benzoxazole Ring Structure. J. Mater. Sci. 2006, 41, 4105–4116. [Google Scholar] [CrossRef]
- Peng, J.; Liu, Y.; Yang, J.; Chen, Z.; Wang, K.; Li, A. Multifunctional Elastic Benzoxazole Derivative Crystals for Advanced Optoelectronic Applications. Sci. China Mater. 2025, 68, 141–148. [Google Scholar] [CrossRef]
- Rajasekhar, S.; Maiti, B.; Chanda, K. A Decade Update on Benzoxazoles, a Privileged Scaffold in Synthetic Organic Chemistry. Synlett 2017, 28, 521–541. [Google Scholar] [CrossRef]
- Basak, S.; Dutta, S.; Maiti, D. Accessing C2-Functionalized 1,3-(Benz)azoles through Transition Metal-Catalyzed C–H Activation. Chem.-Eur. J. 2021, 27, 10533–10557. [Google Scholar] [CrossRef]
- El Alami, A.; El Maraghi, A.; Sdassi, H. Review of Synthesis Process of Benzoxazole and Benzothiazole Derivatives. Syn. Commun. 2024, 54, 769–801. [Google Scholar] [CrossRef]
- Mukai, T.; Hirano, K.; Satoh, T.; Miura, M. Palladium-Catalyzed Direct Benzylation of Azoles with Benzyl Carbonates. Org. Lett. 2010, 12, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, G.; Zhang, Y.; Wang, J. Copper-Catalyzed Direct Benzylation or Allylation of 1,3-Azoles with N-Tosylhydrazones. J. Am. Chem. Soc. 2011, 133, 3296–3299. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Huang, H.; Xie, Y.; Guo, S.; Xia, C. Palladium-Catalyzed Selective C-H Benzylation towards Functionalized Azoles with a Quaternary Carbon Center. Adv. Synth. Catal. 2012, 354, 1692–1700. [Google Scholar] [CrossRef]
- Niu, Z.J.; Li, L.H.; Liu, X.Y.; Liang, Y.M. Transition-Metal-Free Alkylation/Arylation of Benzoxazole via Tf2O-Activated-Amide. Adv. Synth. Catal. 2019, 361, 5217–5222. [Google Scholar] [CrossRef]
- Kumar, R.; Selvam, C.; Kaur, G.; Chakraborti, A.K. Microwave-Assisted Direct Synthesis of 2-Substituted Benzoxazoles from Carboxylic Acids under Catalyst and Solvent-Free Conditions. Synlett 2005, 2005, 1401–1404. [Google Scholar] [CrossRef]
- Mayo, M.S.; Yu, X.; Zhou, X.; Feng, X.; Yamamoto, Y.; Bao, M. Synthesis of Benzoxazoles from 2-Aminophenols and β-Diketones Using a Combined Catalyst of Brønsted Acid and Copper Iodide. J. Org. Chem. 2014, 79, 6310–6314. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Retailleau, P. Elemental Sulfur-Promoted Oxidative Rearranging Coupling between o-Aminophenols and Ketones: A Synthesis of 2-Alkyl benzoxazoles under Mild Conditions. Org. Lett. 2017, 19, 3887–3890. [Google Scholar] [CrossRef]
- Jiang, J.; Li, G.; Zhang, F.; Deng, G.-J. Aniline ortho C−H Sulfuration/Cyclization with Elemental Sulfur for Efficient Synthesis of 2-Substituted Benzothiazoles under Metal-Free Conditions. Adv. Synth. Catal. 2018, 360, 1622–1627. [Google Scholar] [CrossRef]
- Li, Z.; Dong, J.; Wang, J.; Yang, D.-Y.; Weng, Z. Elemental Sulfur-promoted One-pot Synthesis of 2-(2,2,2-trifluoroethyl)benzoxazoles and their Derivatives. Chem. Commun. 2019, 55, 13132–13135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, L.; Liu, Y.; Zhang, Y.; Chen, X.; Luo, Y.; Peng, Y.; Han, S.; Pan, B. Elemental Sulfur-Promoted Benzoxazole/Benzothiazole Formation Using a C=C Double Bond as a One-Carbon Donator. J. Org. Chem. 2021, 86, 14485–14492. [Google Scholar] [CrossRef]
- Chau, T.K.; Ho, N.T.; Ho, T.H.; Nguyen, A.T.; Nguyen, K.D.; Phan, N.T.S.; Le, H.V.; Nguyen, T.T. Synthesis of 2-benzyl Benzoxazoles and Benzothiazoles via Elemental Sulfur Promoted Cyclization of Styrenes with 2-nitrophenols and N,N-dialkyl-3-nitroanilines. Org. Biomol. Chem. 2024, 22, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Bauer, A.; Lemmerer, M.; Maulide, N. Amide Activation: An Emerging Tool for Chemoselective Synthesis. Chem. Soc. Rev. 2018, 47, 7899–7925. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, M.B.; Gnanaprakasam, B. Recent Advances in the Metal-Catalyzed Activation of Amide Bonds. Chem.-Asian J. 2019, 14, 76–93. [Google Scholar] [CrossRef]
- Feng, M.; Zhang, H.; Maulide, N. Challenges and Breakthroughs in Selective Amide Activation. Angew. Chem. Int. Ed. 2022, 61, e202212213. [Google Scholar] [CrossRef]
- Kumar, V.; Dhawan, S.; Bala, R.; Mohite, S.B.; Singh, P.; Karpoormath, R. Cu-Catalysed Transamidation of Unactivated Aliphatic Amides. Org. Biomol. Chem. 2022, 20, 6931–6940. [Google Scholar] [CrossRef]
- Kumar, V.; Dhawan, S.; Bala, R.; Girase, P.S.; Singh, P.; Karpoormath, R. Metal-Free Direct Annulation of 2-Aminophenols and 2-Aminothiophenols with Unactivated Amides through Transamidation: Access to Polysubstituted Benzoxazole and Benzothiazole Derivatives. Tetrahedron 2022, 115, 132794–132802. [Google Scholar] [CrossRef]
- Kaiser, D.; Teskey, C.J.; Adler, P.; Maulide, N. Chemoselective Intermolecular Cross-Enolate-Type Coupling of Amides. J. Am. Chem. Soc. 2017, 139, 16040–16043. [Google Scholar] [CrossRef]
- Fan, T.; Wang, A.; Li, J.Q.; Ye, J.L.; Zheng, X.; Huang, P.Q. Versatile One-Pot Synthesis of Polysubstituted Cyclopent-2-enimines from α,β-Unsaturated Amides: Imino-Nazarov Reaction. Angew. Chem. Int. Ed. 2018, 130, 10509–10513. [Google Scholar] [CrossRef]
- Li, J.; Berger, M.; Zawodny, W.; Simaan, M.; Maulide, N. A Chemoselective α-Oxytriflation Enables the Direct Asymmetric Arylation of Amides. Chem 2019, 5, 1883–1891. [Google Scholar] [CrossRef]
- Weng, Y.; Min, L.; Zeng, X.; Shan, L.; Wang, X.; Hu, Y. General Synthesis of α-Alkyl Ynones from Morpholine Amides and 1-Copper(I) Alkynes Promoted by Triflic Anhydride. Org. Lett. 2020, 22, 8296–8301. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Li, H.; Zheng, W.; Wang, X.; Wang, X.; Hu, Y. Tandem Synthesis of 2-Azaspiro [4.5]deca-1,6,9-trien-8-ones Based on Tf2O-Promoted Activation of N-(2-Propyn-1-yl) Amides. J. Org. Chem. 2022, 88, 525–533. [Google Scholar] [CrossRef]
- Tang, Z.; Yao, Z.; Yu, Y.; Huang, J.; Ma, X.; Zhao, X.; Chang, Z.; Zhao, D. Photoredox-Catalyzed [3+2] annulation of Aromatic Amides with Olefins via Iminium Intermediates. Angew. Chem. Int. Ed. 2024, 136, e202412152. [Google Scholar] [CrossRef]
- Shan, L.; Li, H.; Zheng, W.; Wang, X.; Wang, X.; Hu, Y. Mechanistic Insights into Tf2O-Promoted Electrophilic Activation of 2-Propynamides and a New Synthesis of 2,4-Disubstituted Quinolines. Org. Lett. 2022, 24, 8806–8811. [Google Scholar] [CrossRef]
- Ji, Y.Y.; Zhang, Y.; Hu, Y.Y.; Shao, L.X. N-heterocyclic Carbene-Pd(II)-1-methylimidazole Complex Catalyzed C–H Bond Benzylation of (Benzo) oxazoles with Benzyl Chlorides. Tetrahedron 2015, 71, 6818–6823. [Google Scholar] [CrossRef]
- Shang, R.; Yang, Z.W.; Wang, Y.; Zhang, S.L.; Liu, L. Palladium-Catalyzed Decarboxylative Couplings of 2-(2-Azaaryl) Acetates with Aryl Halides and Triflates. J. Am. Chem. Soc. 2010, 132, 14391–14393. [Google Scholar] [CrossRef]
- Evindar, G.; Batey, R.A. Parallel Synthesis of a Library of Benzoxazoles and Benzothiazoles using Ligand-accelerated Copper-Catalyzed Cyclizations of ortho-Halobenzanilides. J. Org. Chem. 2006, 71, 1802–1808. [Google Scholar] [CrossRef]
- Yi, J.; Lu, X.; Sun, Y.Y.; Xiao, B.; Liu, L. Nickel-Catalyzed Sonogashira Reactions of Non-Activated Secondary Alkyl bromides and Iodides. Angew. Chem. Int. Ed. 2013, 52, 12409–12413. [Google Scholar] [CrossRef]
- Bastug, G.; Eviolitte, C.; Marko, I.E. Functionalized Orthoesters as Powerful Building Blocks for the Efficient Preparation of Heteroaromatic Bicycles. Org. Lett. 2012, 14, 3502–3505. [Google Scholar] [CrossRef]
- Wynne, G.M.; Wren, S.P.; Johnson, P.D.; Price, D.; De Moor, O.; Nugent, G.; Tinsley, J.M.; Storer, R.; Mulvaney, A.; Pye, R.J.; et al. Preparation of Benzimidazoles, Benzoxazoles, Benzothiazoles, Indoles and their Analogs for the Treatment of Muscular Dystrophy and Cachexia. Appl. WO 2007, 2007091106, A2. [Google Scholar]
- Shen, T.Y.; Clark, R.L.; Pessolano, A.A.; Witzel, B.E.; Lanza, T.J. Anti-Inflammatory Oxazole (4,5-B) Pyridines. U.S. Patent 4,038,396, 26 July 1977. [Google Scholar]
- Huang, Y.; Yan, D.; Wang, X.; Zhou, P.; Wu, W.; Jiang, H. Controllable Assembly of the Benzothiazole Framework using a C≡C Triple Bond as a One-Carbon Synthon. Chem. Commun. 2018, 54, 1742–1745. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Base | Tf2O/Base (by Equiv) | Yield of 3aa b (%) |
| 1 | / | 1.2/0 | 41% |
| 2 | 2-F-Pyr | 1.2/1.5 | 87% |
| 3 | 2-Cl-Pyr | 1.2/1.5 | 85% |
| 4 | 2,6-Lutidine | 1.2/1.5 | 68% |
| 5 | DMAP | 1.2/1.5 | 46% |
| 6 | Pyridine | 1.2/1.5 | 65% |
| 7 | 3-F-Pyr | 1.2/1.5 | 80% |
| 8 | 2-Pyridinesulfonic acid | 1.2/1.5 | 30% |
| 9 | 2-Nitropyridine | 1.2/1.5 | 32% |
| 10 | CsF | 1.2/1.5 | 31% |
| 11 | K2CO3 | 1.2/1.5 | 20% |
| 12 | 2-F-Pyr | 1.2/1.2 | 81% |
| 13 | 2-F-Pyr | 1.2/2.0 | 89% |
| 14 | 2-F-Pyr | 1.2/2.5 | 88% |
 | |||||
|---|---|---|---|---|---|
| Entry | 1a/2a (by Equiv) | T (°C) | t (h) | Solvent | Yield of 3aa b (%) |
| 1 | 1.2/1 | 80 | 5 | DCM | 93 |
| 2 | 1.2/1 | 80 | 5 | CHCl3 | 89 |
| 3 | 1.2/1 | 80 | 5 | CH3CN | 63 |
| 4 | 1.2/1 | 80 | 5 | Toluene | 56 |
| 5 | 1.2/1 | 50 | 5 | DCM | 93 |
| 6 | 1.2/1 | 25 | 5 | DCM | 94 |
| 7 | 1.2/1 | 25 | 1 | DCM | 94 |
| 8 | 1.2/1 | 25 | 0.5 | DCM | 86 |
| 9 | 1/1.2 | 25 | 1 | DCM | 75 |
| 10 | 1/1 | 25 | 1 | DCM | 87 |
| 11 | 1.1/1 | 25 | 1 | DCM | 95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Wang, X.; Zhao, F.; Wang, L.; Fu, S. General Synthesis of 2-Substituted Benzoxazoles Based on Tf2O-Promoted Electrophilic Activation of Tertiary Amides. Molecules 2025, 30, 1510. https://doi.org/10.3390/molecules30071510
Li H, Wang X, Zhao F, Wang L, Fu S. General Synthesis of 2-Substituted Benzoxazoles Based on Tf2O-Promoted Electrophilic Activation of Tertiary Amides. Molecules. 2025; 30(7):1510. https://doi.org/10.3390/molecules30071510
Chicago/Turabian StyleLi, Hongchen, Xingyong Wang, Fujun Zhao, Lu Wang, and Songbao Fu. 2025. "General Synthesis of 2-Substituted Benzoxazoles Based on Tf2O-Promoted Electrophilic Activation of Tertiary Amides" Molecules 30, no. 7: 1510. https://doi.org/10.3390/molecules30071510
APA StyleLi, H., Wang, X., Zhao, F., Wang, L., & Fu, S. (2025). General Synthesis of 2-Substituted Benzoxazoles Based on Tf2O-Promoted Electrophilic Activation of Tertiary Amides. Molecules, 30(7), 1510. https://doi.org/10.3390/molecules30071510