
Effect of Functional Group-Modified UiO-66 on the Dehydrogenation of Ammonia Borane




Abstract
1. Introduction
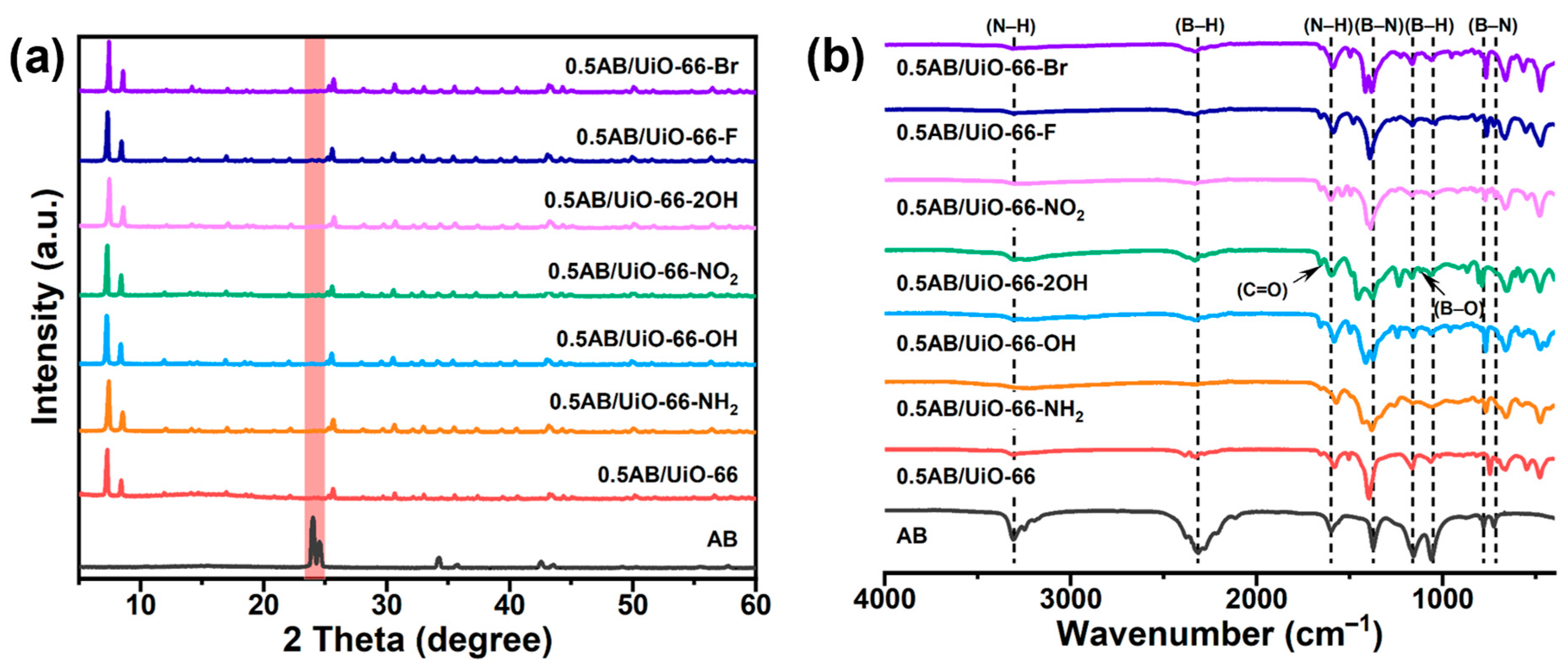
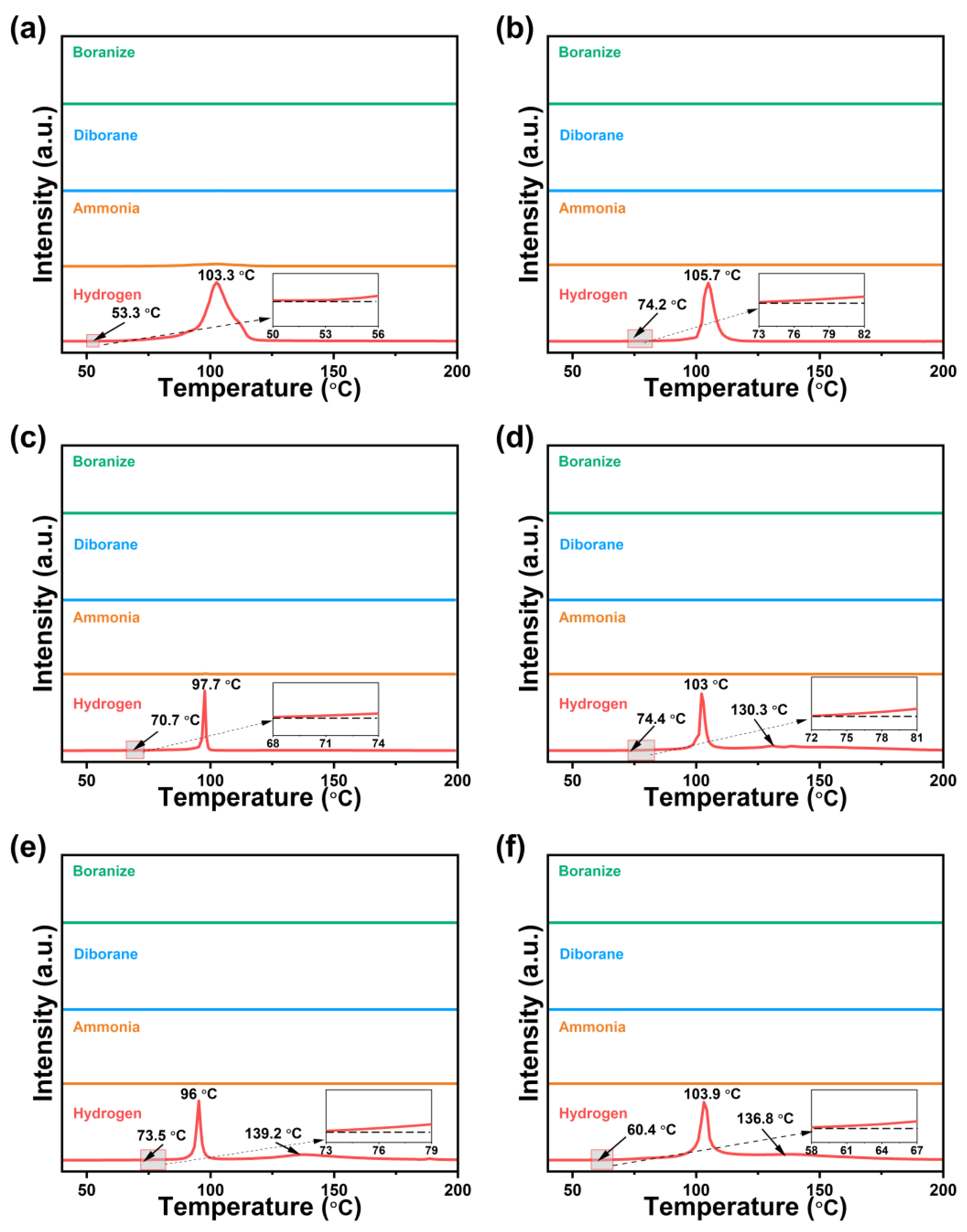
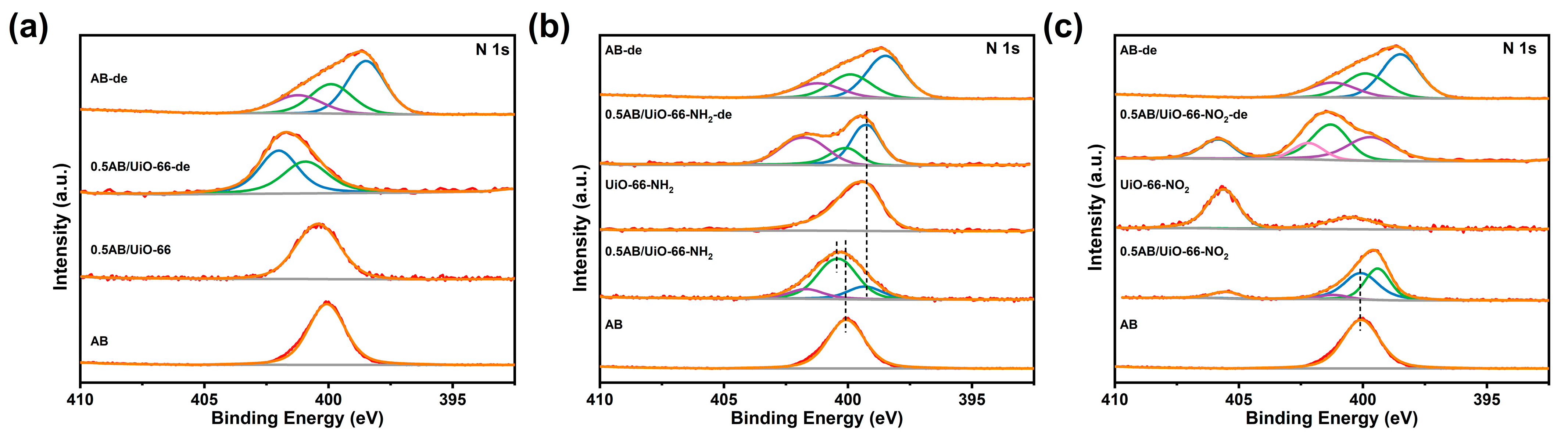
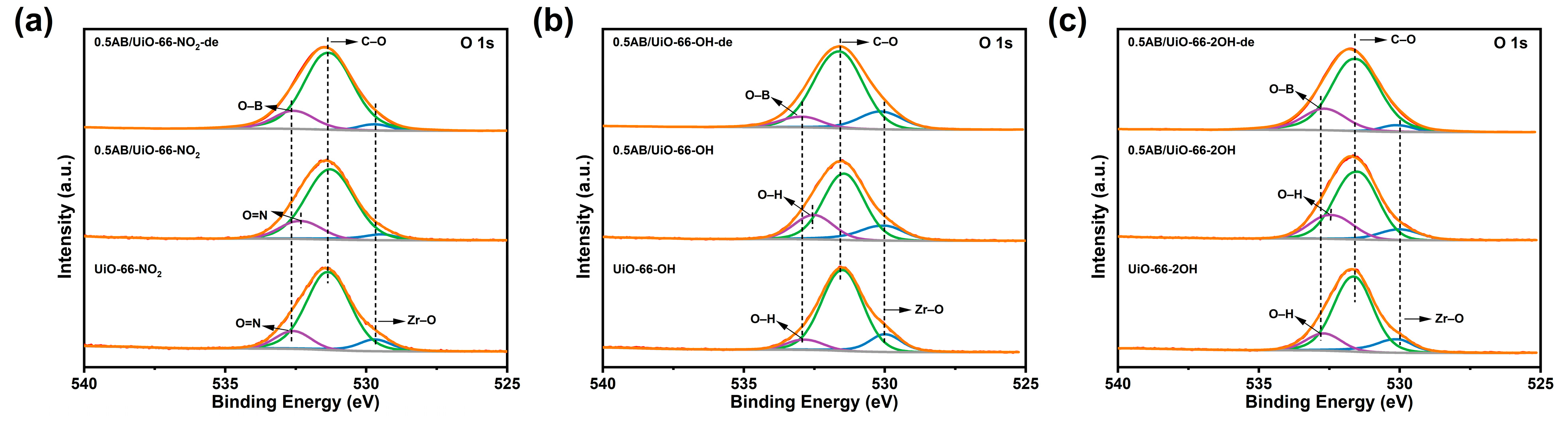
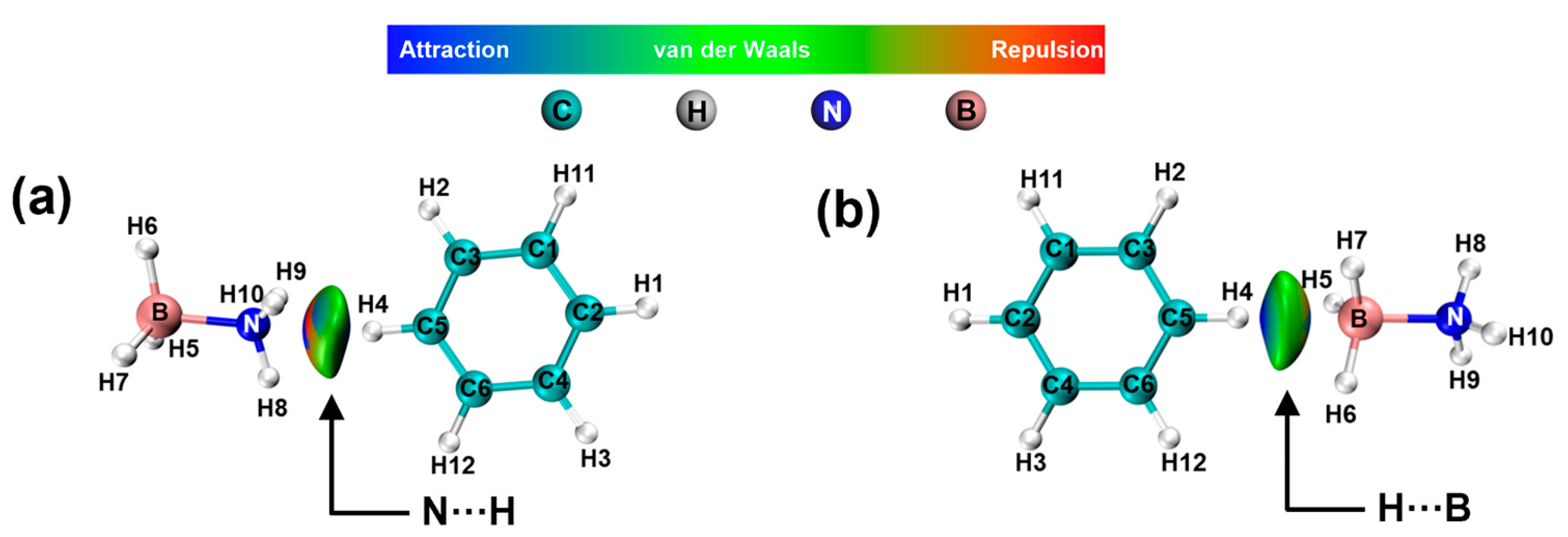
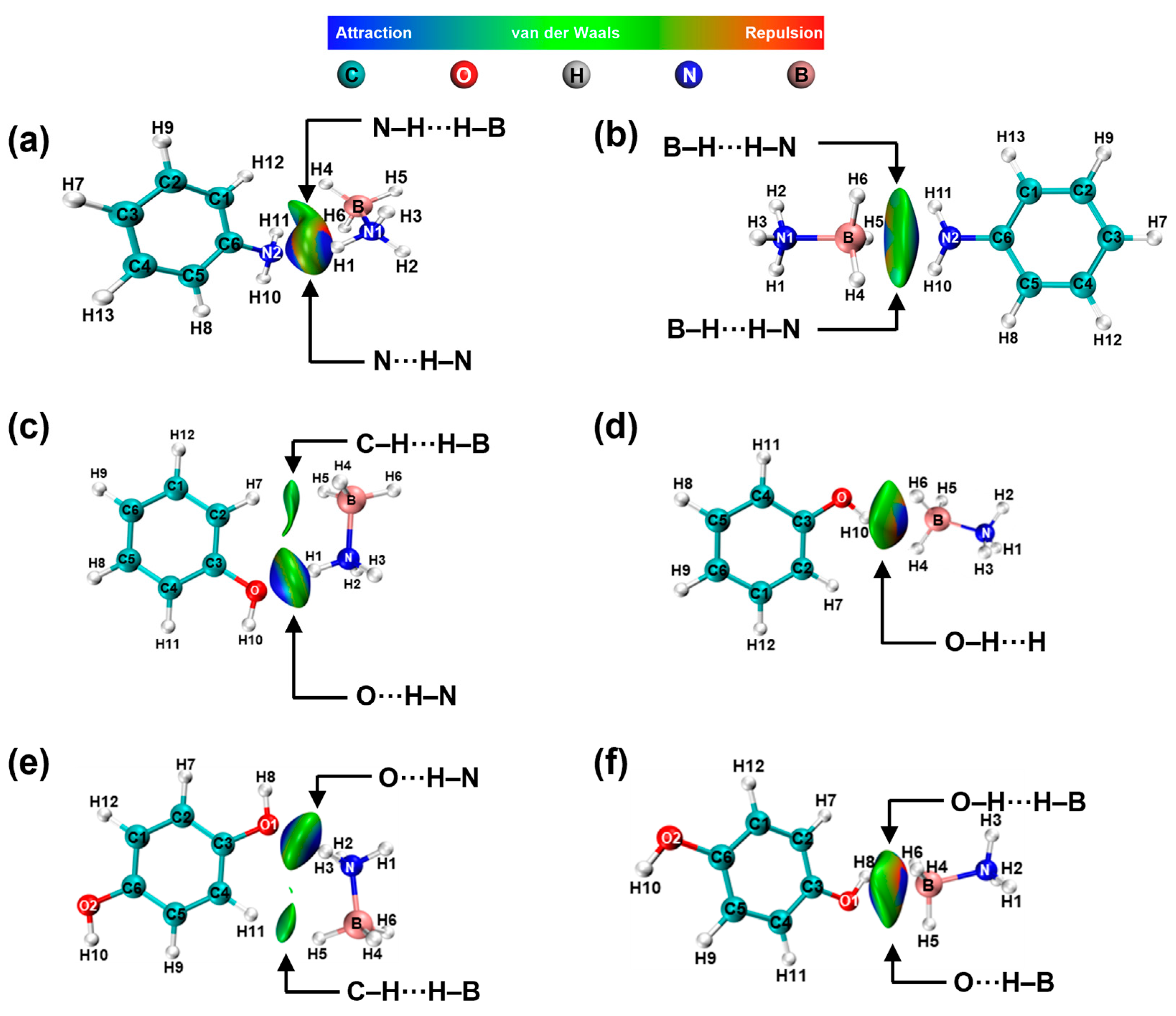
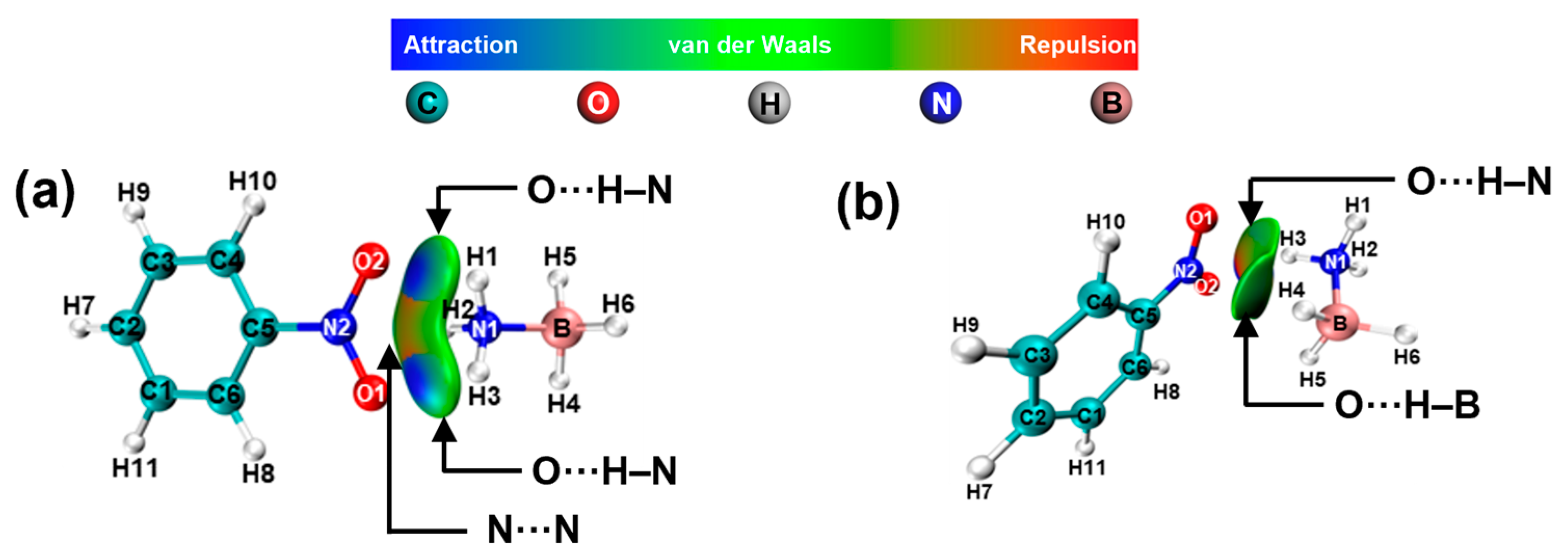
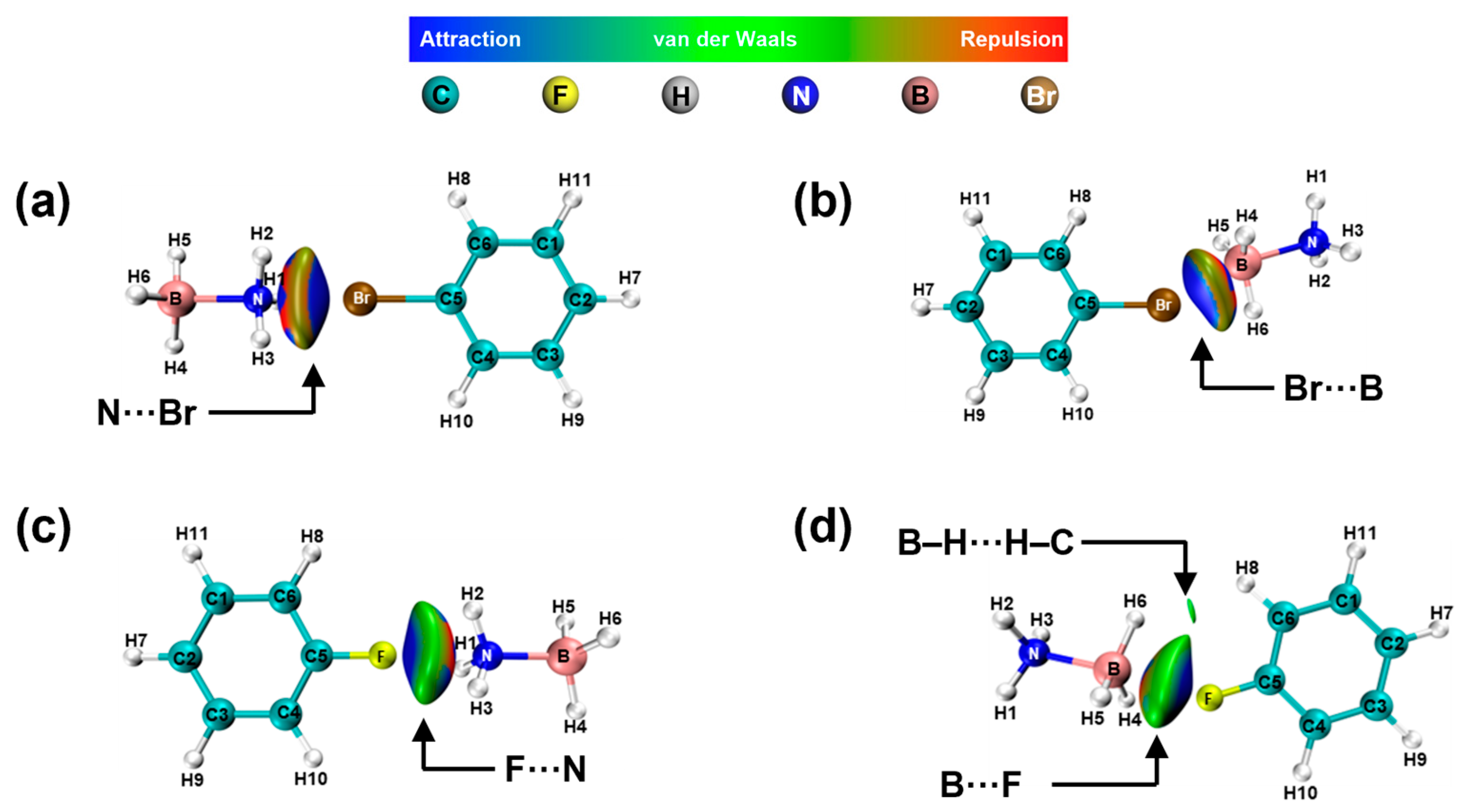
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Instruments and Methods
3.3. Materials Prepared
- Synthesis of UiO-66:
- Synthesis of UiO-66-NH2:
- Synthesis of UiO-66-NO2:
- Synthesis of UiO-66-Br:
- Synthesis of UiO-66-F:
- Synthesis of UiO-66-OH:
- Synthesis of UiO-66-2OH:
- Synthesis of AB/MOF:
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winter, C.-J. Hydrogen energy-Abundant, efficient, clean: A debate over the energy-system-of-change. Int. J. Hydrogen Energy 2009, 34, S1–S52. [Google Scholar] [CrossRef]
- Guan, D.; Wang, B.; Zhang, J.; Shi, R.; Jiao, K.; Li, L.; Wang, Y.; Xie, B.; Zhang, Q.; Yu, J.; et al. Hydrogen society: From present to future. Energy Environ. Sci. 2023, 16, 4926–4943. [Google Scholar]
- Staffell, I.; Scamman, D.; Velazquez Abad, A.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar]
- Wang, Y.; Xue, Y.; Züttel, A. Nanoscale engineering of solid-state materials for boosting hydrogen storage. Chem. Soc. Rev. 2024, 53, 972–1003. [Google Scholar] [CrossRef] [PubMed]
- Sattar, M.A.; Rasul, M.G.; Jahirul, M.I.; Hasan, M.M. An up-to-date review on the progress and challenges of hydrogen storage, and its safety and economic analysis. Sustain. Energy Fuels 2024, 8, 3545–3573. [Google Scholar]
- Rao, P.C.; Kim, Y.; Kim, H.; Son, Y.; Choi, Y.; Na, K.; Yoon, M. Methylbenzyl Naphthalene: Liquid Organic Hydrogen Carrier for Facile Hydrogen Storage and Release. ACS Sustain. Chem. Eng. 2023, 11, 12656–12666. [Google Scholar] [CrossRef]
- Deng, Y.; Guo, Y.; Jia, Z.; Liu, J.C.; Guo, J.; Cai, X.; Dong, C.; Wang, M.; Li, C.; Diao, J.; et al. Few-Atom Pt Ensembles Enable Efficient Catalytic Cyclohexane Dehydrogenation for Hydrogen Production. J. Am. Chem. Soc. 2022, 144, 3535–3542. [Google Scholar]
- Xi, S.; Wang, X.; Tome, K.C.; Zhang, T.; Han, Z.; Gao, M.; Zhou, S.; Yu, H. Effect of Ni and SAPO−34 co−additive on enhancing hydrogen storage performance of MgH2. Int. J. Hydrogen Energy 2021, 46, 23748–23756. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Zhang, H.; Xia, G.; Sun, D.; Yu, X. Heterostructures Built in Metal Hydrides for Advanced Hydrogen Storage Reversibility. Adv. Mater. 2020, 32, 2002647. [Google Scholar] [CrossRef]
- Liu, P.; Lian, J.; Chen, H.; Liu, X.; Chen, Y.; Zhang, T.; Yu, H.; Lu, G.; Zhou, S. In−situ synthesis of Mg2Ni-Ce6O11 catalyst for improvement of hydrogen storage in magnesium. Chem. Eng. J. 2020, 385, 123448. [Google Scholar] [CrossRef]
- Chong, L.; Zeng, X.; Ding, W.; Liu, D.J.; Zou, J. NaBH4 in “Graphene Wrapper:” Significantly Enhanced Hydrogen Storage Capacity and Regenerability through Nanoencapsulation. Adv. Mater. 2015, 27, 5070–5074. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ju, X.; Wan, C.; Li, S. First-principles study of transition metal (Ti, Nb)-doped NaAlH4. Int. J. Hydrogen Energy 2016, 41, 3517–3526. [Google Scholar] [CrossRef]
- Ouyang, L.; Chen, W.; Liu, J.; Felderhoff, M.; Wang, H.; Zhu, M. Enhancing the Regeneration Process of Consumed NaBH4 for Hydrogen Storage. Adv. Energy Mater. 2017, 7, 1700299. [Google Scholar] [CrossRef]
- Xiong, Z.; Yong, C.K.; Wu, G.; Chen, P.; Shaw, W.; Karkamkar, A.; Autrey, T.; Jones, M.O.; Johnson, S.R.; Edwards, P.P.; et al. High-capacity hydrogen storage in lithium and sodium amidoboranes. Nat. Mater. 2008, 7, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Aichouche, A.; Bouhadda, Y.; Bououdina, M.; Benyelloul, K.; Bentria, B. The destabilising effect of alkali metal (Na and K) of hydrazine-borane N2H4BH3 for hydrogen storage: Ab-initio study. Int. J. Hydrogen Energy 2018, 43, 14520–14531. [Google Scholar] [CrossRef]
- Kumar, R.; Karkamkar, A.; Bowden, M.; Autrey, T. Solid-state hydrogen rich boron-nitrogen compounds for energy storage. Chem. Soc. Rev. 2019, 48, 5350–5380. [Google Scholar] [CrossRef]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B-N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef]
- Guan, S.; Yuan, Z.; Zhao, S.; Zhuang, Z.; Zhang, H.; Shen, R.; Fan, Y.; Li, B.; Wang, D.; Liu, B. Efficient Hydrogen Generation from Ammonia Borane Hydrolysis on a Tandem Ruthenium-Platinum-Titanium Catalyst. Angew. Chem. Int. Ed. 2024, 63, e202408193. [Google Scholar] [CrossRef]
- Shen, R.; Liu, Y.; Wen, H.; Wu, X.; Han, G.; Yue, X.; Mehdi, S.; Liu, T.; Cao, H.; Liang, E.; et al. Engineering Bimodal Oxygen Vacancies and Pt to Boost the Activity Toward Water Dissociation. Small 2022, 18, e2105588. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.; Feng, X.; Cai, J.; Yao, W.; Song, J.; Chen, C.; Luo, D. Mechanistic insight into the promoting effect of magnesium nickel hydride on the dehydrogenation of ammonia borane. Int. J. Hydrogen Energy 2018, 43, 1681–1690. [Google Scholar]
- Huang, T.; Zou, J.; Zeng, X.; Wang, J.; Liu, H.; Ding, W. Reversible hydrogen sorption behaviors of the 3NaBH4-(x)YF3-(1-x)GdF3 system: The effect of double rare earth metal cations. Int. J. Hydrogen Energy 2019, 44, 4868–4877. [Google Scholar]
- Yang, H.; Lombardo, L.; Luo, W.; Kim, W.; Züttel, A. Hydrogen storage properties of various carbon supported NaBH4 prepared via metathesis. Int. J. Hydrogen Energy 2018, 43, 7108–7116. [Google Scholar]
- Ma, Z.; Panda, S.; Zhang, Q.; Sun, F.; Khan, D.; Ding, W.; Zou, J. Improving hydrogen sorption performances of MgH2 through nanoconfinement in a mesoporous CoS nano−boxes scaffold. Chem. Eng. J. 2021, 406, 126790. [Google Scholar]
- Demirci, U.B. Mechanistic insights into the thermal decomposition of ammonia borane, a material studied for chemical hydrogen storage. Inorg. Chem. Front. 2021, 8, 1900–1930. [Google Scholar]
- Champet, S.; van den Berg, J.; Szczesny, R.; Godula-Jopek, A.; Gregory, D.H. Nano-inclusion in one step: Spontaneous ice-templating of porous hierarchical nanocomposites for selective hydrogen release. Sustain. Energy Fuels 2019, 3, 396–400. [Google Scholar]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. [Google Scholar]
- Wang, Q.; Astruc, D. State of the art and prospects in metal−organic framework (MOF)−based and MOF−derived nanocatalysis. Chem. Rev. 2020, 120, 1438–1511. [Google Scholar]
- Freund, R.; Zaremba, O.; Arnauts, G.; Ameloot, R.; Skorupskii, G.; Dinca, M.; Bavykina, A.; Gascon, J.; Ejsmont, A.; Goscianska, J.; et al. The current status of MOF and COF applications. Angew. Chem. Int. Ed. 2021, 60, 23975–24001. [Google Scholar]
- Li, Z.; Zhu, G.; Lu, G.; Qiu, S.; Yao, X. Ammonia borane confined by a metal−organic framework for chemical hydrogen storage: Enhancing kinetics and eliminating ammonia. J. Am. Chem. Soc. 2010, 132, 1490–1491. [Google Scholar] [CrossRef]
- Wu, Y.J.; Wang, C.Y. Insight into the catalytic effects of open metal sites in metal−organic frameworks on hydride dehydrogenation via nanoconfinement. ACS Sustain. Chem. Eng. 2019, 7, 16013–16025. [Google Scholar]
- Li, Z.; Liu, W.; Yang, H.; Sun, T.; Liu, K.; Wang, Z.; Niu, C. Improved thermal dehydrogenation of ammonia borane by MOF-5. RSC Adv. 2015, 5, 10746–10750. [Google Scholar]
- Srinivas, G.; Travis, W.; Ford, J.; Wu, H.; Guo, Z.-X.; Yildirim, T. Nanoconfined ammonia borane in a flexible metal–organic framework Fe-MIL-53: Clean hydrogen release with fast kinetics. J. Mater. Chem. A 2013, 1, 4167–4172. [Google Scholar] [CrossRef]
- Srinivas, G.; Ford, J.; Zhou, W.; Yildirim, T. Zn-MOF assisted dehydrogenation of ammonia borane: Enhanced kinetics and clean hydrogen generation. Int. J. Hydrogen Energy 2012, 37, 3633–3638. [Google Scholar]
- Gao, L.; Li, C.Y.; Yung, H.; Chan, K.Y. A functionalized MIL-101(Cr) metal-organic framework for enhanced hydrogen release from ammonia borane at low temperature. Chem. Commun. 2013, 49, 10629–10631. [Google Scholar]
- Peil, S.; Wisser, D.; Stähle, M.; Roßmann, P.K.; Avadhut, Y.S.; Hartmann, M. Hydrogen Release from Ammonia Borane Nanoconfined in Metal−Organic Frameworks with MIL−53 Topology. J. Phys. Chem. C 2021, 125, 9990–10000. [Google Scholar]
- Valero-Pedraza, M.-J.; Cot, D.; Petit, E.; Aguey-Zinsou, K.-F.; Alauzun, J.G.; Demirci, U.B. Ammonia Borane Nanospheres for Hydrogen Storage. ACS Appl. Nano Mater. 2019, 2, 1129–1138. [Google Scholar]
- Yang, H.; Li, Z.; Liu, K.; Meng, F.; Niu, C. Clean Hydrogen Release from Ammonia Borane in a Metal-Organic Framework with Unsaturated Coordinated Tm3+. J. Phys. Chem. C 2015, 119, 2260–2265. [Google Scholar]
- Mishra, S.; Kang, P.-C.; Guo, R.-F.; Wang, C.-Y.; Nebhani, L. Combined Effect of Functionality and Pore Size on Dehydrogenation of Ammonia Borane via Its Nanoconfinement in Polyacrylamide-Grafted Organically Modified Mesoporous Silica. ACS Appl. Energy Mater. 2021, 4, 6585–6598. [Google Scholar] [CrossRef]
- Kim, G.J.; Hunt, S.G.; Hwang, H.T. Effect of maleic acid on onset temperature and H2 release kinetics for thermal dehydrogenation of ammonia borane. Int. J. Hydrogen Energy 2020, 45, 33751–33758. [Google Scholar]
- Chen, W.; Chen, Z.; Chi, Y.; Tian, W. Double Cation—π Directed Two-Dimensional Metallacycle-Based Hierarchical Self-Assemblies for Dual-Mode Catalysis. J. Am. Chem. Soc. 2023, 145, 19746–19758. [Google Scholar]
- Wang, Z.; Hao, A.; Xing, P. Halogen Interaction Effects on Chiral Self-Assemblies on Cyclodipeptide Scaffolds Across Hierarchy. Small 2023, 19, 2302517. [Google Scholar]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09 Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [PubMed]
- Chen, L.W.; Hao, Y.C.; Guo, Y.; Zhang, Q.; Li, J.; Gao, W.Y.; Ren, L.; Su, X.; Hu, L.; Zhang, N.; et al. Metal-organic framework membranes encapsulating gold nanoparticles for direct plasmonic photocatalytic nitrogen fixation. J. Am. Chem. Soc. 2021, 143, 5727–5736. [Google Scholar]
- Zhang, J.; Bai, T.; Huang, H.; Yu, M.H.; Fan, X.; Chang, Z.; Bu, X.H. Metal-organic-framework-based photocatalysts optimized by spatially separated cocatalysts for overall water splitting. Adv. Mater. 2020, 32, 2004747. [Google Scholar]
- He, T.; Xu, X.; Ni, B.; Wang, H.; Long, Y.; Hu, W.; Wang, X. Fast and scalable synthesis of uniform zirconium-, hafnium-based metal-organic framework nanocrystals. Nanoscale 2017, 9, 19209–19215. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | H2 Content (wt.%) | Equivalent of H2 |
|---|---|---|
| 0.5AB/UiO-66 | 1.98 | 0.91 |
| 0.5AB/UiO-66-NH2 | 3.53 | 1.62 |
| 0.5AB/UiO-66-OH | 3.53 | 1.62 |
| 0.5AB/UiO-66-2OH | 3.85 | 1.77 |
| 0.5AB/UiO-66-NO2 | 3.38 | 1.55 |
| 0.5AB/UiO-66-F | 2.87 | 1.32 |
| 0.5AB/UiO-66-Br | 3.05 | 1.4 |
| 10 wt.%AB/Fe-MIL-53 [35] | 1.48 | 2.26 |
| 24 wt.%AB/Al-MIL-53 [35] | 1.61 | 1.03 |
| AB@Cu-BDC [30] | 0.88 | 2.82 |
| AB@Tm(BTC) [37] | 0.97 | 1.94 |
| AB/MOF-5 [31] | 1.28 | 1.41 |
| AB-MIL-101-NH2 [31] | 5.55 | 1.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xi, S.; Xu, D.; Chen, R.; Yao, W.; Wu, W.; Zhang, T.; Yu, L. Effect of Functional Group-Modified UiO-66 on the Dehydrogenation of Ammonia Borane. Molecules 2025, 30, 1487. https://doi.org/10.3390/molecules30071487
Xi S, Xu D, Chen R, Yao W, Wu W, Zhang T, Yu L. Effect of Functional Group-Modified UiO-66 on the Dehydrogenation of Ammonia Borane. Molecules. 2025; 30(7):1487. https://doi.org/10.3390/molecules30071487
Chicago/Turabian StyleXi, Senliang, Dawei Xu, Renzeng Chen, Wenhao Yao, Wenying Wu, Teng Zhang, and Liang Yu. 2025. "Effect of Functional Group-Modified UiO-66 on the Dehydrogenation of Ammonia Borane" Molecules 30, no. 7: 1487. https://doi.org/10.3390/molecules30071487
APA StyleXi, S., Xu, D., Chen, R., Yao, W., Wu, W., Zhang, T., & Yu, L. (2025). Effect of Functional Group-Modified UiO-66 on the Dehydrogenation of Ammonia Borane. Molecules, 30(7), 1487. https://doi.org/10.3390/molecules30071487