
Single-Port Fluorescence Immunoassay for Concurrent Quantification of Live and Dead Bacteria: A Strategy Based on Extracellular Nucleases and DNase I

 ,
,  ,
,  and
and
Abstract
1. Introduction
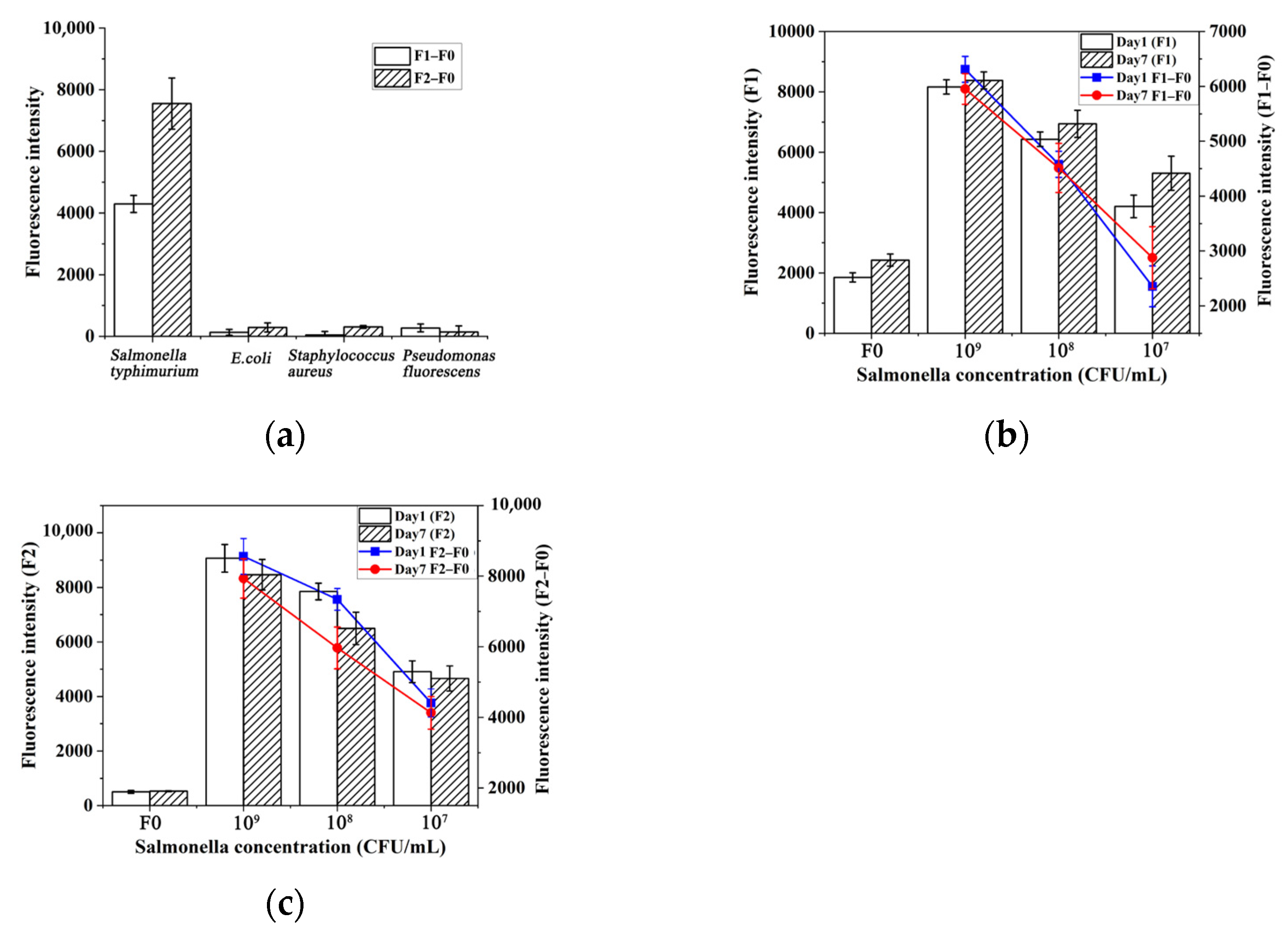
2. Results and Discussion
2.1. Characterization of AuNPs and Apt.-SNA
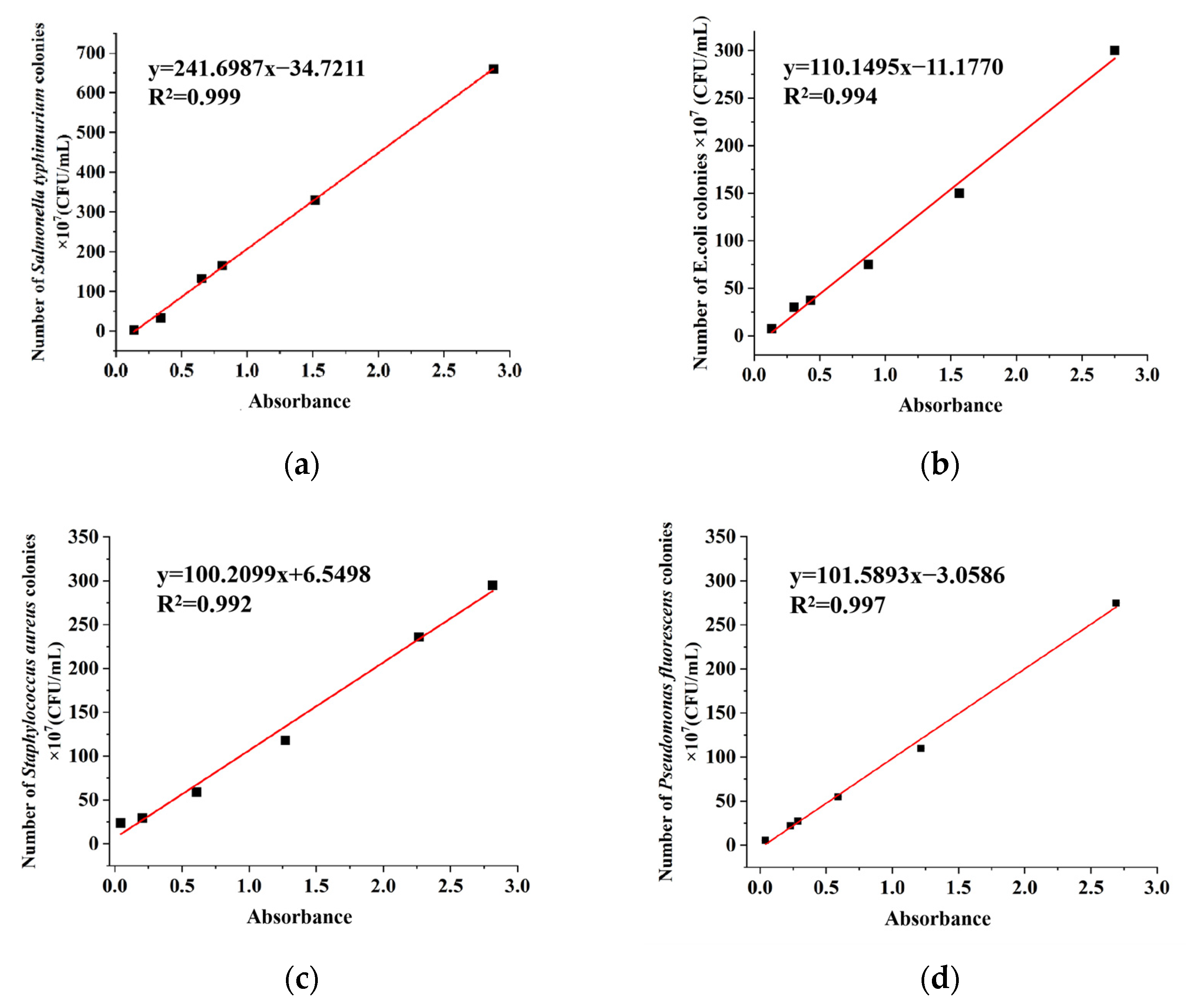
2.2. Linear Curve of Bacterial Colony Number and Absorbance
2.3. Detection Principle and Feasibility
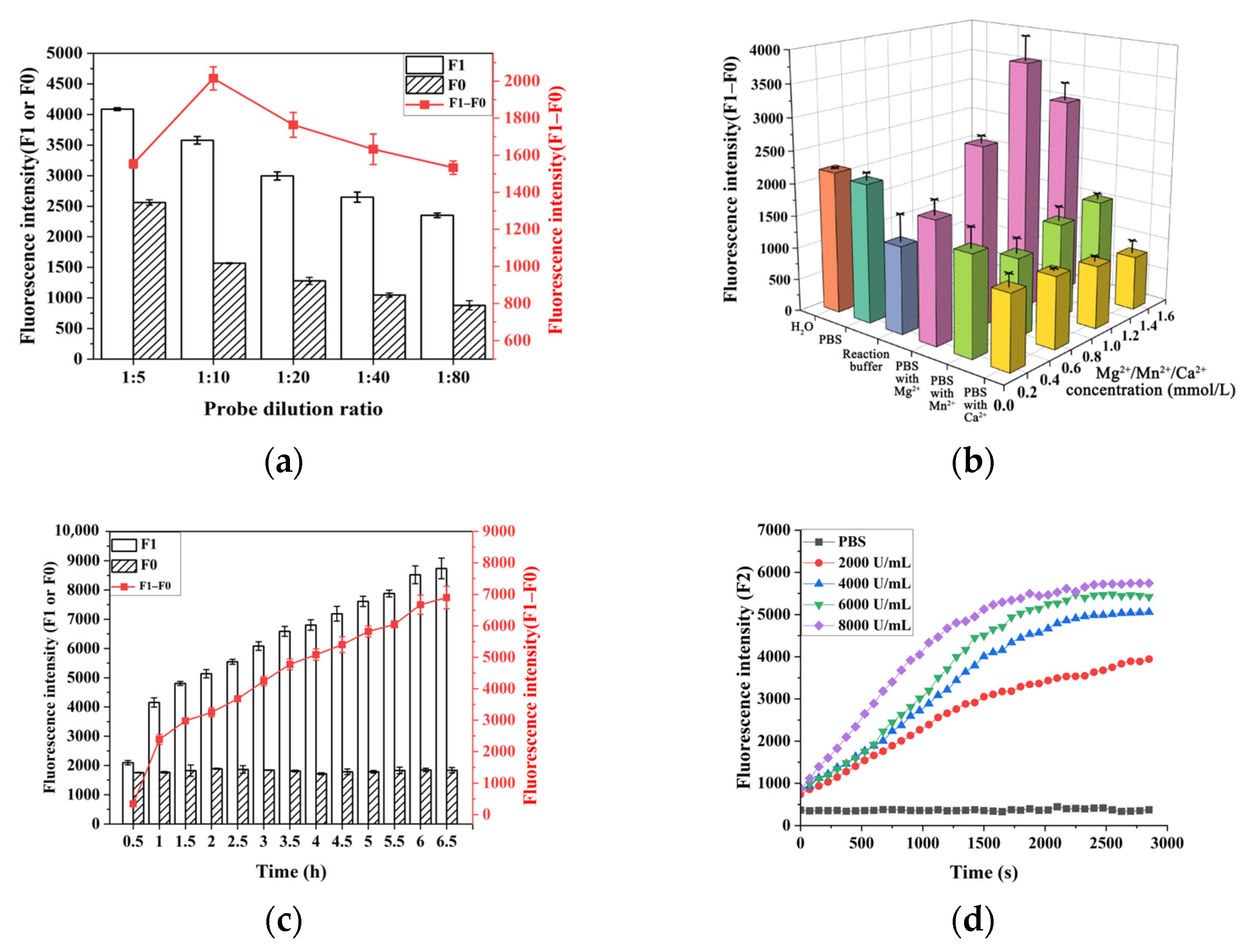
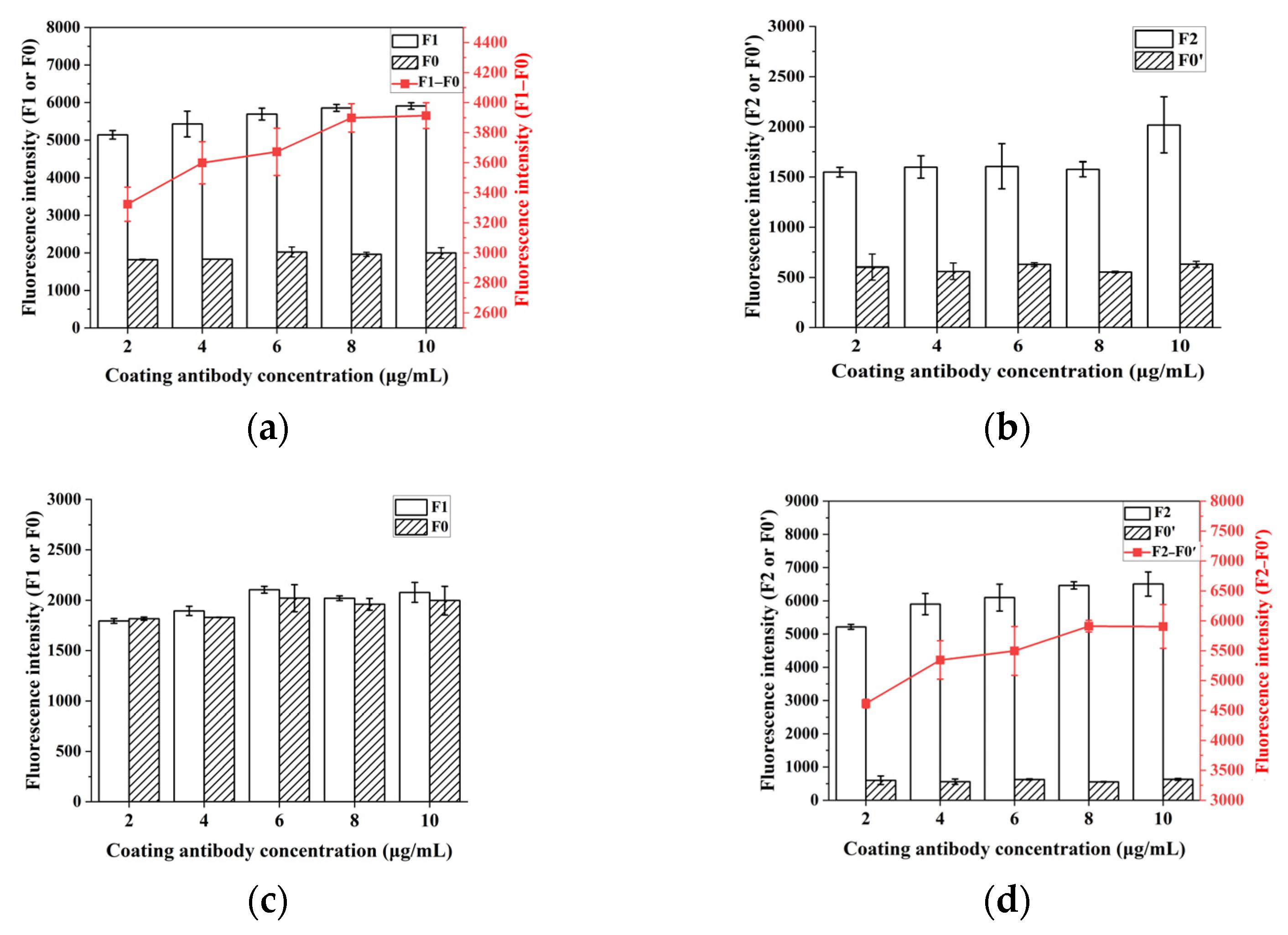
2.4. Optimization of Experimental Conditions
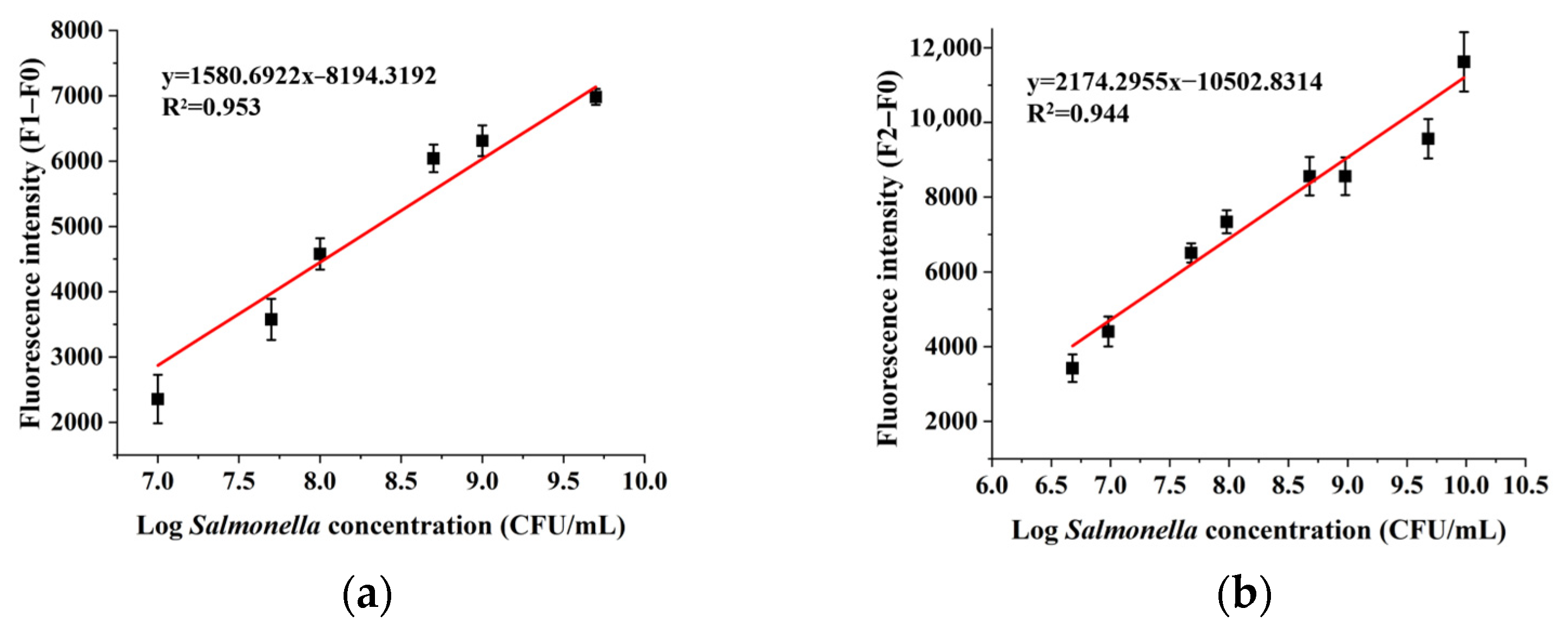
2.5. Sensitivity, Specificity, and Repeatability for the Assay
2.6. Soup Sample Analysis
3. Materials and Methods
3.1. Reagents and Instruments
3.2. Preparation of AuNPs
3.3. Preparation and Functionalization of Apt.-SNA
3.4. Bacterial Culture and Preparation of a Standard Curve for Colony Counts
3.5. Feasibility Verification
3.6. S. Typhimurium Detection
3.7. Sample Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, S.; Shi, X. Microbial Food Safety in China: Past, Present, and Future. Foodborne Pathog. Dis. 2021, 18, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Mather, A.E.; Gilmour, M.W.; Reid, S.W.J.; French, N.P. Foodborne Bacterial Pathogens: Genome-Based Approaches for Enduring and Emerging Threats in a Complex and Changing World. Nat. Rev. Microbiol. 2024, 22, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Subedi, D.; Paudel, M.; Poudel, S.; Koirala, N. Food Safety in Developing Countries: Common Foodborne and Waterborne Illnesses, Regulations, Organizational Structure, and Challenges of Food Safety in the Context of Nepal. Food Front. 2025, 6, 86–123. [Google Scholar] [CrossRef]
- Kuhnert, P.; Boerlin, P.; Frey, J. Target Genes for Virulence Assessment of Escherichia Coli Isolates from Water, Food and the Environment. FEMS Microbiol. Rev. 2000, 24, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, C.; Xing, D. Simultaneous Detection of Salmonella Enterica, Escherichia Coli O157:H7, and Listeria Monocytogenes Using Oscillatory-Flow Multiplex PCR. Microchim. Acta 2011, 173, 503–512. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Fang, Z.; Yu, Y.; Bi, J.; Wang, J.; Dong, Q.; Zhang, H. Persistence of Listeria Monocytogenes ST5 in Ready-to-Eat Food Processing Environment. Foods 2022, 11, 2561. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Chen, X.; Qu, C. Review Controlling Listeria Monocytogenes in Ready-to-Eat Meat and Poultry Products: An Overview of Outbreaks, Current Legislations, Challenges, and Future Prospects. Trends Food Sci. Technol. 2021, 116, 24–35. [Google Scholar] [CrossRef]
- Johnson, R.; Mylona, E.; Frankel, G. Typhoidal Salmonella: Distinctive Virulence Factors and Pathogenesis. Cell. Microbiol. 2018, 20, e12939. [Google Scholar] [CrossRef]
- Ibrahim, D.; Abdelfattah-Hassan, A.; Badawi, M.; Ismail, T.A.; Bendary, M.M.; Abdelaziz, A.M.; Mosbah, R.A.; Mohamed, D.I.; Arisha, A.H.; El-Hamid, M.I.A. Thymol Nanoemulsion Promoted Broiler Chicken’s Growth, Gastrointestinal Barrier and Bacterial Community and Conferred Protection against Salmonella Typhimurium. Sci. Rep. 2021, 11, 7742. [Google Scholar] [CrossRef]
- Salam, F.; Tothill, I.E. Detection of Salmonella Typhimurium Using an Electrochemical Immunosensor. Biosens. Bioelectron. 2009, 24, 2630–2636. [Google Scholar] [CrossRef]
- Katholm, J.; Olesen, L.T.; Petersen, A.; Sigurdsson, S. Evaluation of a New qPCR Test to Identify the Organisms Causing High Total Bacterial Count in Bulk Tank Milk. J. Integr. Agric. 2018, 17, 1241–1245. [Google Scholar] [CrossRef]
- Fan, X.; Li, X.; Zhang, T.; Xu, J.; Shi, Z.; Wu, Z.; Wu, J.; Pan, D.; Du, L. A Novel qPCR Method for the Detection of Lactic Acid Bacteria in Fermented Milk. Foods 2021, 10, 3066. [Google Scholar] [CrossRef]
- Jain, S.; Chattopadhyay, S.; Jackeray, R.; Abid, C.K.V.Z.; Kohli, G.S.; Singh, H. Highly Sensitive Detection of Salmonella Typhi Using Surface Aminated Polycarbonate Membrane Enhanced-ELISA. Biosens. Bioelectron. 2012, 31, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, P.; Zhang, Z.; Chen, Q.; Aguilar, Z.P.; Xu, H.; Yang, L.; Xu, F.; Lai, W.; Xiong, Y.; et al. Rapid and Accurate Detection of Viable Escherichia Coli O157:H7 in Milk Using a Combined IMS, Sodium Deoxycholate, PMA and Real-Time Quantitative PCR Process. Food Control 2014, 36, 119–125. [Google Scholar] [CrossRef]
- Brooks, B.W.; Devenish, J.; Lutze-Wallace, C.L.; Milnes, D.; Robertson, R.H.; Berlie-Surujballi, G. Evaluation of a Monoclonal Antibody-Based Enzyme-Linked Immunosorbent Assay for Detection of Campylobacter Fetus in Bovine Preputial Washing and Vaginal Mucus Samples. Vet. Microbiol. 2004, 103, 77–84. [Google Scholar] [CrossRef]
- Lee, K.-M.; Runyon, M.; Herrman, T.J.; Phillips, R.; Hsieh, J. Review of Salmonella Detection and Identification Methods: Aspects of Rapid Emergency Response and Food Safety. Food Control 2015, 47, 264–276. [Google Scholar] [CrossRef]
- Kumaravel, S.; Jian, S.-E.; Huang, S.-T.; Huang, C.-H.; Hong, W.-Z. Convenient and Ultrasensitive Detection of Live Salmonella Using Ratiometric Electrochemical Molecular Substrates. Anal. Chim. Acta 2022, 1190, 339244. [Google Scholar] [CrossRef]
- Sun, J.; Huang, J.; Li, Y.; Lv, J.; Ding, X. A Simple and Rapid Colorimetric Bacteria Detection Method Based on Bacterial Inhibition of Glucose Oxidase-Catalyzed Reaction. Talanta 2019, 197, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, C.; Bi, W.; Lin, J.; Tan, L.; Gan, N. On-Site Enrichment and Detection of Live Salmonella Typhimurium Using a Bioluminescence Sensor Coupled with a Hyperbranched Aptamer Probe-Labelled Stir-Bars Array. Sens. Actuators B Chem. 2022, 364, 131862. [Google Scholar] [CrossRef]
- Jhelum, H.; Sori, H.; Sehgal, D. A Novel Extracellular Vesicle-Associated Endodeoxyribonuclease Helps Streptococcus Pneumoniae Evade Neutrophil Extracellular Traps and Is Required for Full Virulence. Sci. Rep. 2018, 8, 7985. [Google Scholar] [CrossRef]
- Doke, M.; Fukamachi, H.; Morisaki, H.; Arimoto, T.; Kataoka, H.; Kuwata, H. Nucleases from Prevotella Intermedia Can Degrade Neutrophil Extracellular Traps. Mol. Oral. Microbiol. 2017, 32, 288–300. [Google Scholar] [CrossRef]
- Arazna, M.; Pruchniak, M.P.; Demkow, U. Neutrophil Extracellular Traps in Bacterial Infections: Strategies for Escaping from Killing. Respir. Physiol. Neurobiol. 2013, 187, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Binnenkade, L.; Kreienbaum, M.; Thormann, K.M. Characterization of ExeM, an Extracellular Nuclease of Shewanella Oneidensis MR-1. Front. Microbiol. 2018, 9, 1761. [Google Scholar] [CrossRef]
- Tam, K.; Torres, V.J. Staphylococcus Aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Xu, K.; Zhang, M.; Niu, J.; Zhao, T.; Wang, X.; Jia, Y.; Li, J.; Yu, Z.; He, L.; et al. 5′-Nucleotidase Is Dispensable for the Growth of Salmonella Typhimurium but Inhibits the Bactericidal Activity of Macrophage Extracellular Traps. Arch. Microbiol. 2023, 205, 20. [Google Scholar] [CrossRef]
- Liao, C.; Zhang, M.; Cheng, X.; Li, Q.; Mao, F.; Wang, X.; Yu, C.; Yu, Z.; Jia, Y.; Li, J.; et al. Identification and Characterization of the Nuclease Activity of the Extracellular Proteins from Salmonella Enterica Serovar Typhimurium. Curr. Microbiol. 2020, 77, 3651–3660. [Google Scholar] [CrossRef] [PubMed]
- Machado, I.; Garrido, V.; Hernandez, L.I.; Botero, J.; Bastida, N.; San-Roman, B.; Grilló, M.-J.; Hernandez, F.J. Rapid and Specific Detection of Salmonella Infections Using Chemically Modified Nucleic Acid Probes. Anal. Chim. Acta 2019, 1054, 157–166. [Google Scholar] [CrossRef]
- Barbau-Piednoir, E.; Mahillon, J.; Pillyser, J.; Coucke, W.; Roosens, N.H.; Botteldoorn, N. Evaluation of Viability-qPCR Detection System on Viable and Dead Salmonella Serovar Enteritidis. J. Microbiol. Methods 2014, 103, 131–137. [Google Scholar] [CrossRef]
- Wu, W.; Li, J.; Pan, D.; Li, J.; Song, S.; Rong, M.; Li, Z.; Gao, J.; Lu, J. Gold Nanoparticle-Based Enzyme-Linked Antibody-Aptamer Sandwich Assay for Detection of Salmonella Typhimurium. ACS Appl. Mater. Interfaces 2014, 6, 16974–16981. [Google Scholar] [CrossRef]
- Joshi, R.; Janagama, H.; Dwivedi, H.P.; Senthil Kumar, T.M.A.; Jaykus, L.-A.; Schefers, J.; Sreevatsan, S. Selection, Characterization, and Application of DNA Aptamers for the Capture and Detection of Salmonella Enterica Serovars. Mol. Cell. Probes 2009, 23, 20–28. [Google Scholar] [CrossRef]
- Li, C.; Li, D.; Wan, G.; Xu, J.; Hou, W. Facile Synthesis of Concentrated Gold Nanoparticles with Low Size-Distribution in Water: Temperature and pH Controls. Nanoscale Res. Lett. 2011, 6, 440. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhong, L.; Cheng, Y.; Yu, H.; Xie, Y.; Yao, W.; Guo, Y.; Kawasaki, H. A Study on the PEG-Assisted Stability of Spherical Nucleic Acid Constructed by the Freezing Method. Colloids Surf. A Physicochem. Eng. Asp. 2024, 686, 133349. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, N.; Li, J.; Ye, J.; Ma, S.; Le, G. Ultrasensitive Chemiluminescent Immunoassay of Salmonella with Silver Enhancement of Nanogold Labels. Luminescence 2011, 26, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Zheng, S.; Kim, T.S.; Kim, S.K. Analysis of DNA Coverage Using Enzymatic Cleavage of Fluorescent Labels. BioChip J. 2011, 5, 39–46. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrichment Time | Added Viable Concentration (CFU/mL) | Added Dead Concentration (CFU/mL) | Results of Plate Counting Method (CFU/mL) | Measurement Results of Step 1 (CFU/mL) | Measurement Results of Step 2 (CFU/mL) |
|---|---|---|---|---|---|
| 8 h | 104 | 105 | 8.4 × 106 | 1.02 × 106 | 3.63 × 105 |
| 16 h | 104 | 8.6 × 107 | 6.84 × 107 | 2.03 × 106 | |
| 103 | 5 × 107 | 1.36 × 107 | 7.1 × 105 | ||
| 24 h | 104 | 3.7 × 1010 | 1.44 × 109 | 1.29 × 106 | |
| 103 | 9.1 × 108 | 3.68 × 108 | 1.57 × 106 | ||
| 102 | 1.4 × 108 | 5.28 × 108 | 1.35 × 106 |
| Name | 3′ Modified Group | Sequence (5′ to 3′) | 5′ Modified Group |
|---|---|---|---|
| Apt. | HS | TAT GGC GGC GTC ACC CGA CGG GGA CTT GAC ATT ATG ACA G TTTTTTTTTT | FAM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Dong, H.; Yu, H.; Yuan, S.; Kawasaki, H.; Guo, Y.; Yao, W. Single-Port Fluorescence Immunoassay for Concurrent Quantification of Live and Dead Bacteria: A Strategy Based on Extracellular Nucleases and DNase I. Molecules 2025, 30, 1374. https://doi.org/10.3390/molecules30061374
Wang Y, Dong H, Yu H, Yuan S, Kawasaki H, Guo Y, Yao W. Single-Port Fluorescence Immunoassay for Concurrent Quantification of Live and Dead Bacteria: A Strategy Based on Extracellular Nucleases and DNase I. Molecules. 2025; 30(6):1374. https://doi.org/10.3390/molecules30061374
Chicago/Turabian StyleWang, Yuhan, Han Dong, Hang Yu, Shaofeng Yuan, Hideya Kawasaki, Yahui Guo, and Weirong Yao. 2025. "Single-Port Fluorescence Immunoassay for Concurrent Quantification of Live and Dead Bacteria: A Strategy Based on Extracellular Nucleases and DNase I" Molecules 30, no. 6: 1374. https://doi.org/10.3390/molecules30061374
APA StyleWang, Y., Dong, H., Yu, H., Yuan, S., Kawasaki, H., Guo, Y., & Yao, W. (2025). Single-Port Fluorescence Immunoassay for Concurrent Quantification of Live and Dead Bacteria: A Strategy Based on Extracellular Nucleases and DNase I. Molecules, 30(6), 1374. https://doi.org/10.3390/molecules30061374