

Natural Flavonoids from Licorice as Potent Inhibitors of β-Glucuronidase Elucidated Through Computational Studies

Abstract
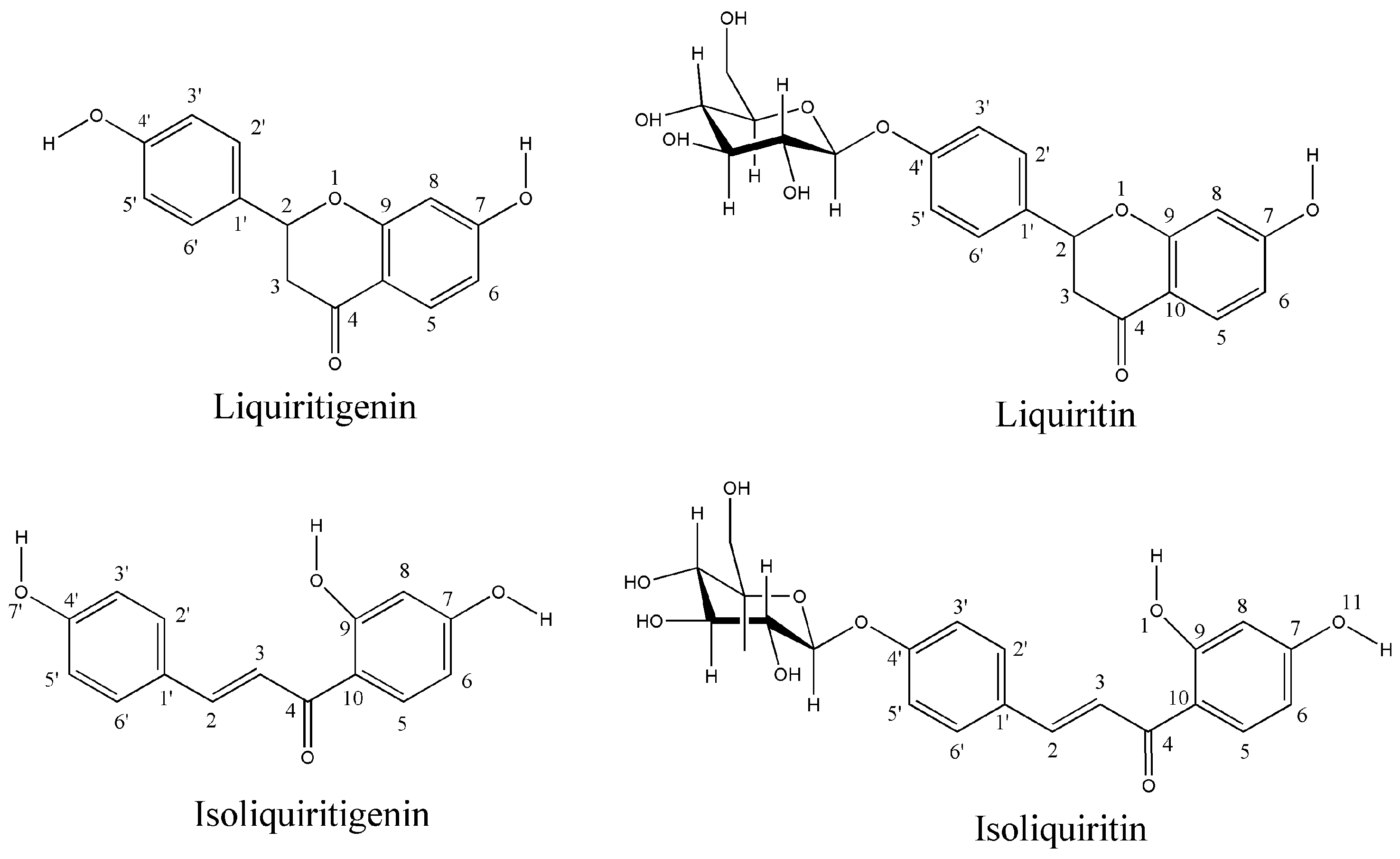
1. Introduction
2. Results and Discussion
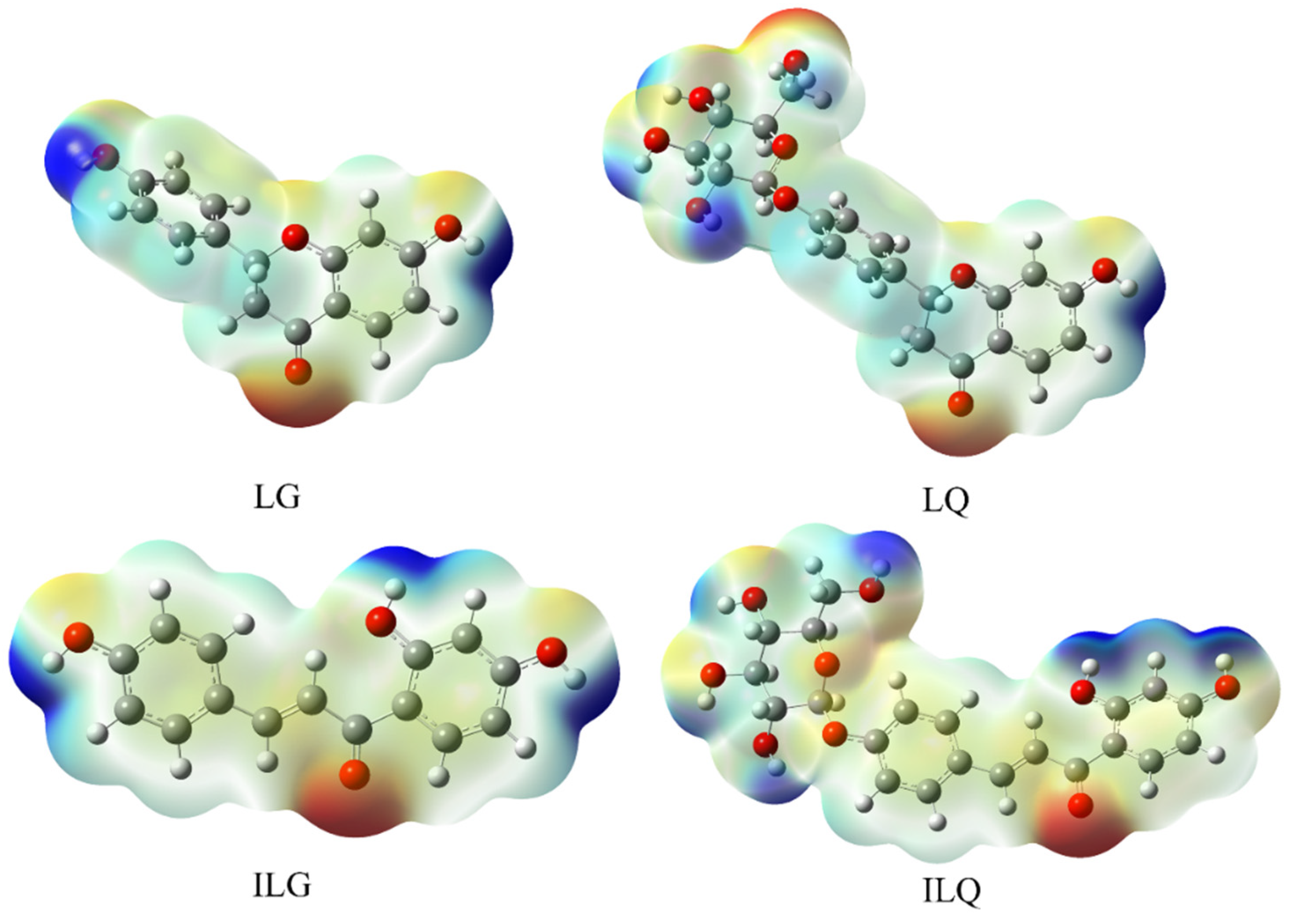
2.1. Density Functional Theory (DFT) Calculations
Molecular Electrostatic Potential (MEP)
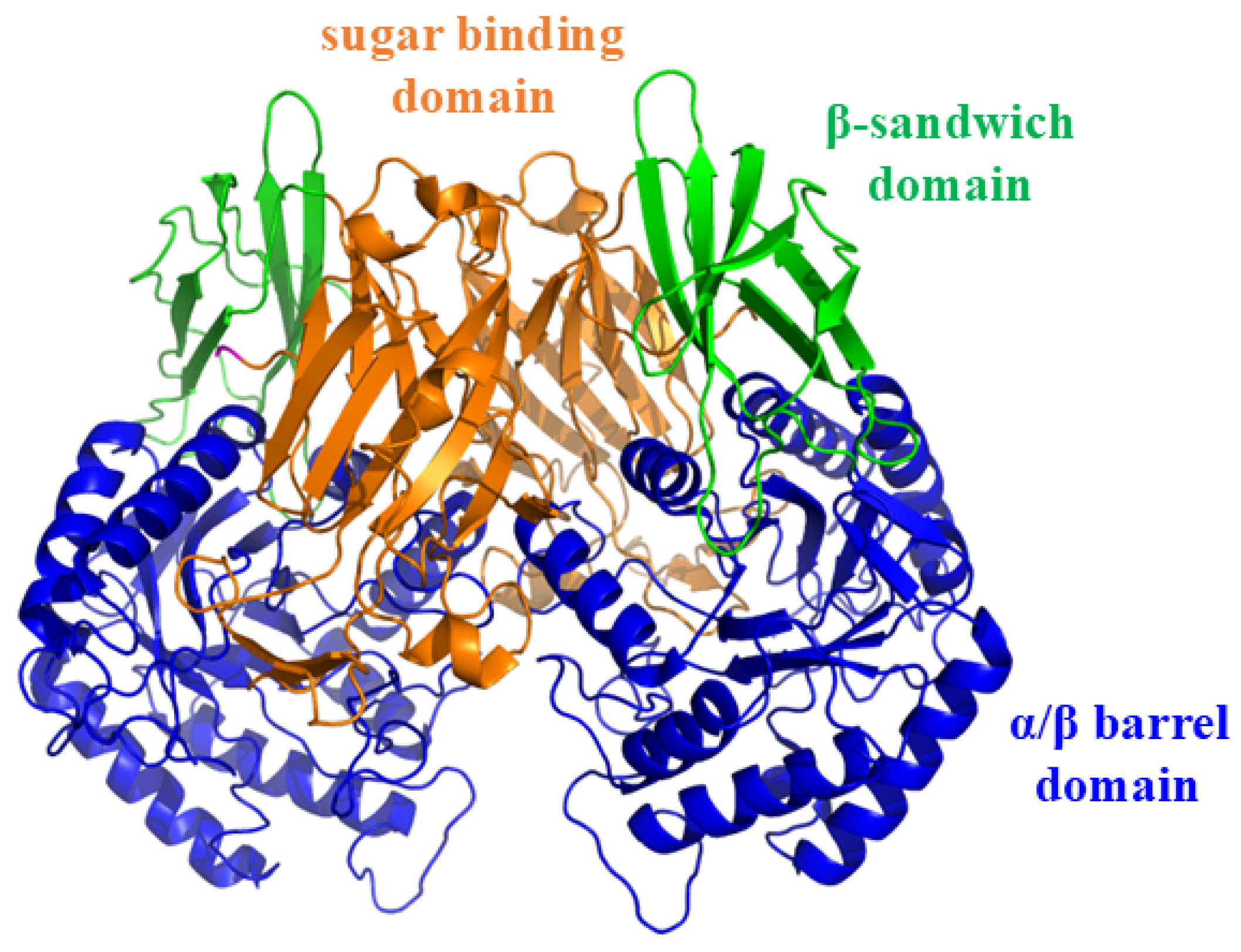
2.2. Molecular Docking Study
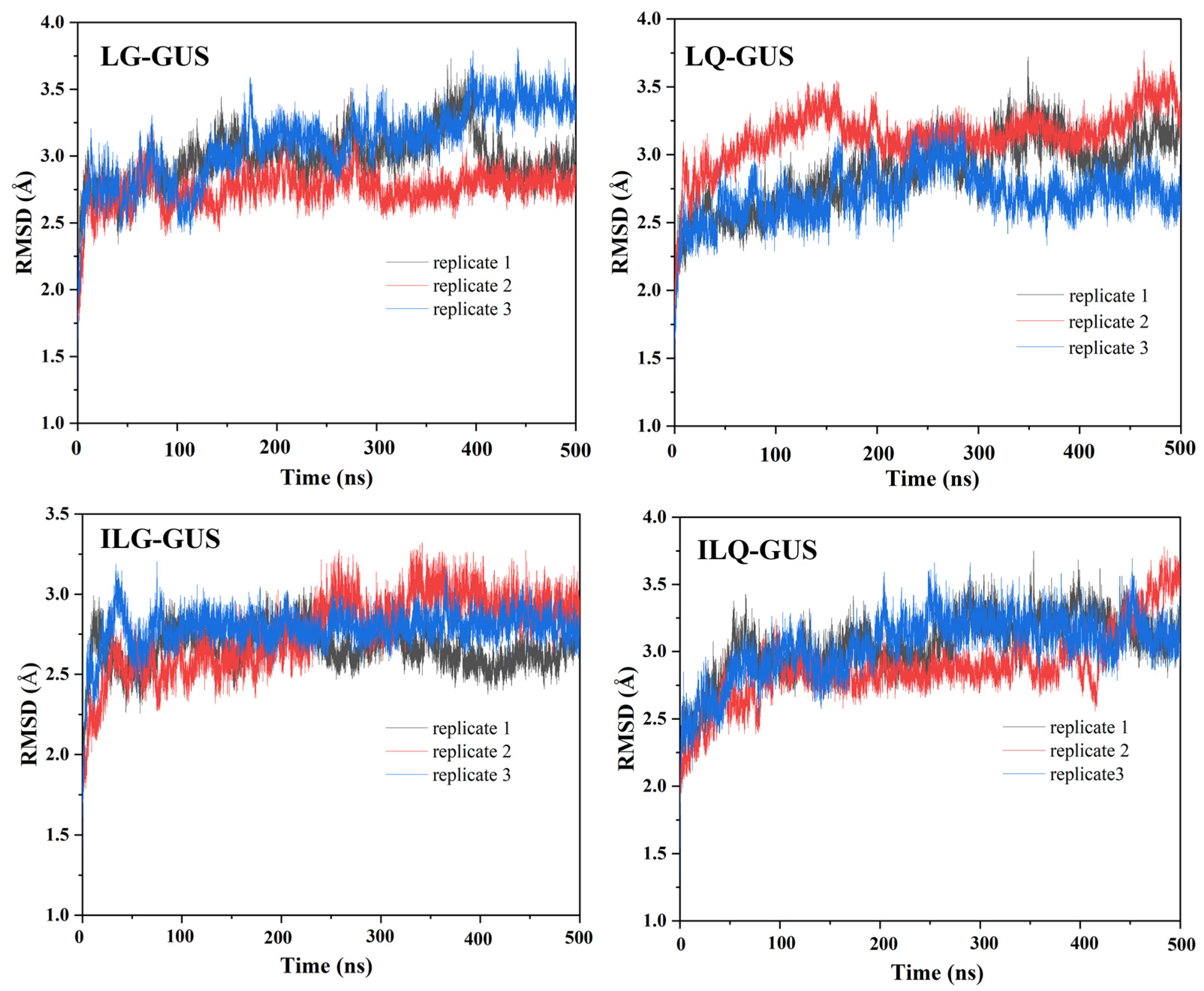
2.3. MD Simulation
2.4. Binding Free Energy
2.5. Free Energy Decomposition
3. Computational Methods
3.1. DFT Calculations
3.2. Molecular Docking
3.3. MD Simulation Method
3.4. Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 11. [Google Scholar] [CrossRef]
- De Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef] [PubMed]
- Awolade, P.; Cele, N.; Kerru, N.; Gummidi, L.; Oluwakemi, E.; Singh, P. Therapeutic significance of β-glucuronidase activity and its inhibitors: A review. Eur. J. Med. Chem. 2020, 187, 46. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Tseng, C.; Yang, C.; Tzeng, C.; Cheng, T.; Leu, Y.; Chuang, Y.; Wang, J.; Lu, Y.; Chen, Y.; et al. Specific Inhibition of Bacterial β-Glucuronidase by Pyrazolo[4,3-c]quinoline Derivatives via a pH-Dependent Manner To Suppress Chemotherapy-Induced Intestinal Toxicity. J. Med. Chem. 2017, 60, 9222–9238. [Google Scholar] [CrossRef]
- Wallace, B.D.; Wang, H.W.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.A.; Mani, S.; et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science 2010, 330, 831–835. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wallace, B.D.; Venkatesh, M.K.; Mani, S.; Redinbo, M.R. Molecular Insights into Microbial β-Glucuronidase Inhibition to Abrogate CPT-11 Toxicity. Mol. Pharmacol. 2013, 84, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.D.; Roberts, A.B.; Pollet, R.M.; Ingle, J.D.; Biernat, K.A.; Pellock, S.J.; Venkatesh, M.K.; Guthrie, L.; O’Neal, S.K.; Robinson, S.J.; et al. Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem. Biol. 2015, 22, 1238–1249. [Google Scholar] [CrossRef]
- Pellock, S.J.; Creekmore, B.C.; Walton, W.G.; Mehta, N.; Biernat, K.A.; Cesmat, A.P.; Ariyarathna, Y.; Dunn, Z.D.; Li, B.; Jin, J.; et al. Gut Microbial β-Glucuronidase Inhibition via Catalytic Cycle Interception. ACS Cent. Sci. 2018, 4, 868–879. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Pellock, S.J.; Biernat, K.A.; Walton, W.G.; Wallace, B.D.; Creekmore, B.C.; Letertre, M.M.; Swann, J.R.; Wilson, I.D.; Roques, J.R.; et al. Targeted inhibition of gut bacterial β-glucuronidase activity enhances anticancer drug efficacy. Proc. Natl. Acad. Sci. USA 2020, 117, 7374–7381. [Google Scholar] [CrossRef]
- Cheng, K.W.; Tseng, C.H.; Tzeng, C.C.; Leu, Y.L.; Cheng, T.C.; Wang, J.Y.; Chang, J.M.; Lu, Y.C.; Cheng, C.M.; Chen, I.J.; et al. Pharmacological inhibition of bacterial β-glucuronidase prevents irinotecan-induced diarrhea without impairing its antitumor efficacy in vivo. Pharmacol. Res. 2019, 139, 41–49. [Google Scholar] [CrossRef]
- Challa, A.P.; Hu, X.; Zhang, Y.Q.; Hymes, J.; Wallace, B.D.; Karavadhi, S.; Sun, H.M.; Patnaik, S.; Hall, M.D.; Shen, M. Virtual Screening for the Discovery of Microbiome β-Glucuronidase Inhibitors to Alleviate Cancer Drug Toxicity. J. Chem. Inf. Model. 2022, 62, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.M.; Wang, P.; Ge, G.B.; Dai, Z.R.; Wu, D.C.; Zou, L.W.; Dou, T.Y.; Zhang, T.Y.; Yang, L.; Hou, J. Structure-activity relationships of flavonoids as natural inhibitors against E. coli β-glucuronidase. Food Chem. Toxicol. 2017, 109, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Yang, W.; Yan, Z.X.; Zhang, Q.W.; Yan, R. Prenylflavonoids sanggenon C and kuwanon G from mulberry Morus alba as potent broad-spectrum bacterial β-glucuronidase inhibitors: Biological evaluation and molecular docking studies. J. Funct. Food. 2018, 48, 210–219. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, L.; Wang, P.P.; Tang, Y.Q.; Wu, D.C.; Zhang, C.L.; Zhou, Q.; Yan, R.; Hou, J. Discovery of a naturally occurring broad-spectrum inhibitor against gut bacterial β-glucuronidases from Ginkgo biloba. Food Funct. 2021, 12, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.G.; Yan, J.K.; Sun, C.P.; Li, J.X.; Ning, J.; Wang, C.; Huo, X.K.; Zhao, W.Y.; Yu, Z.L.; Feng, L.; et al. Amentoflavone from Selaginella tamariscina as a potent inhibitor of gut bacterial β-glucuronidase: Inhibition kinetics and molecular dynamics stimulation. Chem.-Biol. Interact. 2021, 340, 8. [Google Scholar] [CrossRef]
- Batiha, G.E.; Beshbishy, A.M.; El-Mleeh, A.; Abdel-Daim, M.M.; Devkota, H.P. Traditional Uses, Bioactive Chemical Constituents, and Pharmacological and Toxicological Activities of Glycyrrhiza glabra L. (Fabaceae). Biomolecules 2020, 10, 19. [Google Scholar] [CrossRef]
- Wahab, S.; Annadurai, S.; Abullais, S.S.; Das, G.; Ahmad, W.; Ahmad, M.F.; Kandasamy, G.; Vasudevan, R.; Ali, M.S.; Amir, M. Glycyrrhiza glabra (Licorice): A Comprehensive Review on Its Phytochemistry, Biological Activities, Clinical Evidence and Toxicology. Plants 2021, 10, 36. [Google Scholar] [CrossRef]
- Jiang, M.Y.; Zhao, S.J.; Yang, S.S.; Lin, X.; He, X.G.; Wei, X.Y.; Song, Q.; Li, R.; Fu, C.M.; Zhang, J.M.; et al. An “essential herbal medicine”-licorice: A review of phytochemicals and its effects in combination preparations. J. Ethnopharmacol. 2020, 249, 14. [Google Scholar] [CrossRef]
- Cheng, M.Z.; Zhang, J.Q.; Yang, L.; Shen, S.J.; Li, P.; Yao, S.; Qu, H.; Li, J.Y.; Yao, C.L.; Wei, W.L.; et al. Recent advances in chemical analysis of licorice (Gan-Cao). Fitoterapia 2021, 149, 12. [Google Scholar] [CrossRef]
- Qin, Y.F.; Hang, X.M.; Kang, A.; Tang, Y.P.; Jiang, J.Q. Research progress on irinotecan induced metabolic dysregulation in microbiota gut-liver axis and Chinese materia medica intervention. Chin. Tradit. Herbal Drugs 2017, 48, 4114–4119. [Google Scholar]
- Yue, S.J.; Qin, Y.F.; Kang, A.; Tao, H.J.; Zhou, G.S.; Chen, Y.Y.; Jiang, J.Q.; Tang, Y.P.; Duan, J.A. Total Flavonoids of Glycyrrhiza uralensis Alleviates Irinotecan-Induced Colitis via Modification of Gut Microbiota and Fecal Metabolism. Front. Immunol. 2021, 12, 628358. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.P.; Yan, J.K.; Yi, J.; Zhang, X.Y.; Yu, Z.L.; Huo, X.K.; Liang, J.H.; Ning, J.; Feng, L.; Wang, C.; et al. The study of inhibitory effect of natural flavonoids toward β-glucuronidase and interaction of flavonoids with β-glucuronidase. Int. J. Biol. Macromol. 2020, 143, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Guo, H.; Zhou, J.; Wang, Y.W.; Yan, H.; Jin, R.Y.; Tang, Y.P. Evodiamine and Rutaecarpine as Potential Anticancer Compounds: A Combined Computational Study. Int. J. Mol. Sci. 2022, 23, 13. [Google Scholar] [CrossRef]
- Ritu, J.; Mehak, D.; Alka, K.; Anil Kumar, C. Relevance of Molecular Docking Studies in Drug Designing. Curr. Bioinform. 2020, 15, 270–278. [Google Scholar]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Jain, S.; Drendel, W.B.; Hen, Z.-W.C.; Mathews, F.S.; Sly, W.S.; Grubb, J.H. Structure of human β-glucuronidase reveals candidate lysosomal targeting and active-site motifs. Nat. Struct. Biol. 1996, 3, 375–381. [Google Scholar] [CrossRef]
- Sun, C.P.; Tian, X.G.; Feng, L.; Wang, C.; Li, J.X.; Huo, X.K.; Zhao, W.Y.; Ning, J.; Yu, Z.L.; Deng, S.; et al. Inhibition of gut bacterial β-glucuronidase by chemical components from black tea: Inhibition interactions and molecular mechanism. Arab. J. Chem. 2021, 14, 103457. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. 1999, 17, 57–61. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A New Lamarckian Genetic Algorithm for Flexible Ligand-Receptor Docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges—The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.Z.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. AMBER 2024; University of California: San Francisco, CA, USA, 2024. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.H.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. Parallelization of CPPTRAJ Enables Large Scale Analysis of Molecular Dynamics Trajectory Data. J. Comput. Chem. 2018, 39, 2110–2117. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Lee, M.S.; Salsbury, F.R.; Olson, M.A. An efficient hybrid explicit/implicit solvent method for biomolecular simulations. J. Comput. Chem. 2004, 25, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Xu, D. Molecular dynamics investigations suggest a non-specific recognition strategy of 14-3-3σ protein by tweezer: Implication for the inhibition mechanism. Front. Chem. 2019, 7, 237. [Google Scholar] [CrossRef]
- Wang, J.Y.; Chen, Q.; Wang, M.; Zhong, C. The opening/closure of the P-loop and hinge of BCR-ABL1 decodes the low/high bioactivities of dasatinib and axitinib. Phys. Chem. Chem. Phys. 2017, 19, 22444–22453. [Google Scholar] [CrossRef]
- Tse, A.; Verkhivker, G.M. Molecular Dynamics Simulations and Structural Network Analysis of c-Abl and c-Src Kinase Core Proteins: Capturing Allosteric Mechanisms and Communication Pathways from Residue Centrality. J. Chem. Inf. Model. 2015, 55, 1645–1662. [Google Scholar] [CrossRef]
- Honig, B.; Nicholls, A. Classical electrostatics in biology and chemistry. Science 1995, 268, 1144–1149. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. In Annual Review of Biophysics; Dill, K.A., Ed.; Annual Reviews: Palo Alto, CA, USA, 2019; Volume 48, pp. 275–296. [Google Scholar]
- Gaillard, T.; Simonson, T. Pairwise decomposition of an MMGBSA energy function for computational protein design. J. Comput. Chem. 2014, 35, 1371–1387. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | LG | LQ | ILG | ILQ |
|---|---|---|---|---|
| Ecorr | 0.24 | 0.41 | 0.23 | 0.41 |
| ZPVE | −879.50 | −1490.08 | −879.48 | −1490.01 |
| Etot | −879.48 | −1490.05 | −879.46 | −1489.98 |
| H | −879.48 | −1490.05 | −879.46 | −1489.98 |
| G | −879.54 | −1490.14 | −879.53 | −1490.07 |
| Total Dipole Moment μ | 2.21 | 4.53 | 1.96 | 5.29 |
| Polarizability α | 169.70 | 253.58 | 198.97 | 284.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Xue, Y.; Yan, H.; Zhou, J.; Long, X.; Tang, Y. Natural Flavonoids from Licorice as Potent Inhibitors of β-Glucuronidase Elucidated Through Computational Studies. Molecules 2025, 30, 1324. https://doi.org/10.3390/molecules30061324
Liu J, Xue Y, Yan H, Zhou J, Long X, Tang Y. Natural Flavonoids from Licorice as Potent Inhibitors of β-Glucuronidase Elucidated Through Computational Studies. Molecules. 2025; 30(6):1324. https://doi.org/10.3390/molecules30061324
Chicago/Turabian StyleLiu, Jingli, Yingying Xue, Hao Yan, Jing Zhou, Xu Long, and Yuping Tang. 2025. "Natural Flavonoids from Licorice as Potent Inhibitors of β-Glucuronidase Elucidated Through Computational Studies" Molecules 30, no. 6: 1324. https://doi.org/10.3390/molecules30061324
APA StyleLiu, J., Xue, Y., Yan, H., Zhou, J., Long, X., & Tang, Y. (2025). Natural Flavonoids from Licorice as Potent Inhibitors of β-Glucuronidase Elucidated Through Computational Studies. Molecules, 30(6), 1324. https://doi.org/10.3390/molecules30061324