3.1. General Information
The 1H- and 13C-NMR analysis was performed on a 400 MHz Bruker Avance NEO 400 spectrometer (Bruker Switzerland AG, Faellanden, Switzerland) by using deuterochloroform as a solvent. Prior to use, the solvent was treated by filtration through aluminum oxide to remove acid impurities. Chemical shifts (δ) are indicated in parts per million (ppm) from tetramethylsilane (TMS) using the solvent resonance as an internal standard. The coupling constants (J) are reported in Hertz (Hz) with the following abbreviations to describe the multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; dt, doublet of triplets; AB q, AB-quartet; m, multiplet. Regarding 13C-NMR, multiple carbon nuclei contributing to a signal are indicated in parentheses after the chemical shift value. A Nicolet Avatar FT-IR (E.S.P.) spectrometer (Thermo Scientific, Madison, WI, USA) was used to record infrared spectra with sodium chloride windows (NaCl) for liquids or potassium bromide pellets (KBr) for solid compounds. The peaks are described as follows: s, strong; vs, very strong; m, medium; w, weak; br, broad. A Bruker OTOF-Q Compact ESI mass spectrometer (Bruker Daltonic, Bremen, Germany) was used to record the high-resolution mass spectra. An Autopol V automatic Polarimeter from Rudolph Research Analytical (Hacketstown, NJ, USA) was used for the optical activity measurements utilizing a 40T-2.5-100-0.7 TempTrol polarimetric cell with a 2.5 mm inside diameter, 100 mm optical length, and 0.7 mL volume, with c (concentration) referring to g sample/100 mL. A Büchi m-560 melting point apparatus (Büchi, Uster, Switzerland) was used to determine melting points. Silica plates from SiliCycle (Québec, QC, Canada) were used to perform TLC monitoring with the use of a 4% PMA solution in methanol to develop the plates. The silica gel impregnated with boric acid was prepared as follows: Boric acid (4 g) was dissolved in methanol (100 mL), followed by the addition of silica gel (55 g). The resulting slurry was swirled for a few minutes, the methanol evaporated off, and the resulting silica preparation dried in vacuo for 6 h at 40 °C.
All solvents were purchased from Sigma-Aldrich (Steinheim, Germany) and were used without purification unless otherwise stated. They include deuterated chloroform (99.8% D), dichloromethane (99.8%), diethyl ether (≥99.8%), ethyl acetate (≥99.7%), ethanol (≥99.8%), hexane (>99%), methanol (99.9%), and tetrahydrofuran (THF) (99.9%), which was dried over sodium wire in the presence of benzophenone under a dry nitrogen atmosphere before use. Dichloromethane was kept over molecular sieves under dry nitrogen after it was brought to use. All chemicals were used without further purification. The following chemicals were purchased from Sigma-Aldrich: acetone oxime (98%), benzyl bromide (98%), boric acid (≥99.5%), DMAP (4-dimethylaminopyridine, >99%), EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, >99%), hydrochloric acid (37%), (S)-ibuprofen (99%), magnesium sulfate (≥99.5%), palladium (10%) on activated charcoal catalyst, perchloric acid (>70%), phosphomolybdic acid, sodium bicarbonate (≥99.0%), sodium hydride (60% dispersion in mineral oil), sodium sulfate (≥99%), (R)-solketal (98%, 98% ee), (S)-solketal (98%, 99% ee), and vinyl dodecanoate (≥99%). The following chemicals were obtained from TCI Europe (Zwinderecht, Belgium): vinyl decanoate (>99%), vinyl hexanoate (>99%), and vinyl octanoate (>99%). Novozymes Denmark (Bagsvaerd, Denmark) donated the immobilized Candida antarctica lipase B (CAL-B) as a gift. Ethyl esters of EPA (98%) and DHA (≥95%) were donated as gifts from Pronova Biopharma (Sandefjord, Norway). They were hydrolyzed to their corresponding free acids [
1]. (S)-Naproxen was obtained from Prof. Thorsteinn Loftsson at the Faculty of Pharmaceutical Sciences at the University of Iceland (Reykjavik, Iceland). The silica gel for the column chromatography (40–63 µm, 0.060–0.300, F60) was obtained from SiliCycle. The TLC plates used for monitoring the reactions were dipped into a methanol solution of phosphomolybdic acid (PMA) for developing the spots.
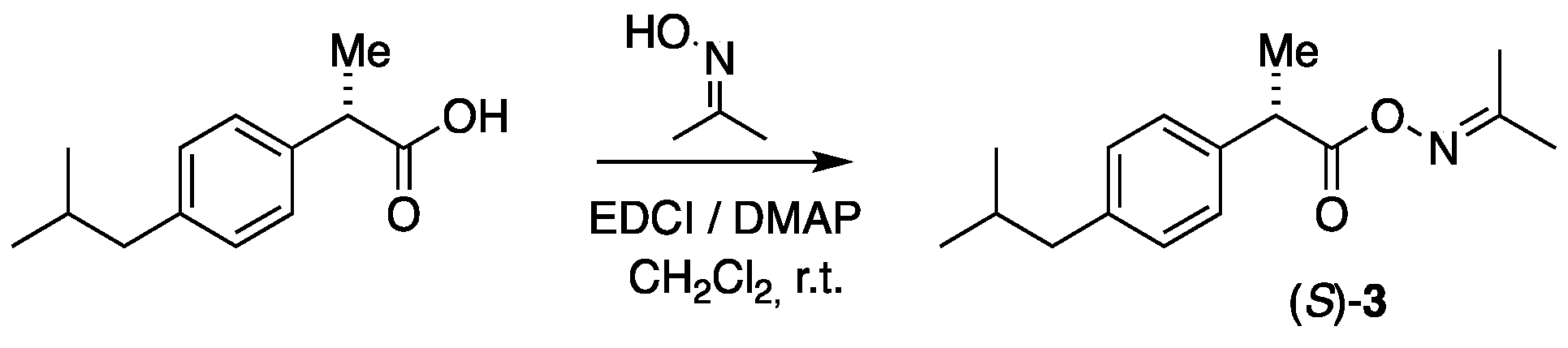
3.2. Activation of Drugs as Oximes
3.2.1. Synthesis of (S)-Propan-2-one-O-(2-(4-isobutylphenyl)propanoyl oxime, (S)-3
To a solution of (S)-ibuprofen (94 mg, 0.456 mmol), DMAP (16 mg, 0.131 mmol), and EDCI (105 mg, 0.553 mmol) in CH2Cl2 (2 mL) were added to acetoxime (34 mg, 0.465 mmol), and the solution was stirred on a magnetic stirrer at room temperature for 3–4 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using ethyl acetate/petroleum ether (3:2) as an eluent. The solvent was removed in vacuo on a rotary evaporator, and the crude product was applied to a silica gel flash chromatography using ethyl acetate/petroleum ether (1:1) as an eluent, which produced the product (S)-3 as a slightly yellow liquid in a 96% yield (115 mg, 0.440 mmol). = −7.18 (c. 7.3, CH2Cl2). IR (NaCl, νmax/cm−1): 3058 (vs), 2958 (vs), 2931 (vs), 2853 (vs), 1758 (vs), 1653 (s). 1H NMR (400 MHz, CDCl3) δH: 7.27–7.17 (m, 2H, Ibu-2,6), 7.14–7.01 (m, 2H, Ibu-3,5), 3.79 (q, J = 7.2 Hz, 1H, CHCH3), 2.44 (d, J = 7.2 Hz, 2H, CH2CH), 1.99 (s, 3H, NC(CH3)2), 1.83 (nonet, J = 6.7 Hz, 1H, CH(CH3)2), 1.83 (s, 3H, NC(CH3)2), 1.56 (d, J = 7.2 Hz, 3H, CHCH3), 0.88 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 171.9, 164.4 (N=C), 140.7, 137.5 (2), 129.4 (2), 127.3, 45.1, 44.3, 30.3, 22.5 (2), 22.1, 18.5, 16.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C16H23NO2Na 284.1621; found, 284.1618.
3.2.2. Synthesis of (S)-Propan-2-one-O-(2-(6-methoxynaphthalen-2-yl)propanoyl oxime, (S)-4
The same procedure was followed as described for (S)-3 using (S)-naproxen (105 mg, 0.456 mmol), DMAP (14 mg, 0.115 mmol), EDCI (109 mg, 0.569 mmol) and acetoxime (33 mg, 0.451 mol) in CH2Cl2 (2 mL) were added acetoxime (33 mg, 0.451 mmol). Purification on silica gel flash chromatography using ethyl acetate/petroleum ether (1:1) as an eluent, followed by recrystallization from n-hexane, produced the product (S)-4 as a white solid in a quantitative yield (130 mg, 0.456 mmol). M.p. 42.3–43.4 °C. = −12.3 (c. 16.5, CH2Cl2). IR (NaCl, νmax/cm−1): 3052 (vs), 2956 (vs), 2932 (vs), 2863 (vs), 2848 (vs), 1753 (vs), 1652 (s). 1H NMR (400 MHz, CDCl3) δH: 7.72–7.67 (m, 3H, Nap-1,4,8), 7.44 (dd, J = 8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.11 (d, J = 2.5 Hz, 1H, Nap-5), 3.96 (q, J = 7.2 Hz, 1H, CHCH3), 3.91 (s, 3H, OCH3), 1.99 (s, 3H, NC(CH3)2), 1.83 (s, 3H, NC(CH3)2), 1.65 (d, J = 7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 171.9, 164.4, 157.8, 135.4, 133.8, 129.4, 129.0, 127.3, 126.4, 126.1, 119.1, 105.7, 55.4, 44.6, 22.1, 18.7, 17.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C17H19NO3Na 308.1257; found, 308.1251.
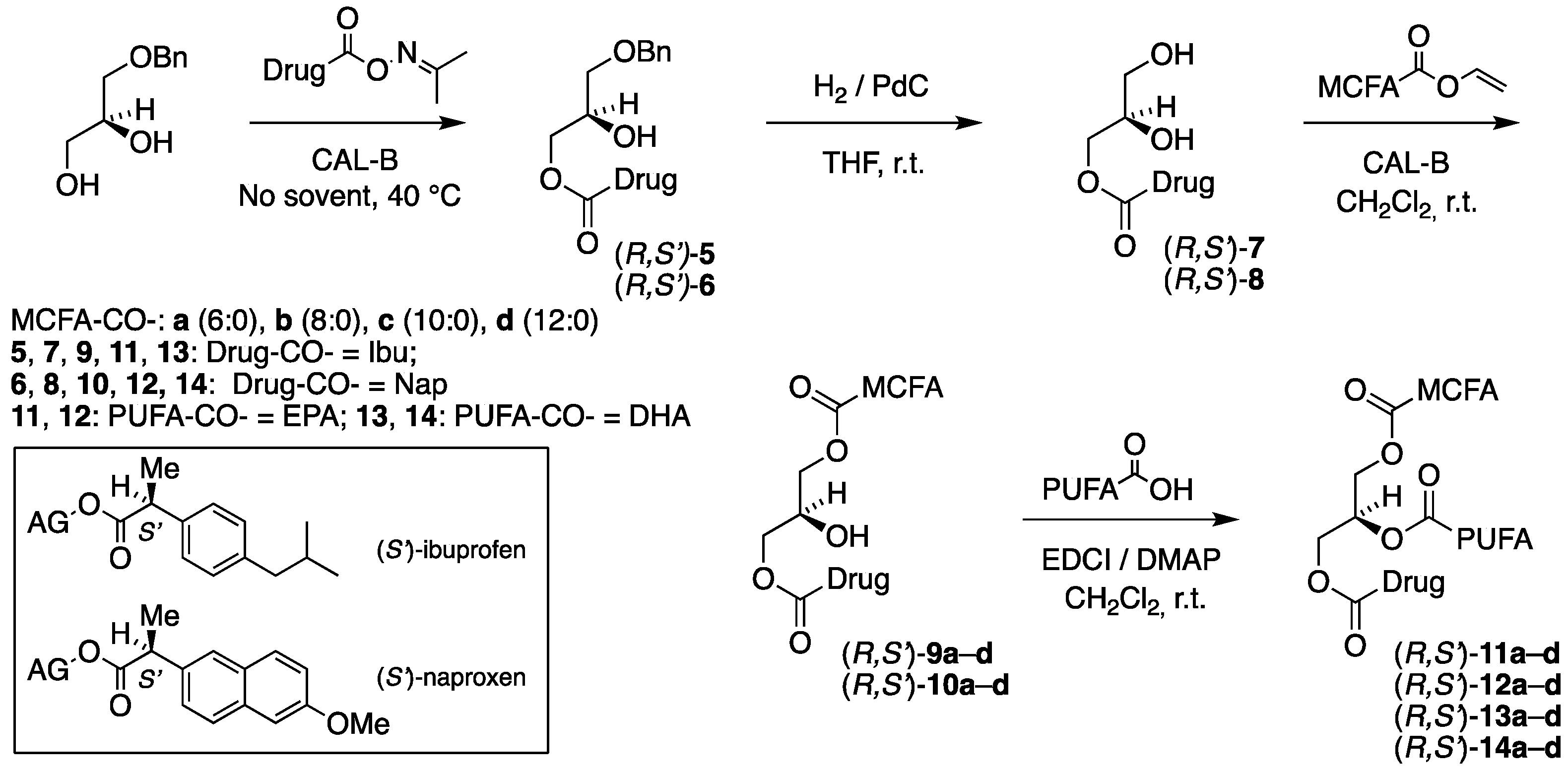
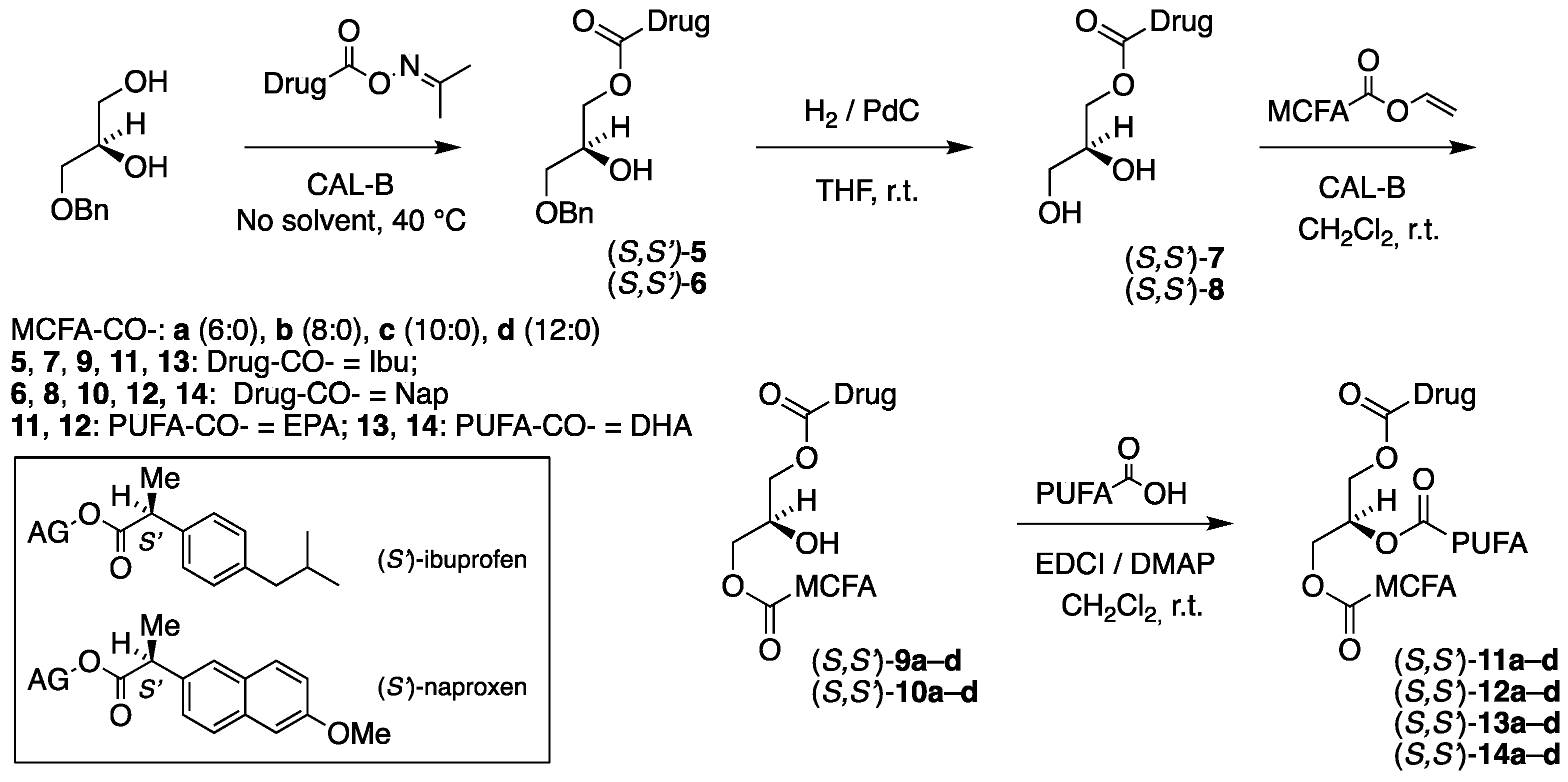
3.3. Enzymatic Coupling of the Drugs: Synthesis of (R,S′)-5, (S,S′)-5, (R,S′)-6, and (S,S′)-6
3.3.1. Synthesis of 1-O-Benzyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S′)-5
Immobilized CAL-B (40 mg) was added to a mixture of 1-O-benzyl-sn-glycerol (100 mg, 0.549 mmol) and ibuprofen acetoxime ester (S)-3 (163 mg, 0.659 mmol). The resulting mixture was stirred at 40 °C for 31 h in a nitrogen atmosphere. The lipase preparation was removed by filtration, and the solvent was distilled off in vacuo on a rotary evaporator. The concentrate was applied to a 4% boric-acid-impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (3:2) as an eluent. The first fraction from the column was contaminated with some oxime starting material, and repeated chromatography was required. The product (R,S′)-5 was produced as a colorless liquid in an 86% yield (175 mg, 0.472 mmol) from the combined fractions. = +25.0 (c. 14.0, CH2Cl2). IR (NaCl, νmax/cm−1): 3458 (br s), 3089 (s), 3462 (br s), 3089 (s), 3062 (s), 3028 (s), 2954 (vs), 2925 (vs), 2868 (vs), 1736 (vs), 1607. 1H NMR (400 MHz, CDCl3) δH: 7.38–7.27 (m, 5H, Ph-H), 7.21–7.16 (m, 2H, Ibu-2,6), 7.10–7.06 (m, 2H, Ibu-3,5), 4.47 (s, 2H, CH2Ph), 4.18 (dd, J = 11.4, 4.7 Hz, 1H, CH2 sn-3), 4.13 (dd, J = 11.4, 6.1 Hz, 1H, CH2 sn-3), 3.99–3.95 (m, 1H, CH sn-2), 3.72 (q, J = 7.2 Hz, 1H, CHCH3), 3.44 (dd, J = 9.6, 4.5 Hz, 1H, CH2 sn-1), 3.37 (dd, J = 9.6, 5.9 Hz, 1H, CH2 sn-1), 2.44 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 1.84 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.49 (d, J = 7.2 Hz, 3H, CHCH3), 0.89 (d, J = 6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9, 140.8, 137.8, 137.7, 129.5 (2), 128.6 (2), 128.0 (2), 127.8 (2), 127.3, 73.6, 70.8, 69.0, 65.7, 45.2, 30.3, 22.5, 18.53 (2), 18.47 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O4Na 393.2036; found, 393.2030.
3.3.2. Synthesis of 3-O-Benzyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S′)-5
The same procedure was followed as described for (R,S′)-5 using 3-O-benzyl-sn-glycerol (100 mg, 0.549 mmol), ibuprofen acetoxime ester (S)-3 (163 mg, 0.659 mmol), and immobilized CAL-B (45 mg). Purification on a 4% boric-acid-impregnated flash silica gel column using petroleum ether/ethyl acetate (3:2) as an eluent produced the product (S,S′)-5 as a colorless liquid in a 92% yield (187 mg, 0.505 mmol). As before, the first fraction from the column was contaminated with some oxime starting material and required repeated chromatography. = +20.7 (c. 11.0, CH2Cl2). IR (NaCl, νmax/cm−1): 3458 (br s), 3089 (s), 3062 (s), 3028 (s), 2954 (vs), 2925 (vs), 2865 (vs), 1740 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.37–7.27 (m, 5H, Ph-H), 7.18 (d, J = 8.1 Hz, 2H, Ibu-2,6), 7.08 (d, J = 8.1 Hz, 2H, Ibu-3,5), 4.47 (s, 2H, CH2Ph), 4.16 (d, J = 5.2 Hz, 2H, CH2 sn-1), 3.98–3.94 (m, 1H, CH sn-2), 3.72 (q, J = 7.2 Hz, 1H, CHCH3), 3.42 (dd, J = 9.6, 4.5 Hz, 1H, CH2 sn-3), 3.36 (dd, J = 9.6, 5.9 Hz, 1H, CH2 sn-3), 2.44 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 1.84 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.49 (d, J = 7.2 Hz, 3H, CHCH3), 0.89 (d, J = 6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9, 140.8, 137.9, 137.7, 129.5 (2), 128.6 (2), 128.0 (2), 127.9 (2), 127.3, 73.6, 70.8, 69.0, 65.6, 45.2, 30.3, 22.5, 18.52 (2), 18.46 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O4Na 393.2036; found, 393.2031.
3.3.3. Synthesis of 1-O-Benzyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (R,S′)-6
The same procedure was followed as described for (R,S′)-5 using 1-O-benzyl-sn-glycerol (100 mg, 0.549 mmol), naproxen acetoxime ester (S)-4 (172 mg, 0.604 mmol), and immobilized CAL-B (38 mg). Purification on a 4% boric-acid-impregnated flash silica gel column using petroleum ether/ethyl acetate (3:2) as an eluent resulted in a first fraction contaminated with the starting material that, as before, required repeated chromatography. Recrystallization of the combined fractions from n-hexane produced the product (R,S′)-6 as a white solid in a 69% yield (149 mg, 0.378 mmol). M.p. 51.7–52.1 °C. = +53.8 (c. 1.9, CH2Cl2). IR (NaCl, νmax/cm−1): 3538 (br s), 3057 (s), 2973 (vs), 2936 (vs), 2909 (vs), 2864 (vs), 1719 (vs), 1632 (s), 1605 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71–7.65 (m, 3H, Nap-1,4,8), 7.38 (dd, J = 8.4, 1.9 Hz, 1H, Nap-3), 7.34–7.22 (m, 5H, Ph-H), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J = 2.5 Hz, 1H, Nap-5), 4.40 (s, 2H, CH2Ph), 4.19 (dd, J = 11.5, 4.8 Hz, 1H, CH2 sn-3), 4.14 (dd, J = 11.5, 6.0 Hz, 1H, CH2 sn-3), 4.00–3.87 (m, 1H, CH sn-2), 3.91 (s, 3H, OCH3), 3.87 (q, J = 7.2 Hz, 1H, CHCH3), 3.40 (dd, J = 9.6, 4.4 Hz, 1H, CH2 sn-1), 3.32 (dd, J = 9.6, 6.0 Hz, 1H, CH2 sn-1), 2.31 (d, J = 5.2 Hz, 1H, OH), 1.58 (d, J = 7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.8, 157.9, 137.8, 135.6, 133.9, 129.4, 129.1, 128.6, 128.0 (2), 127.8 (2), 127.4, 126.3, 129.1, 119.2, 105.8, 73.6, 70.9, 69.0, 65.8, 55.5, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C24H26O5Na 417.1672; found, 417.1663.
3.3.4. Synthesis of 3-O-Benzyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (S,S′)-6
The same procedure was followed as described for (R,S′)-5 using 3-O-benzyl-sn-glycerol (76 mg, 0.417 mmol), naproxen acetoxime ester (S)-4 (172 mg, 0.439 mmol), and immobilized CAL-B (42 mg). Purification on a 4% boric-acid-impregnated flash silica gel column using petroleum ether/ethyl acetate (3:2) as an eluent resulted in a first fraction contaminated with the starting material that, as before, required repeated chromatography. Recrystallization of the combined fractions from n-hexane produced the product (S,S′)-6 as a white solid in a 65% yield (107 mg, 0.272 mmol). M.p. 63.2–63.5 °C. = +47.7 (c. 1.7, CH2Cl2). IR (NaCl, νmax/cm−1): 3540 (br s), 3053 (s), 2972 (vs), 2940 (vs), 2904 (vs), 2862 (vs), 1718 (vs), 1630 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71–7.64 (m, 3H, Nap-1,4,8), 7.38 (dd, J = 8.5, 1.9 Hz, 1H, Nap-3), 7.34–7.21 (m, 5H, Ph-H), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J = 2.5 Hz, 1H, Nap-5), 4.37 (s, 2H, CH2Ph), 4.17 (d, J = 5.5 Hz, 2H, CH2 sn-1), 4.00–3.90 (m, 1H, CH sn-2), 3.91 (s, 3H, OCH3), 3.88 (q, J = 7.2 Hz, 1H, CHCH3), 3.37 (dd, J = 9.6, 4.4 Hz, 1H, CH2 sn-3), 3.31 (dd, J = 9.6, 6.2 Hz, 1H, CH2 sn-3), 2.32 (d, J = 4.8 Hz, 1H, OH), 1.58 (d, J = 7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.8, 157.8, 137.8, 135.6, 133.9, 129.4, 129.1, 128.6, 128.0 (2), 127.8 (2), 127.4, 126.3, 126.1, 119.2, 105.8, 73.5, 70.8, 68.9, 65.7, 55.5, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C24H26O5Na 417.1672; found, 417.1671.
3.4. Removal of the Benzyl Protective Group: Synthesis of (R,S′)-7, (S,S′)-7, (R,S′)-8, and (S,S′)-8
3.4.1. Synthesis of 3-[(S)-2-(4-Isobutylphenyl)propanoyl]-sn-glycerol, (R,S′)-7
A Pd/C catalyst (8 mg) was added to a 25 mL flame-dried two-necked round-bottom flask equipped with a magnetic stirrer under nitrogen atmosphere at room temperature. The flask was sealed with a septum, and a solution of 1-O-benzyl-3-[(S)-2-(4-isobutylphenyl)-propanoyl]-sn-glycerol (R,S′)-5 (40 mg, 0.108 mmol) dissolved in dry THF (3.2 mL) was added with a syringe, followed by n-hexane (5.2 mL). A balloon filled with hydrogen gas that was mounted on a syringe was then stuck through the septum. Through stirring, the nitrogen atmosphere was replaced with hydrogen from the balloon by blowing it through the system. Then, a tiny drop of perchloric acid was added, and the solution was stirred vigorously at room temperature while being monitored with TLC. When the reaction had proceeded to an end, according to the TLC (approximately 12 min), the flask was promptly opened, and the acid was neutralized by adding NaHCO3 (s). Then, the solution was filtered, and the solvent was removed in vacuo on a rotary evaporator. The crude product was applied to a 4% boric-acid-impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (2:3) as an eluent to produce the product (R,S′)-7 as a pale-yellow oil in a 98% yield (30 mg, 0.107 mmol). = +42.9 (c. 3.5, CH2Cl2). IR (NaCl, νmax/cm−1): 3423 (br s), 3063 (s), 3025 (s), 2954 (vs), 2923 (vs), 2867 (vs), 1740 (vs) 1H NMR (400 MHz, CDCl3) δH: 7.19 (d, J = 8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J = 8.1 Hz, 2H, H-4,6 Ibu), 4.22 (dd, J = 11.6, 4.6 Hz, 1H, CH2 sn-3), 4.10 (dd, J = 11.4, 6.1 Hz, 1H, CH2 sn-3), 3.87–3.80 (m, 1H, CH sn-2), 3.74 (q, J = 7.2 Hz, 1H, CHCH3), 3.57 (dd, J = 11.5, 4.0 Hz, 1H, CH2 sn-1), 3.45 (dd, J = 11.5, 5.6 Hz, 1H, CH2 sn-1), 3.10–2.75 (bm, 2H, OH), 2.45 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 1.85 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.51 (d, J = 7.2 Hz, 3H, CHCH3), 0.89 (d, J = 6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 140.8, 137.4, 129.5 (2), 127.1 (2), 70.2, 65.4, 63.2, 45.1, 45.1, 30.2, 22.4 (2), 18.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C16H24O4Na 303.1567; found, 303.1563.
3.4.2. Synthesis of 1-[(S)-2-(4-Isobutylphenyl)propanoyl]-sn-glycerol, (S,S′)-7
The same procedure was followed as described for (R,S′)-7 using Pd/C (15 mg), 3-O-benzyl-1-[(S)-2-(4-isobutylphenyl)-propanoyl]-sn-glycerol (S,S′)-5 (50 mg, 0.135 mmol), THF (4.0 mL) and n-hexane (6.5 mL). Purification on a 4% boric-acid-impregnated flash silica gel column using petroleum ether/ethyl acetate (2:3) as an eluent produced the product (S,S′)-7 as a colorless liquid in a 93% yield (35 mg, 0.125 mmol). = +33.9 (c. 2.0, CH2Cl2). IR (NaCl, νmax/cm−1): 3455 (br s), 3060 (s), 3028 (s), 2954 (vs), 2925 (vs), 2865 (vs), 1742 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.19 (d, J = 8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J = 8.1 Hz, 2H, H-4,6 Ibu), 4.22 (dd, J = 11.6, 4.6 Hz, 1H, CH2 sn-1), 4.10 (dd, J = 11.4, 6.2 Hz, 1H, CH2 sn-1), 3.87–3.80 (m, 1H, CH sn-2), 3.74 (q, J = 7.2 Hz, 1H, CHCH3), 3.57 (dd, J = 11.5, 4.0 Hz, 1H, CH2 sn-3), 3.45 (dd, J = 11.5, 5.6 Hz, 1H, CH2 sn-3), 2.45 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 2.35–2.19 (bs, 1H, OH), 1.82–1.92 (bs, 1H, OH), 1.85 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.51 (d, J = 7.2 Hz, 3H, CHCH3), 0.89 (d, J = 6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 140.8, 137.5, 129.5 (2), 127.1 (2), 70.2, 65.4, 63.2, 45.1, 45.1, 30.2, 22.4 (2), 18.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C16H24O4Na 303.1567; found, 303.1569.
3.4.3. Synthesis of 3-[(S)-2-(6-Methoxynaphthalen-2-yl)]-sn-glycerol, (R,S′)-8
The same procedure was followed as described for (R,S′)-7 using 1-O-benzyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (R,S′)-6 (130 mg, 0.330 mmol), THF (9.5 mL), n-hexane (16.5 mL) and Pd/C catalyst (25 mg). Purification on a 4% boric-acid-impregnated flash silica gel column using petroleum ether/ethyl acetate (2:3) as an eluent, followed by recrystallization from n-hexane, produced the product (R,S′)-8 as white, thin, needle-like crystals in a 93% yield (93 mg, 0.306 mmol). M.p. 42.7–43.4 °C. = +43.5 (c. 2.2, CH2Cl2). IR (NaCl, νmax/cm−1): 3459 (br), 3058 (vs), 2980 (vs), 2940 (vs), 2878 (vs), 1732 (vs), 1634 (s), 1606 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71–7.61 (m, 3H, Nap-1,4,8), 7.38 (dd, J = 8.6, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J = 2.5 Hz, 1H, Nap-5), 4.23–4.07 (m, 2H, CH2 sn-3), 3.90 (s, 3H, OCH3), 3.96–3.84 (m, 1H, CH sn-2), 3.81 (q, J = 7.2 Hz, 1H, CHCH3), 3.57 (m, 1H, CH2 sn-1), 3.45 (m, 1H, CH2 sn-1), 1.59 (d, J = 7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 157.9, 135.4, 133.9, 129.4, 129.0, 127.4, 126.1 (2), 119.3, 105.8, 70.2, 65.7, 63.3, 55.4, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C17H20O5Na 327.1203; found, 327.1201.
3.4.4. Synthesis of 1-[(S)-2-(6-Methoxynaphthalen-2-yl)]-sn-glycerol, (S,S′)-8
The same procedure was followed as described for (R,S′)-7 using 3-O-benzyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (S,S′)-6 (126 mg, 0.319 mmol), THF (9.5 mL), n-hexane (15 mL) and Pd/C catalyst (17 mg). Purification on a 4% boric-acid-impregnated flash silica gel column using petroleum ether/ethyl acetate (2:3) as an eluent, followed by recrystallization from n-hexane, produced the product (S,S′)-8 as white solid in a 99% yield (96 mg, 0.315 mmol). M.p. 58.9–59.7 °C. = +34.3 (c. 1.0, CH2Cl2). IR (NaCl, νmax/cm−1): 3455 (br s), 3056 (vs), 2982 (vs), 2945 (vs), 2874 (vs), 1734 (vs), 1633 (s), 1605 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71–7.61 (m, 3H, Nap-1,4,8), 7.39 (dd, J = 8.6, 1.5 Hz, 1H, Nap-3), 7.15 (dd, J = 8.9, 2.4 Hz, 1H, Nap-7), 7.12 (d, J = 2.4 Hz, 1H, Nap-5), 4.18 (d, J = 4.9 Hz, 2H, CH2 sn-1), 3.92 (s, 3H, OCH3), 3.92–3.88 (m, 1H, CHCH3), 3.88–3.82 (m, 1H, CH sn-2), 3.56 (dd, J = 11.1, 4.9 Hz, 1H, CH2 sn-3), 3.45 (dd, J = 11.1, 5.6 Hz, 1H, CH2 sn-3), 2.30–184 (m, 2H, OH), 1.59 (d, J = 7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 157.9, 135.4, 133.9, 129.4, 129.1, 127.5, 126.1 (2), 119.3, 105.8, 70.2, 65.7, 63.3, 55.5, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C17H20O5Na 327.1203; found, 327.1212.
3.5. The Enzymatic Coupling of the MCFAs: Synthesis of (R,S′)-9a, (S,S′)-9a, (R,S′)-10a, and (S,S′)-10a
3.5.1. Synthesis of 1-Hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S′)-9a
Immobilized CAL-B (18 mg) was added to a solution of 3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (R,S′)-7 (37 mg, 0.132 mmol), and vinyl hexanoate (21 mg, 0.145 mmol) in CH2Cl2 (3.5 mL). The resulting mixture was stirred at room temperature for 7 h. The lipase preparation was separated by filtration, and the solvent was removed in vacuo on a rotary evaporator. The concentrate was applied to a 4% boric-acid-impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (7:3) as an eluent. This produced the product (R,S′)-9a as a colorless liquid in an 80% yield (40 mg, 0.106 mmol). = +22.7 (c. 3.0, CH2Cl2). IR (NaCl, νmax/cm−1): 3321 (br s), 2956 (vs), 2931 (vs), 2870 (vs), 1740 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.19 (d, J = 8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J = 8.1 Hz, 2H, H-4,6 Ibu), 4.21–3.94 (m, 5H, CH2 sn-1/3, CH sn-2), 3.74 (q, J = 7.2 Hz, 1H, CHCH3), 2.44 (d, J = 6.8 Hz, 2H, CH2CH(CH3)2), 2.34–2.27 (m, 2H, CH2COO SFA), 1.84 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.67–1.57 (m, 2H, CH2CH2COO), 1.50 (d, J = 7.2 Hz, 3H, CHCH3), 1.37–1.22 (m, 4H, CH2), 0.90 (t, J = 6.9 Hz, 3H, CH2CH3), 0.89 (d, J = 6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9 (C=O Ibu), 174.0 (C=O SFA), 140.9, 137.6, 129.6 (2), 127.2 (2), 68.5, 65.5, 65.0, 45.2 (2), 34.2, 31.4, 30.3, 24.7, 22.5 (2), 22.4, 18.5, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H34O5Na 401.2298; found, 401.2300.
3.5.2. Synthesis of 3-Hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S′)-9a
Immobilized CAL-B (17 mg) was added to a solution of 1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (S,S′)-7 (25 mg, 0.089 mmol), and vinyl hexanoate (14 mg, 0.098 mmol) in CH2Cl2 (2 mL). The resulting mixture was stirred at room temperature for 7 h. The lipase preparation was separated by filtration, and the solvent was removed in vacuo on a rotary evaporator. The concentrate was applied to a 4% boric-acid-impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (4:1) as an eluent. This produced the product (S,S′)-9a as a colorless liquid in a 94% yield (32 mg, 0.085 mmol). = +21.0 (c. 0.4, CH2Cl2). IR (NaCl, νmax/cm−1): 3465 (br s), 2975 (vs), 2941 (vs), 2864 (vs), 2834 (vs), 1738 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.20 (d, J = 8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J = 8.1 Hz, 2H, H-4,6 Ibu), 4.21–3.97 (m, 5H, CH2 sn-1/3, CH sn-2), 3.74 (q, J = 7.2 Hz, 1H, CHCH3), 2.44 (d, J = 6.8 Hz, 2H, CH2CH(CH3)2), 2.39–2.28 (m, 1H, OH), 2.34–2.27 (m, 2H, CH2COO SFA), 1.84 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.68–1.55 (m, 2H, CH2CH2COO), 1.51 (d, J = 7.2 Hz, 3H, CHCH3), 1.38–1.24 (m, 4H, CH2), 0.90 (t, J = 6.9 Hz, 3H, CH2CH3), 0.89 (d, J = 6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9 (C=O Ibu), 174.0 (C=O SFA), 140.9, 137.6, 129.6 (2), 127.2 (2), 68.5, 65.4, 65.0, 45.2, 45.2, 34.2, 31.4, 30.3, 24.7, 22.5 (2), 22.4, 18.5, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H34O5Na 401.2298; found, 401.2294.
3.5.3. Synthesis of 1-Hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (R,S′)-10a
Immobilized CAL-B (15 mg) was added to a solution of 3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (R,S′)-8 (27 mg, 0.089 mmol), and vinyl hexanoate (14 mg, 0.098 mmol) in CH2Cl2 (2.4 mL). The resulting mixture was stirred at room temperature for 2 h after which more CAL-B (5 mg) was added to speed up the reaction. After a further 3.5 h of reaction, TLC monitoring indicated a complete reaction. The lipase preparation was separated by filtration, and the solvent was removed in vacuo on a rotary evaporator. The concentrate was applied to a 4% boric-acid-impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (7:3) as an eluent. This produced the product (R,S′)-10a as a colorless liquid in a 92% yield (33 mg, 0.082 mmol). = +22.6 (c. 2.5, CH2Cl2). IR (NaCl, νmax/cm−1): 3459 (br s), 2946 (vs), 2930 (vs), 2870 (vs), 1740 (vs), 1635 (s), 1605 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.86–7.58 (m, 3H, Nap-1,4,8), 7.47–7.30 (m, 1H, Nap-3), 7.19–7.04 (m, 2H, Nap-5,7), 4.33–3.62 (m, 9H, CH2 sn-1/3, CH sn-2, OCH3, CHCH3), 2.24–2.19 (m, 3H, OH, CH2COO), 1.61–1.52 (m, 5H, CH2CH2COO, CHCH3), 1.32–1.23 (m, 4H, CH2), 0.87 (t, J = 6.7 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Nap), 173.3 (C=O SFA), 157.9, 135.4, 133.9, 129.4, 129.1, 127.4, 126.10, 126.07, 119.2, 105.8, 68.4, 65.6, 65.0, 55.4, 45.4, 34.1, 31.4, 24.7, 22.4, 18.5, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O6Na 425.1935; found, 425.1933.
3.5.4. Synthesis of 3-Hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (S,S′)-10a
Immobilized CAL-B (15 mg) was added to a solution of 1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (S,S′)-8 (33 mg, 0.108 mmol), and vinyl hexanoate (28 mg, 0.198 mmol) in CH2Cl2 (3 mL). The resulting mixture was stirred at room temperature for 3 h after which more CAL-B (5 mg) was added to speed up the reaction. After a further 5.5 h of reaction, TLC monitoring indicated a complete reaction. The lipase preparation was separated by filtration, and the solvent was removed in vacuo on a rotary evaporator. The concentrate was applied to a 4% boric-acid-impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (7:3) as an eluent. This produced the product (S,S′)-10a as a colorless liquid in a 70% yield (30 mg, 0.075 mmol). = +21.5 (c. 0.6, CH2Cl2). IR (NaCl, νmax/cm−1): 3466 (br s), 2969 (vs), 2972 (vs), 1735 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.73–7.68 (m, 2H, Nap-4,8), 7.66 (d, J = 1.9 Hz, 1H, Nap-1), 7.39 (dd, J = 8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.11 (d, J = 2.5 Hz, 1H, Nap-5), 4.21–3.98 (m, 5H, CH2 sn-1/3, CH sn-2), 3.91 (s, 3H, OCH3), 3.90 (q, J = 7.2 Hz, 1H, CHCH3), 2.28 (t, J = 7.6 Hz, 2H, CH2COO), 1.61–1.57 (m, 2H, CH2CH2COO), 1.59 (d, J = 7.2 Hz, 3H, CHCH3), 1.32–1.23 (m, 4H, CH2), 0.88 (t, J = 6.9 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Nap), 174.8 (C=O Nap), 174.0 (C=O SFA), 157.9, 135.4, 133.9, 129.4, 129.1, 127.5, 126.2, 126.1, 119.3, 105.8, 68.5, 65.6, 65.0, 55.5, 45.5, 34.2, 31.4, 24.7, 22.4, 18.6, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O6Na 425.1935; found, 425.1939.
3.6. Coupling of EPA: Synthesis of (R,S′)-11a, (S,S′)-11a, (R,S′)-12a and (S,S′)-12a
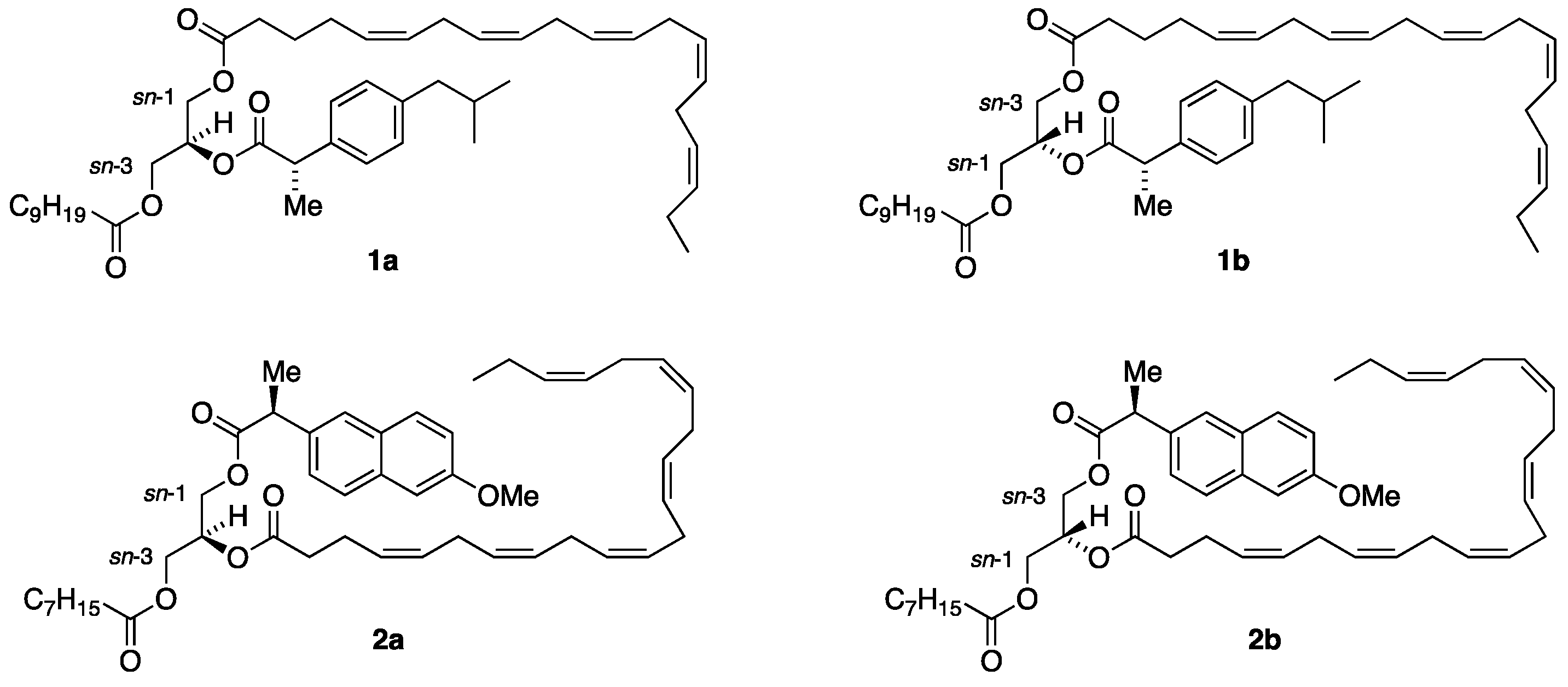
3.6.1. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-Eicosa-5,8,11,14,17-pentaenoyl]-1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S′)-11a
To a solution of 1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (R,S′)-9a (15 mg, 0.040 mmol) and EPA as a free acid (13 mg, 0.044 mmol) in CH2Cl2 (2 mL) were added DMAP (6 mg, 0.043 mmol) and EDCI (12 mg, 0.058 mmol). The solution was stirred on a magnetic stirrer at room temperature for 23 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (9:1) as an eluent, which produced the product (R,S′)-11a as a yellow oil, in a 96% yield (26 mg, 0.039 mmol). = +8.29 (c. 2.8, CH2Cl2). IR (NaCl, νmax/cm−1): 3012 (vs), 2958 (vs), 2927 (vs), 2871 (vs), 1744 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J = 8.1 Hz, 2H, Ibu-2,6), 7.08 (d, J = 8.1 Hz, 2H, Ibu-3,5), 5.40–5.28 (m, 10H, =CH), 5.23–5.17 (m, 1H, CH sn-2), 4.29 (dd, J = 11.9, 4.4 Hz, 1H, CH2 sn-1/3), 4.21 (dd, J = 11.9, 5.4 Hz, 1H, CH2 sn-1/3), 4.14 (dd, J = 11.9, 5.8 Hz, 1H, CH2 sn-1/3), 4.00 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J = 7.1 Hz, 1H, CHCH3), 2.89–2.77 (m, 8H, =CHCH2CH=), 2.44 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 2.31–2.20 (m, 4H, CH2COO EPA, CH2COO SFA), 2.13–2.03 (m, 4H, CH2CH2CH= and =CHCH2CH3), 1.84 (nonet, J = 6.9 Hz, 1H, CH(CH3)2), 1.69–1.62 (m, 2H, CH2CH2COO EPA), 1.62–1.55 (m, 2H, CH2CH2COO SFA), 1.49 (d, J = 7.1 Hz, 3H, CHCH3), 1.33–1.21 (m, 4H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH3 EPA), 0.89 (d, J = 6.7 Hz, 6H, CH(CH3)2), 0.88 (t, J = 7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.6 (C=O EPA), 140.8, 137.4, 132.2, 129.5 (2), 129.1, 129.0, 128.7, 128.5, 128.4, 128.3, 128.2, 128.0, 127.3 (2), 127.2, 69.1, 62.4, 62.1, 45.2, 45.1, 34.7, 34.1, 33.7, 31.4, 26.7, 26.4 (3), 25.8, 24.8, 24.7, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C42H62O6Na 685.4439; found, 685.4412.
3.6.2. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-Eicosa-5,8,11,14,17-pentaenoyl]-3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S′)-11a
To a solution of 3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (S,S′)-9a (11 mg, 0.029 mmol) and EPA as a free acid (10 mg, 0.032 mmol) in CH2Cl2 (1.5 mL) were added DMAP (4 mg, 0.031 mmol) and EDCI (8 mg, 0.042 mmol). The solution was stirred on a magnetic stirrer at room temperature for 25 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as an eluent, which produced the product (S,S′)-11a as a yellow oil, in an 89% yield (17 mg, 0.026 mmol). = +8.27 (c. 2.2, CH2Cl2). IR (NaCl, νmax/cm−1): 3013 (vs), 2970 (vs), 2873 (vs), 2829 (vs), 1744 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J = 8.1 Hz, 2H, Ibu-2,6), 7.08 (d, J = 8.1 Hz, 2H, Ibu-3,5), 5.40–5.26 (m, 10H, =CH), 5.23–5.17 (m, 1H, CH sn-2), 4.29 (dd, J = 11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.19 (dd, J = 11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.12 (dd, J = 11.9, 6.1 Hz, 1H, CH2 sn-1/3), 4.05 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J = 7.1 Hz, 1H, CHCH3), 2.87–2.77 (m, 8H, =CHCH2CH=), 2.44 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 2.37–2.20 (m, 4H, CH2COO EPA, CH2COO SFA), 2.13–2.02 (m, 4H, CH2CH2CH= and =CHCH2CH3), 1.85 (nonet, J = 6.9 Hz, 1H, CH(CH3)2), 1.70–1.54 (m, 4H, CH2CH2COO EPA, CH2CH2COO SFA), 1.49 (d, J = 7.1 Hz, 3H, CHCH3), 1.33–1.21 (m, 4H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH3 EPA), 0.89 (d, J = 6.8 Hz, 6H, CH(CH3)2), 0.88 (t, J = 7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.7 (C=O EPA), 140.8, 137.5, 132.2, 129.5 (2), 129.1, 129.0, 128.7, 128.5, 128.4, 128.3, 128.2, 128.0, 127.3 (2), 127.2, 69.0, 62.6, 62.1, 45.2, 45.1, 34.7, 34.1, 33.7, 31.4, 26.7, 25.8 (3), 25.7, 24.9, 24.7, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C42H62O6Na 685.4439; found, 685.4436.
3.6.3. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-Eicosa-5,8,11,14,17-pentaenoyl]-1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (R,S′)-12a
To a solution of 1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (R,S′)-10a (10 mg, 0.025 mmol) and EPA as a free acid (8 mg, 0.027 mmol) in CH2Cl2 (1.3 mL) were added DMAP (3 mg, 0.027 mmol) and EDCI (8 mg, 0.037 mmol). The solution was stirred on a magnetic stirrer at room temperature for 24 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as an eluent, which produced the product (R,S′)-12a as a yellow oil, in an 80% yield (14 mg, 0.020 mmol). = +9.29 (c. 1.4, CH2Cl2). IR (NaCl, νmax/cm−1): 3013 (vs), 2970 (vs), 2940 (vs), 2853 (vs), 1743 (vs), 1635 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72–7.66 (m, 2H, Nap-4,8), 7.64 (d, J = 1.9 Hz, 1H, Nap-1), 7.37 (dd, J = 8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J = 2.5 Hz, 1H, Nap-5), 5.48–5.27 (m, 10H, =CH), 5.20 (m, 1H, CH sn-2), 4.30 (dd, J = 11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.22 (dd, J = 11.9, 4.4 Hz, 1H CH2 sn-1/3), 4.16 (dd, J = 11.9, 6.0 Hz, 1H, CH2 sn-1/3), 4.03 (dd, J = 11.9, 5.8 Hz, 1H, CH2 sn-1/3), 3.90 (s, 3H, OCH3), 3.86 (q, J = 7.2 Hz, 1H, CHCH3), 2.90–2.75 (m, 8H, =CHCH2CH=), 2.24 (t, J = 7.5 Hz, 2H, CH2COO EPA), 2.12–2.04 (m, 2H, CH2COO SFA), 2.08 (td, J = 7.4, 1.6 Hz, 2H, CH2CH2CH=), 2.05–1.96 (m, 2H, =CHCH2CH3), 1.83–1.75 (m, 2H, CH2CH2COO EPA), 1.60–1.51 (m, 5H, CH2CH2COO SFA and CHCH3), 1.34–1.21 (m, 4H, CH2), 0.98 (t, J = 7.5 Hz, 3H, CH3 EPA), 0.88 (t, J = 7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.2 (C=O Nap), 173.3 (C=O SFA), 172.6 (C=O EPA), 157.9, 135.3, 133.9, 132.2, 129.4, 129.1, 129.0, 128.7, 128.6, 128.5, 128.4, 128.3, 128.2, 128.0, 127.3, 127.2, 126.3, 126.1, 119.2, 105.7, 69.0, 62.5, 62.1, 55.4, 45.5, 34.1, 33.6, 31.4, 26.4, 25.8 (3), 25.7, 24.6, 24.2, 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C43H58O7Na 709.4075; found, 709.4059.
3.6.4. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-Eicosa-5,8,11,14,17-pentaenoyl]-3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (S,S′)-12a
To a solution of 3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (S,S′)-10a (11 mg, 0.027 mmol) and EPA as a free acid (9 mg, 0.030 mmol) in CH2Cl2 (1.3 mL) were added DMAP (4 mg, 0.029 mmol) and EDCI (8 mg, 0.040 mmol). The solution was stirred on a magnetic stirrer at room temperature for 30 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as an eluent, which produced the product (S,S′)-12a as a yellow oil, in a 95% yield (18 mg, 0.026 mmol). = +12.4 (c. 1.5, CH2Cl2). IR (NaCl, νmax/cm−1): 3012 (vs), 2962 (vs), 2934 (vs), 2873 (vs), 1743 (vs), 1635 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72–7.66 (m, 2H, Nap-4,8), 7.65 (d, J = 1.9 Hz, 1H, Nap-1), 7.37 (dd, J = 8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J = 2.5 Hz, 1H, Nap-5), 5.44–5.28 (m, 10H, =CH), 5.24 (m, 1H, CH sn-2), 4.30 (dd, J = 11.9, 4.1 Hz, 1H, CH2 sn-1/3), 4.20 (dd, J = 11.9, 4.4 Hz, 1H CH2 sn-1/3), 4.13 (dd, J = 11.9, 6.3 Hz, 1H, CH2 sn-1/3), 4.06 (dd, J = 11.9, 5.8 Hz, 1H, CH2 sn-1/3), 3.91 (s, 3H, OCH3), 3.86 (q, J = 7.2 Hz, 1H, CHCH3), 2.89–2.75 (m, 8H, =CHCH2CH=), 2.24 (t, J = 7.5 Hz, 2H, CH2COO EPA), 2.19–2.11 (m, 2H, CH2COO SFA), 2.07 (td, J = 7.4, 1.4 Hz, 2H, CH2CH2CH=), 2.05–1.97 (m, 2H, =CHCH2CH3), 1.85–1.71 (m, 2H, CH2CH2COO EPA), 1.60–1.53 (m, 2H, CH2CH2COO SFA), 1.58 (d, J = 7.2 Hz, 3H, CHCH3), 1.34–1.21 (m, 4H, CH2), 0.98 (t, J = 7.5 Hz, 3H, CH3 EPA), 0.88 (t, J = 6.9 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.2 (C=O Nap), 173.3 (C=O SFA), 172.6 (C=O EPA), 157.9, 135.4, 133.9, 132.2, 129.4, 129.1, 129.0, 128.44, 128.36, 128.3, 128.2, 128.0, 127.3, 127.2, 126.3, 126.1, 119.2, 105.7, 69.0, 62.7, 62.1, 55.4, 45.4, 34.1, 33.6, 31.4, 29.9, 26.6, 25.78, 25.75 (3), 25.7, 24.8, 24.6, 22.4, 20.7, 18.5, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C43H58O7Na 709.4059; found, 709.4059.
3.7. Coupling of DHA: Synthesis of (R,S′)-13a, (S,S′)-13a, (R,S′)-14a and (S,S′)-14a
3.7.1. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-Docosa-4,7,10,13,16,19-hexaenoyl]-1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S′)-13a
To a solution of 1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (R,S′)-9a (15 mg, 0.040 mmol) and DHA as a free acid (15 mg, 0.044 mmol) in CH2Cl2 (2 mL) were added DMAP (6 mg, 0.043 mmol) and EDCI (12 mg, 0.058 mmol). The solution was stirred on a magnetic stirrer at room temperature for 23 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (9:1) as an eluent, which produced the product (R,S′)-13a as a yellow oil, in a 79% yield (22 mg, 0.032 mmol). = +6.90 (c. 1.0, CH2Cl2). IR (NaCl, νmax/cm−1): 3013 (vs), 2954 (vs), 2925 (vs), 2854 (vs), 1743 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J = 7.8 Hz, 2H, Ibu-2,6), 7.08 (d, J = 7.8 Hz, 2H, Ibu-3,5), 5.50–5.24 (m, 12H, =CH), 5.23–5.17 (m, 1H, CH sn-2), 4.29 (dd, J = 11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.21 (dd, J = 11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.14 (dd, J = 11.9, 5.7 Hz, 1H, CH2 sn-1/3), 4.01 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J = 7.2 Hz, 1H, CHCH3), 2.89–2.79 (m, 10H, =CHCH2CH=), 2.44 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 2.37–2.19 (m, 6H, CH2CH2COO DHA, CH2COO SFA), 2.08 (quint., J = 7.6 Hz, 2H, =CHCH2CH3), 1.83 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.62–1.56 (m, 2H, CH2CH2COO SFA), 1.49 (d, J = 7.2 Hz, 3H, CHCH3), 1.36–1.13 (m, 4H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH3 DHA), 0.89 (d, J = 6.4 Hz, 6H, CH(CH3)2), 0.88 (t, J = 7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.9 (C=O DHA), 140.8, 137.4, 132.2, 129.5 (2), 128.7, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6 (2), 127.3, 127.2, 69.00, 62.5, 62.1, 45.2, 45.1, 34.2, 34.1, 31.4, 30.3, 25.8 (3), 25.7, 25.5, 24.7, 22.7, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C44H64O6Na 711.4595; found, 711.4577.
3.7.2. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-Docosa-4,7,10,13,16,19-hexaenoyl]-3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S′)-13a
To a solution of 3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (S,S′)-9a (11 mg, 0.029 mmol) and DHA as a free acid (11 mg, 0.032 mmol) in CH2Cl2 (1.5 mL) were added DMAP (4 mg, 0.031 mmol) and EDCI (8 mg, 0.042 mmol). The solution was stirred on a magnetic stirrer at room temperature for 25 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as an eluent, which produced the product (S,S′)-13a as a yellow oil, in a 65% yield (13 mg, 0.019 mmol). = +8.22 (c. 0.9, CH2Cl2). IR (NaCl, νmax/cm−1): 3013 (vs), 2972 (vs), 2874 (vs), 1748 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J = 7.8 Hz, 2H, Ibu-2,6), 7.08 (d, J = 7.8 Hz, 2H, Ibu-3,5), 5.46–5.27 (m, 12H, =CH), 5.23 (tt, J = 6.0, 4.3 Hz, 1H, CH sn-2), 4.29 (dd, J = 11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.19 (dd, J = 11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.12 (dd, J = 11.9, 6.1 Hz, 1H, CH2 sn-1/3), 4.05 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J = 7.2 Hz, 1H, CHCH3), 2.90–2.80 (m, 10H, =CHCH2CH=), 2.44 (d, J = 7.2 Hz, 2H, CH2CH(CH3)2), 2.39–2.18 (m, 6H, CH2CH2COO DHA, CH2COO SFA), 2.13–2.03 (m, 2H, =CHCH2CH3), 1.84 (nonet, J = 6.8 Hz, 1H, CH(CH3)2), 1.64–1.57 (m, 2H, CH2CH2COO SFA), 1.49 (d, J = 7.2 Hz, 3H, CHCH3), 1.36–1.23 (m, 4H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH3 DHA), 0.89 (d, J = 6.4 Hz, 6H, CH(CH3)2), 0.88 (t, J = 7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.2 (C=O DHA), 140.9, 137.5, 132.2, 129.5 (2), 128.7, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6 (2), 127.3, 127.2, 69.1, 62.5, 62.1, 45.2, 45.1, 34.2, 34.1, 31.4, 30.3, 25.8 (3), 25.8, 25.7, 24.7, 22.8, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C44H64O6Na 711.4595; found, 711.4581.
3.7.3. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-Docosa-4,7,10,13,16,19-hexaenoyl]-1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (R,S′)-14a
To a solution of 1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (R,S′)-10a (10 mg, 0.025 mmol) and DHA as a free acid (9 mg, 0.027 mmol) in CH2Cl2 (1.3 mL) were added DMAP (3 mg, 0.027 mmol) and EDCI (8 mg, 0.037 mmol). The solution was stirred on a magnetic stirrer at room temperature for 24 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as an eluent, which produced the product (R,S′)-14a as a yellow oil, in an 84% yield (15 mg, 0.021 mmol). = +8.00 (c. 1.5, CH2Cl2). IR (NaCl, νmax/cm−1): 3009 (vs), 2979 (vs), 2941 (vs), 2837 (vs), 1740 (vs), 1634 (s), 1609 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72–7.66 (m, 2H, Nap-4,8), 7.65 (d, J = 1.9 Hz, 1H, Nap-1), 7.37 (dd, J = 8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J = 2.5 Hz, 1H, Nap-5), 5.44–5.24 (m, 12H, =CH), 5.21 (tt, J = 5.9, 4.5 Hz, 1H, CH sn-2), 4.30 (dd, J = 11.9, 4.4 Hz, 1H, CH2 sn-1/3), 4.21 (dd, J = 11.9, 4.4 Hz, 1H, CH2 sn-1/3), 4.16 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 4.03 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.91 (s, 3H, OCH3), 3.87 (q, J = 7.2 Hz, 1H, CHCH3), 2.89–2.76 (m, 10H, =CHCH2CH=), 2.31–2.16 (m, 6H, CH2CH2COO DHA, =CHCH2CH3), 2.13–2.03 (m, 2H, CH2COO SFA), 1.63–1.50 (m, 2H, CH2CH2COO SFA), 1.58 (d, J = 7.1 Hz, 3H, CHCH3), 1.33–1.20 (m, 4H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH3 DHA), 0.88 (t, J = 7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.2 (C=O Nap), 173.3 (C=O SFA), 172.1 (C=O DHA), 157.9, 135.3, 133.9, 132.2, 129.5, 129.4, 129.1, 128.7, 128.47 (2), 128.45, 128.4, 128.3, 128.23, 128.16, 128.0, 127.8, 127.3, 127.2, 126.3, 126.1, 119.2, 105.8, 69.2, 62.5, 62.1, 55.5, 45.5, 34.1, 34.0, 31.4, 25.8 (3), 25.73, 25.70, 24.6, 22.7, 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C45H60O7Na 735.4231; found, 735.4210.
3.7.4. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-Docosa-4,7,10,13,16,19-hexaenoyl]-3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (S,S′)-14a
To a solution of 3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (S,S′)-10a (11 mg, 0.027 mmol) and DHA as a free acid (10 mg, 0.030 mmol) in CH2Cl2 (1.3 mL) were added DMAP (4 mg, 0.029 mmol) and EDCI (8 mg, 0.037 mmol). The solution was stirred on a magnetic stirrer at room temperature for 30 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by using Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as an eluent, which produced the product (S,S′)-14a as a yellow oil, in a 79% yield (15 mg, 0.021 mmol). = +12.6 (c. 0.5, CH2Cl2). IR (NaCl, νmax/cm−1): 3012 (vs), 2977 (vs), 2941 (vs), 2878 (vs), 2834 (vs), 1741 (vs), 1635 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72–7.66 (m, 2H, Nap-4,8), 7.65 (d, J = 1.9 Hz, 1H, Nap-1), 7.37 (dd, J = 8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J = 8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J = 2.5 Hz, 1H, Nap-5), 5.44–5.24 (m, 12H, =CH), 5.21 (tt, J = 6.0, 4.4 Hz, 1H, CH sn-2), 4.30 (dd, J = 11.9, 4.2 Hz, 1H, CH2 sn-1/3), 4.19 (dd, J = 11.9, 4.5 Hz, 1H, CH2 sn-1/3), 4.13 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 4.03 (dd, J = 11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.91 (s, 3H, OCH3), 3.85 (q, J = 7.1 Hz, 1H, CHCH3), 2.89–2.77 (m, 10H, =CHCH2CH=), 2.30–2.16 (m, 6H, CH2CH2COO DHA, =CHCH2CH3), 2.10–2.03 (m, 2H, CH2COO SFA), 1.60–1.52 (m, 2H, CH2CH2COO SFA), 1.58 (d, J = 7.1 Hz, 3H, CHCH3), 1.33–1.20 (m, 4H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH3 DHA), 0.88 (t, J = 7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Nap), 173.3 (C=O SFA), 172.2 (C=O DHA), 157.9, 135.3, 133.9, 132.2, 129.5, 129.4, 129.1, 128.7, 128.5 (2), 128.5, 128.4, 128.3, 128.2, 128.0, 127.8, 127.4, 127.2, 126.3, 126.1, 119.2, 105.8, 69.1, 62.7, 62.1, 55.5, 45.4, 34.1, 34.0, 31.4, 25.8 (3), 25.74, 25.71, 24.6, 22.7, 22.4, 20.7, 18.5, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C45H60O7Na 735.4231; found, 735.4217.




{kind=link}
{kind=link}
{kind=link}
{kind=link}