
Enhancing the Thermostability of Bacillus licheniformis Alkaline Protease 2709 by Computation-Based Rational Design


Abstract
1. Introduction
2. Results
2.1. Three-Dimensional Structure Prediction of AprE 2709 (WT) Using AlphaFold 3 and SWISS-MODEL
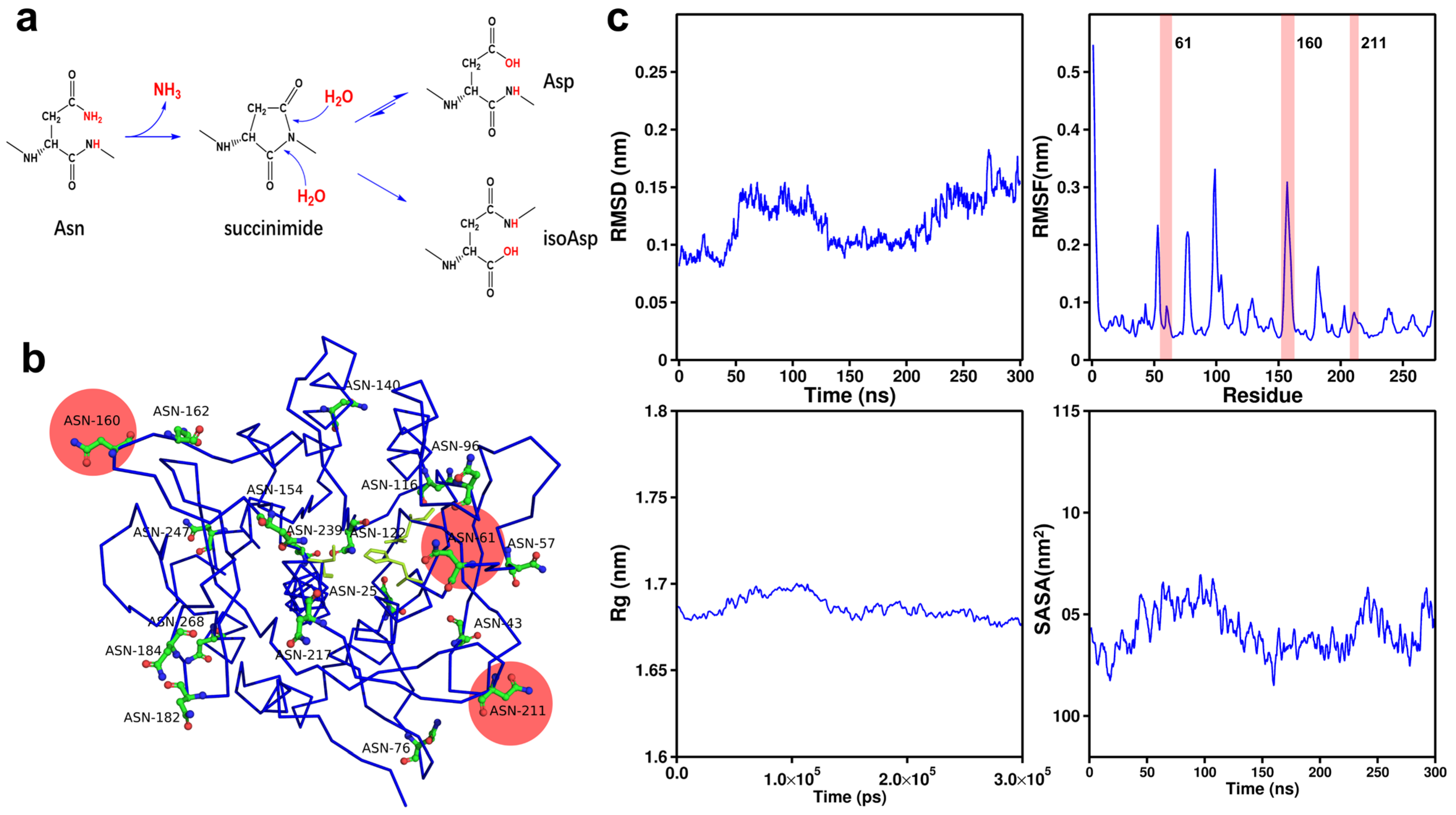
2.2. Identification of Potential Mutation Sites in AprE 2709 (WT) Through Amino Acid Fluctuation and Spatial Position Analysis
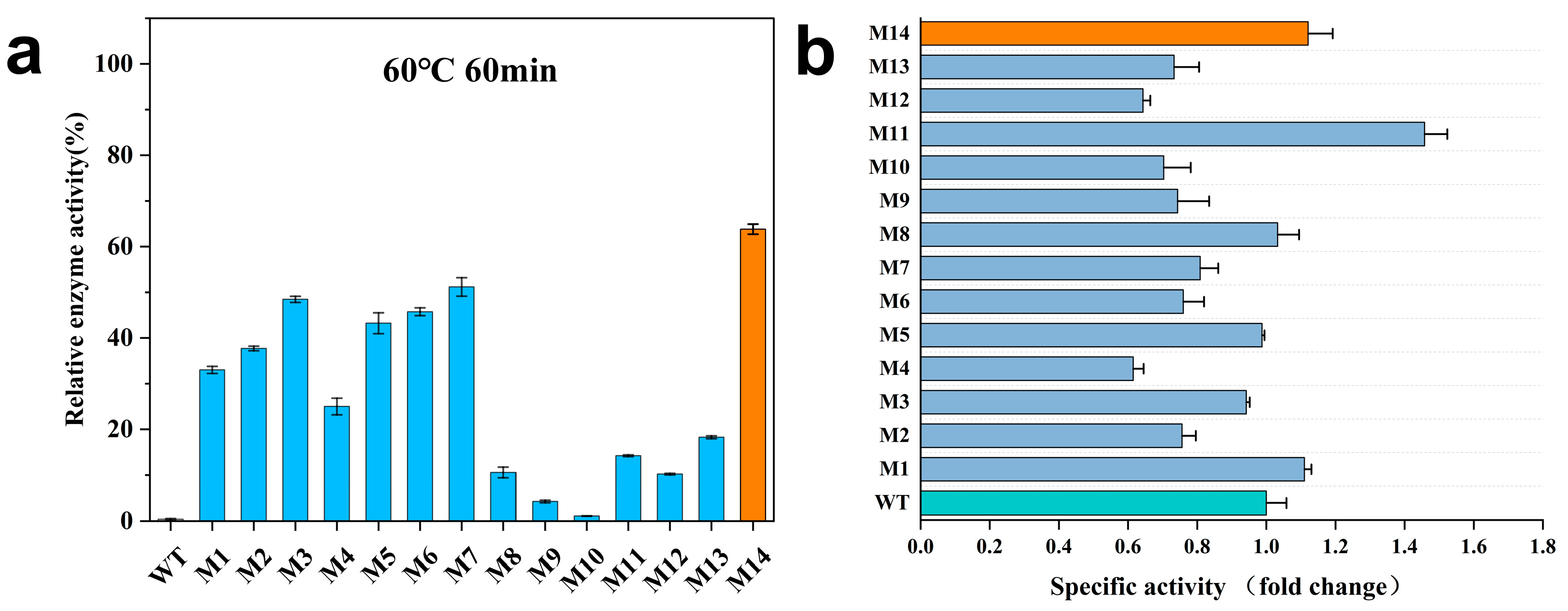
2.3. Biochemical Characterization of AprE 2709 Mutants
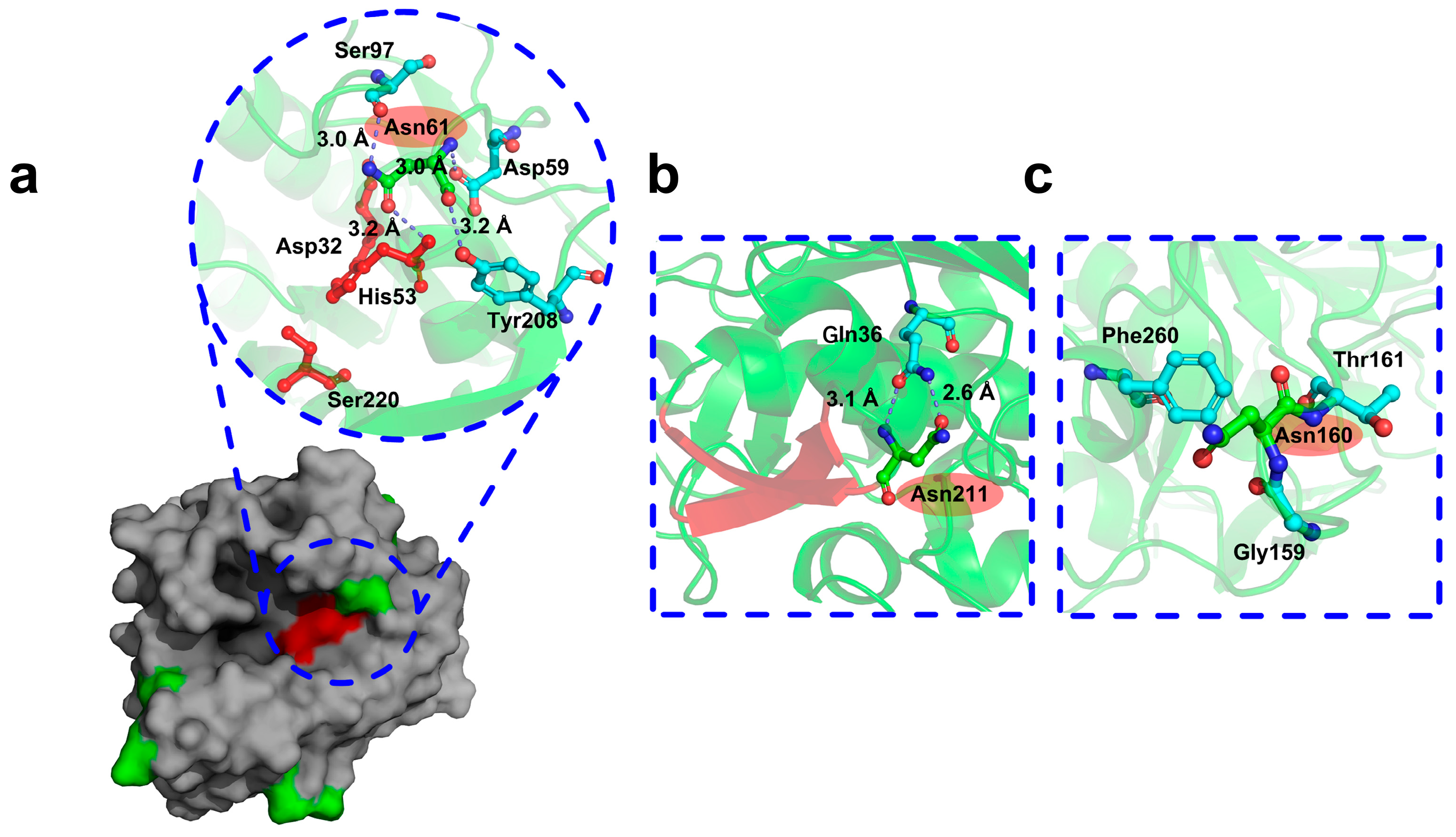
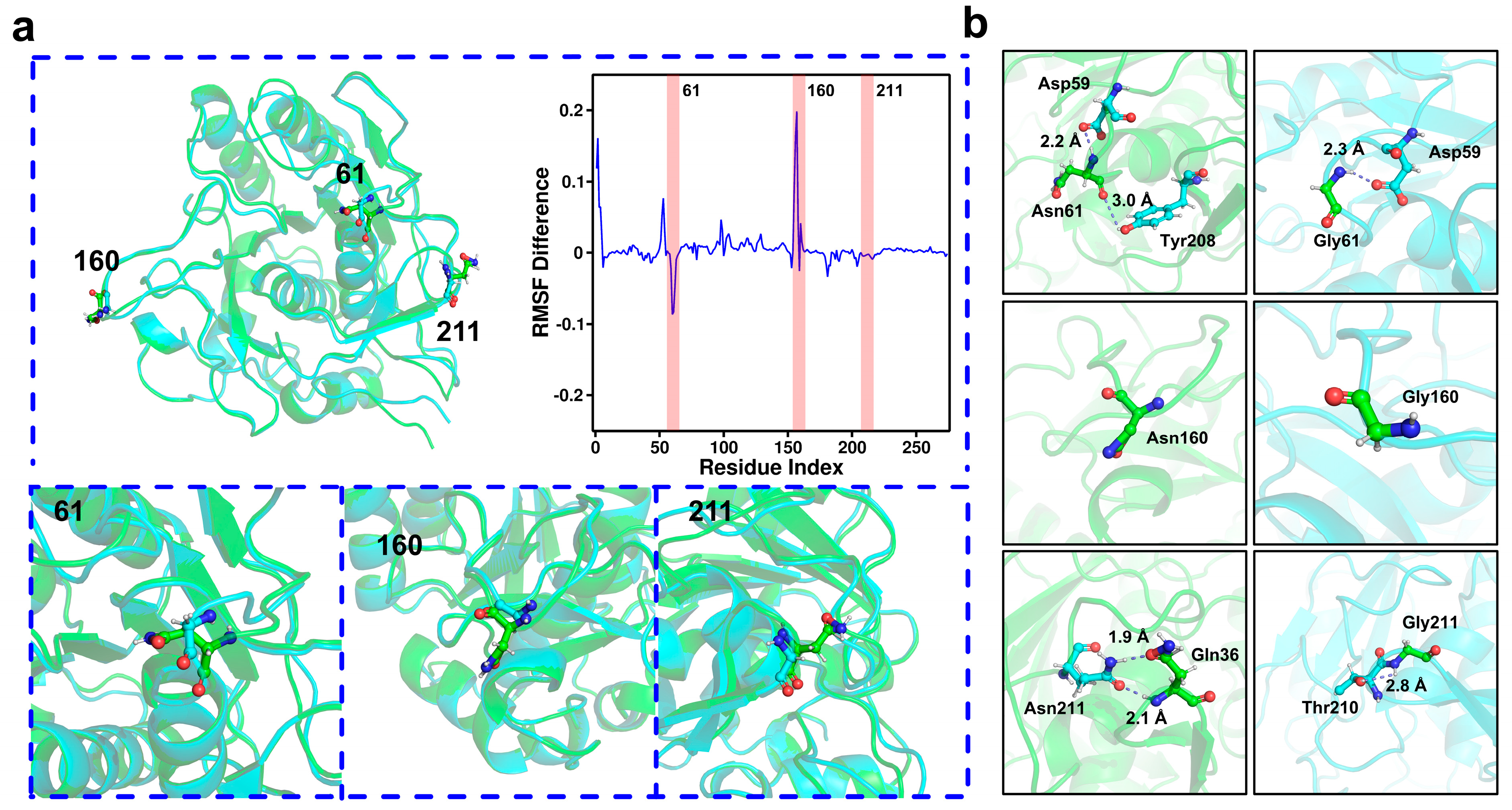
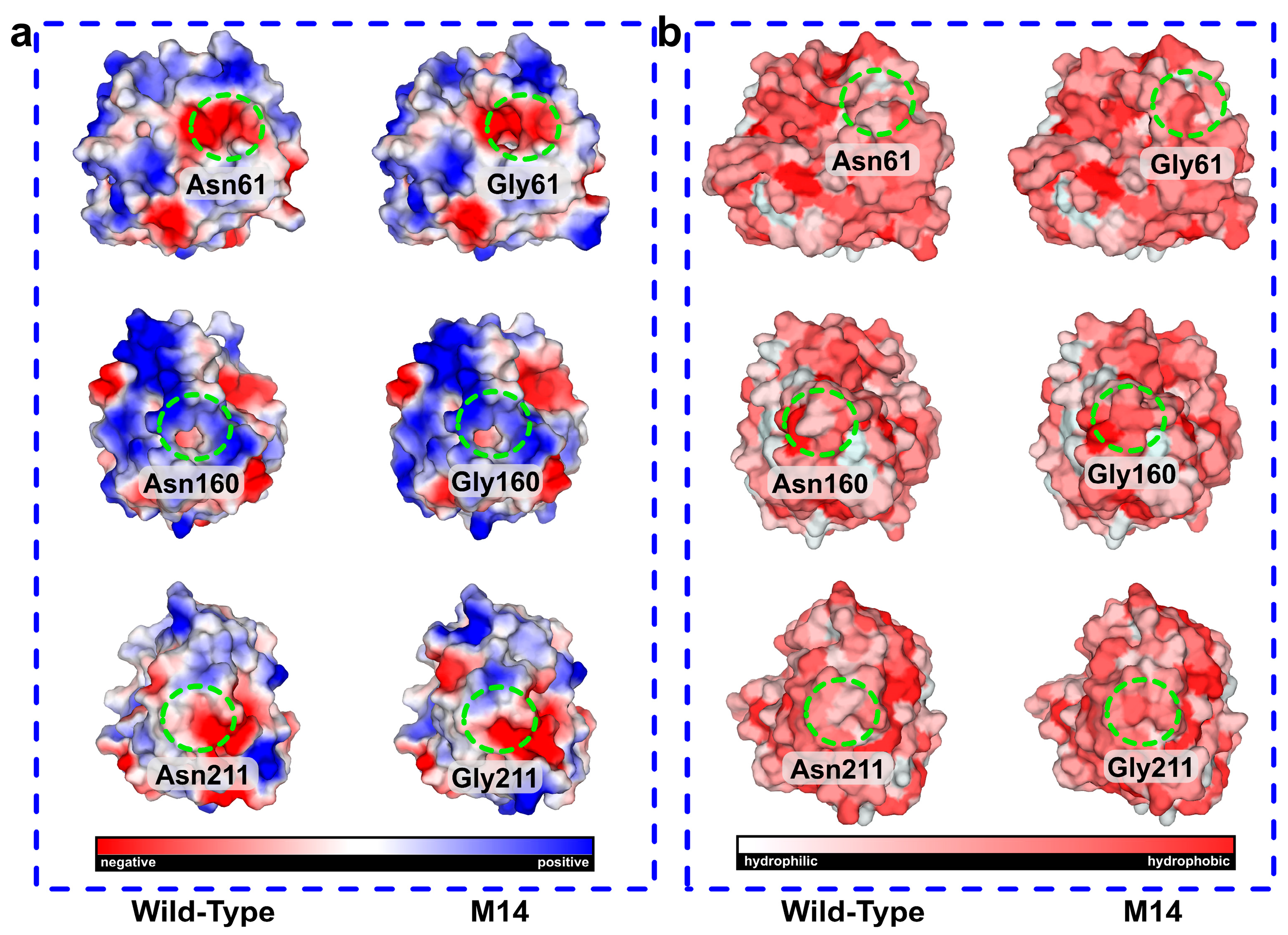
2.4. Molecular Dynamics Simulation and Structural Analysis of the Triple Mutant AprE 2709 (N61G/N160G/N211G) for Enhanced Stability
2.5. Mechanisms Underlying the Stability of AprE 2709 (N61G/N160G/N211G) Revealed by Molecular Dynamics Simulations
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Plasmid and Strains
3.3. Protein Structure Prediction and Evaluation of AprE 2709
3.4. Gene Cloning and Mutant Construction
3.5. Heterologous Expression of AprE 2709 Wild-Type and Mutants
3.6. Enzyme Activity
3.7. Enzymatic Properties of AprE 2709 Wild-Type and Mutants
3.8. Enzymatic Kinetic Assays
3.9. Molecular Dynamics Simulation
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mokashe, N.; Chaudhari, B.; Patil, U. Operative utility of salt–stable proteases of halophilic and halotolerant bacteria in the biotechnology sector. Int. J. Biol. Macromol. 2018, 117, 493–522. [Google Scholar] [CrossRef] [PubMed]
- Rozanov, A.S.; Shekhovtsov, S.V.; Bogacheva, N.V.; Pershina, E.G.; Ryapolova, A.V.; Bytyak, D.S.; Peltek, S.E. Production of subtilisin proteases in bacteria and yeast. Vavilov J. Genet. Breed. 2021, 25, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hammami, A.; Hamdi, M.; Abdelhedi, O.; Jridi, M.; Nasri, M.; Bayoudh, A. Surfactant and oxidant stable alkaline proteases from Bacillus invictae: Characterization and potential applications in chitin extraction and as a detergent additive. Int. J. Biol. Macromol. 2017, 96, 272–281. [Google Scholar] [CrossRef]
- Matkawala, F.; Nighojkar, S.; Kumar, A.; Nighojkar, A. Microbial alkaline serine proteases: Production, properties and applications. World J. Microbiol. Biotechnol. 2021, 37, 63. [Google Scholar] [CrossRef]
- Zhou, C.; Yang, G.; Zhang, L.; Zhang, H.; Zhou, H.; Lu, F. Construction of an alkaline protease overproducer strain based on Bacillus licheniformis 2709 using an integrative approach. Int. J. Biol. Macromol. 2021, 193, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.X.; Zhang, H.T.; Fang, H.L.; Sun, Y.Q.; Zhou, H.Y.; Yang, G.C.; Lu, F.P. Transcriptome based functional identification and application of regulator AbrB on alkaline protease synthesis in 2709. Int. J. Biol. Macromol. 2021, 166, 1491–1498. [Google Scholar] [CrossRef]
- Zhou, C.; Kong, Y.; Zhang, N.; Qin, W.; Li, Y.; Zhang, H.; Yang, G.; Lu, F. Regulator DegU can remarkably influence alkaline protease AprE biosynthesis in Bacillus licheniformis 2709. Int. J. Biol. Macromol. 2024, 266, 130818. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, H.; Yuan, F.; Chai, H.; Wang, H.; Liu, F.; Li, Y.; Zhang, H.; Lu, F. Development and application of a CRISPR/Cas9 system for Bacillus licheniformis genome editing. Int. J. Biol. Macromol. 2019, 122, 329–337. [Google Scholar] [CrossRef]
- Zhou, C.; Zhou, H.; Li, D.; Zhang, H.; Wang, H.; Lu, F. Optimized expression & enhanced production of alkaline protease by genetically modified Bacillus licheniformis 2709. Microb. Cell Factories 2020, 19, 45. [Google Scholar]
- Zhou, C.; Yang, G.; Meng, P.; Qin, W.; Li, Y.; Lin, Z.; Hui, W.; Zhang, H.; Lu, F. Identification and engineering of the aprE regulatory region and relevant regulatory proteins in Bacillus licheniformis 2709. Microb. Cell Factories 2024, 172, 110310. [Google Scholar] [CrossRef]
- Ashraf, N.M.; Krishnagopal, A.; Hussain, A.; Kastner, D.; Sayed, A.M.M.; Mok, Y.K.; Swaminathan, K.; Zeeshan, N. Engineering of serine protease for improved thermostability and catalytic activity using rational design. Int. J. Biol. Macromol. 2019, 126, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, G.; Wei, P.; Song, C.; Xu, Q.; Ma, M.; Ma, J.; Song, P.; Zhang, S. Rational engineering of a metalloprotease to enhance thermostability and activity. Enzym. Microb. Technol. 2022, 159, 110123. [Google Scholar] [CrossRef]
- Peng, Z.; Miao, Z.; Ji, X.; Zhang, G.; Zhang, J. Engineering flexible loops to enhance thermal stability of keratinase for efficient keratin degradation. Sci. Total Environ. 2022, 818, 157161. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, J.; Fang, Z.; Gu, L.; Liao, X.; Du, G.; Chen, J. Enhanced thermostability of keratinase by computational design and empirical mutation. J. Ind. Microbiol. Biotechnol. 2013, 40, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Nakayoshi, T.; Kitamura, Y.; Kurimoto, E.; Oda, A.; Ishikawa, Y. Identification of the Most Impactful Asparagine Residues for γS- Crystallin Aggregation by Deamidation. Biochemistry 2023, 62, 1679–1688. [Google Scholar] [CrossRef]
- Shi, Y.; Rhodes, N.R.; Abdolvahabi, A.; Kohn, T.; Cook, N.P.; Marti, A.A.; Shaw, B.F. Deamidation of Asparagine to Aspartate Destabilizes Cu, Zn Superoxide Dismutase, Accelerates Fibrillization, and Mirrors ALS-Linked Mutations. J. Am. Chem. Soc. 2013, 135, 15897–15908. [Google Scholar] [CrossRef]
- Hains, P.G.; Truscott, R.J. Age-dependent deamidation of lifelong proteins in the human lens. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3107–3114. [Google Scholar] [CrossRef]
- Pace, A.L.; Wong, R.L.; Zhang, Y.T.; Kao, Y.H.; Wang, Y.J. Asparagine deamidation dependence on buffer type, pH, and temperature. J. Pharm. Sci. 2013, 102, 1712–1723. [Google Scholar] [CrossRef]
- Rahimzadeh, M.; Khajeh, K.; Mirshahi, M.; Khayatian, M.; Schwarzenbacher, R. Probing the role of asparagine mutation in thermostability of Bacillus KR-8104 α-amylase. Int. J. Biol. Macromol. 2012, 50, 1175–1182. [Google Scholar] [CrossRef]
- Declerck, N.; Machius, M.; Wiegand, G.; Huber, R.; Gaillardin, C. Probing structural determinants specifying high thermostability in Bacillus licheniformis α-amylase. J. Mol. Biol. 2000, 301, 1041–1057. [Google Scholar] [CrossRef]
- Bhanuramanand, K.; Ahmad, S.; Rao, N.M. Engineering deamidation-susceptible asparagines leads to improved stability to thermal cycling in a lipase. Biochem. Biophys. Res. Commun. 2014, 23, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Bandi, S.; Singh, S.M.; Shah, D.D.; Upadhyay, V.; Mallela, K.M.G. 2D NMR Analysis of the Effect of Asparagine Deamidation Versus Methionine Oxidation on the Structure, Stability, Aggregation, and Function of a Therapeutic Protein. Mol. Pharmacist. 2019, 17, 4406–4416. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.S.; Yan, R.X.; Lin, B.Y.; Li, R.K.; Ye, X.Y. Improving thermostability of Bacillus amyloliquefaciens alpha-amylase by multipoint mutations. Biochem. Biophys. Res. Commun. 2023, 653, 69–75. [Google Scholar] [CrossRef]
- Zhao, J.-F.; Wang, Z.; Gao, F.-L.; Lin, J.-P.; Yang, L.-R.; Wu, M.-B. Enhancing the thermostability of Rhizopus oryzae lipase by combined mutation of hot-spots and engineering a disulfide bond. RSC Adv. 2018, 8, 41247–41254. [Google Scholar] [CrossRef]
- Aghaeepoor, M.; Akbarzadeh, A.; Mirzaie, S.; Hadian, A.; Aval, S.J.; Dehnavi, E. Selective reduction in glutaminase activity of L-Asparaginase by asparagine 248 to serine mutation: A combined computational and experimental effort in blood cancer treatment. Int. J. Biol. Macromol. 2018, 120, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Kamble, A.; Singh, R.; Singh, H. Structural and Functional Characterization of Eubacterium proteus Phytase: A Comprehensive In-Silico Study. Mol. Biotechnol. 2025, 67, 588–616. [Google Scholar] [CrossRef]
- Min, K.; Kim, H.; Park, H.J.; Lee, S.; Jung, Y.J.; Yoon, J.H.; Lee, J.S.; Park, K.; Yoo, Y.J.; Joo, J.C. Improving the catalytic performance of xylanase from Bacillus circulans through structure-based rational design. Bioresour. Technol. 2021, 340, 125737. [Google Scholar] [CrossRef]
- Amatto, I.V.d.S.; da Rosa-Garzon, N.G.; de Oliveira Simões, F.A.; Santiago, F.; da Silva Leite, N.P.; Martins, J.R.; Cabral, H. Enzyme engineering and its industrial applications. Biotechnol. Appl. Biochem. 2022, 69, 389–409. [Google Scholar] [CrossRef]
- Su, H.H.; Peng, F.; Xu, P.; Wu, X.L.; Zong, M.H.; Yang, J.G.; Lou, W.Y. Enhancing the thermostability and activity of uronate dehydrogenase from LBA4404 by semi-rational engineering. Bioresour. Bioprocess. 2019, 6, 36. [Google Scholar] [CrossRef]
- Lv, K.; Li, X.; Chen, K.; Wu, B.; He, B.; Schenk, G. Improving the Catalytic Efficiency of a GH5 Processive Endoglucanase by a Combinatorial Strategy Using Consensus Mutagenesis and Loop Engineering. ACS Catal. 2024, 14, 6856–6867. [Google Scholar] [CrossRef]
- Lee, C.Y.; Yu, K.O.; Kim, S.W.; Han, S.O. Enhancement of the thermostability and activity of mesophilic EngD by DNA recombination with CelE. J. Biosci. Bioeng. 2010, 109, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, J.R.; Tang, Q.Y.; Lan, D.M.; Wang, Y.H. Improving the Catalytic Activity and Thermostability of MAS1 Lipase by Alanine Substitution. Mol. Biotechnol. 2018, 60, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Deng, F.; Chen, Y.; Xu, M.; Ma, F.; Shi, L. Electrostatic and hydrophobic interaction cooperative nanochaperone regulates protein folding. Aggregate 2024, 5, e429. [Google Scholar] [CrossRef]
- Baldwin, R.L.; Rose, G.D. How the hydrophobic factor drives protein folding. Proc. Natl. Acad. Sci. USA 2016, 113, 12462–12466. [Google Scholar] [CrossRef]
- Yi, Z.L.; Pei, X.Q.; Wu, Z.L. Introduction of glycine and proline residues onto protein surface increases the thermostability of endoglucanase CelA from Clostridium thermocellum. Bioresour. Technol. 2011, 102, 3636–3638. [Google Scholar] [CrossRef]
- Li, Y.; Wei, L.; Zhu, Z.; Li, S.; Wang, J.W.; Xin, Q.; Wang, H.; Lu, F.; Qin, H.M. Rational design to change product specificities and thermostability of cyclodextrin glycosyltransferase from Paenibacillus sp. RSC Adv. 2017, 23, 13726–13732. [Google Scholar] [CrossRef]
- Lee, J.M.; Moon, S.Y.; Kim, Y.R.; Kim, K.W.; Lee, S.J.; Kong, I.S. Improvement of thermostability and halo stability of β-1,3-1,4-glucanase by substituting hydrophobic residue for Lys. Int. J. Biol. Macromol. 2017, 94, 594–602. [Google Scholar] [CrossRef]
- Gaines, J.C.; Clark, A.H.; Regan, L.; O’Hern, C.S. Packing in protein cores. J. Phys. Condens. Matter 2017, 29, 293001. [Google Scholar] [CrossRef]
- Nguyen, C.; Young, J.T.; Slade, G.G.; Oliveira, R.J.; McCully, M.E. A Dynamic Hydrophobic Core and Surface Salt Bridges Thermostabilize a Designed Three-Helix Bundle. Biophys. J. 2019, 116, 621–632. [Google Scholar] [CrossRef]
- Taylor, T.J.; Vaisman, I.I. Discrimination of thermophilic & mesophilic proteins. BMC Struct. Biol. 2010, 10, S5. [Google Scholar]
- Cui, X.; Yuan, X.; Li, S.Y.; Hu, X.L.; Zhao, J.; Zhang, G.M. Simultaneously improving the specific activity and thermostability of α-amylase BLA by rational design. Bioprocess Biosyst. Eng. 2022, 45, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Aderinwale, T.; Bharadwaj, V.; Christoffer, C.; Terashi, G.; Zhang, Z.; Jahandideh, R.; Kagaya, Y.; Kihara, D. Real-time structure search and structure classification for AlphaFold protein models. Commun. Biol. 2022, 5, 316. [Google Scholar] [CrossRef]
- Tang, X.M.; Shen, W.; Lakay, F.M.; Shao, W.L.; Wang, Z.X.; Prior, B.A.; Zhuge, J. Cloning and over-expression of an alkaline protease from Bacillus licheniformis. Biotechnol. Lett. 2004, 26, 975–979. [Google Scholar] [CrossRef]
- Hiraga, K.; Nishikata, Y.; Namwong, S.; Tanasupawat, S.; Takada, K.; Oda, K. Purification and characterization of serine proteinase from a Halophilic bacterium, sp. RF2-5. Biosci. Biotechnol. Biochem. 2005, 69, 38–44. [Google Scholar] [CrossRef] [PubMed]
- GB/T 23527-2009; Proteinase Preparations. National Standards of the People’s Republic of China: Beijing, China, 2009.
- Siu, S.W.I.; Pluhackova, K.; Böckmann, R.A. Optimization of the OPLS–AA Force Field for Long Hydrocarbons. J. Chem. Theory Comput. 2012, 8, 1459–1470. [Google Scholar] [CrossRef]
- Fyta, M.; Netz, R.R. Ionic force field optimization based on single-ion and ion-pair solvation properties: Going beyond st and ard mixing rules. Chin. J. Chem. Phys. 2012, 136, 124103. [Google Scholar] [CrossRef]
- Kusalik, P.G.; Svishchev, I.M. The Spatial Structure in Liquid Water. Science 1994, 265, 1219–1221. [Google Scholar] [CrossRef]
- Hess, B. P–LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Ullah, S.; Khan, S.U.; Khan, A.; Junaid, M.; Rafiq, H.; Htar, T.T.; Zhao, Y.X.; Shah, S.A.A.; Wadood, A. Prospect of Anterior Gradient 2 homodimer inhibition via repurposing FDA–approved drugs using structure–based virtual screening. Mol. Divers. 2022, 26, 1399–1409. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Asparagine Sites (19 in Total) | RMSF Values (nm) |
|---|---|
| 76 | 0.2127 |
| 160 | 0.1723 |
| 182 | 0.1631 |
| 184 | 0.107 |
| 96 | 0.105 |
| 43 | 0.098 |
| 154 | 0.0977 |
| 239 | 0.0898 |
| 116 | 0.0858 |
| 61 | 0.0849 |
| 211 | 0.0832 |
| 25 | 0.0753 |
| 162 | 0.0703 |
| 217 | 0.0592 |
| 140 | 0.0584 |
| 57 | 0.0579 |
| 247 | 0.0475 |
| 268 | 0.046 |
| 122 | 0.0443 |
| Enzymes | Mutated Sites | T1/2 a (min) |
|---|---|---|
| WT | - | 19 |
| M1 | N61S | 29 |
| M2 | N160S | 39 |
| M3 | N211S | 54 |
| M4 (M1 + M2) | N61S/N160S | 30 |
| M5 (M2 + M3) | N160S/N211S | 40 |
| M6 (M1 + M3) | N61S/N211S | 41 |
| M7 (M1 + M2 + M3) | N61S/N160S/N211S | 62 |
| M8 | N61G | 23 |
| M9 | N160G | 15 |
| M10 | N211G | 14 |
| M11 (M8 + M9) | N61G/N160G | 35 |
| M12 (M9 + M10) | N160G/N211G | 18 |
| M13 (M8 + M10) | N61G/N211G | 28 |
| M14 (M8 + M9 + M10) | N61G/N160G/N211G | 74 |
| Types | Km (mM) | kcat (min−1) | kcat/Km (mM−1·min−1) | Optimal pH | Optimum Temperature |
|---|---|---|---|---|---|
| WT | 0.57 | 9.42 | 16.29 | 10 | 50 °C |
| M1 | 0.15 | 4.82 | 32.11 | 11 | 60 °C |
| M2 | 0.65 | 5.25 | 7.99 | 11 | 60 °C |
| M3 | 0.06 | 5.42 | 82.19 | 11 | 60 °C |
| M4 | 1.09 | 5.52 | 5.06 | 11 | 60 °C |
| M5 | 0.05 | 6.97 | 148.32 | 11 | 60 °C |
| M6 | 0.04 | 4.65 | 116.41 | 11 | 60 °C |
| M7 | 0.50 | 5.19 | 10.31 | 11 | 60 °C |
| M8 | 0.55 | 7.86 | 14.22 | 11 | 60 °C |
| M9 | 0.28 | 6.40 | 22.28 | 11 | 60 °C |
| M10 | 0.21 | 7.37 | 33.75 | 11 | 60 °C |
| M11 | 0.77 | 6.12 | 7.89 | 11 | 60 °C |
| M12 | 0.16 | 5.92 | 35.15 | 11 | 60 °C |
| M13 | 0.28 | 6.46 | 22.34 | 11 | 60 °C |
| M14 | 0.25 | 6.53 | 25.42 | 11 | 60 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Zhao, G.; Lu, J.; Wang, L.; Shi, Y.; Zhang, J. Enhancing the Thermostability of Bacillus licheniformis Alkaline Protease 2709 by Computation-Based Rational Design. Molecules 2025, 30, 1160. https://doi.org/10.3390/molecules30051160
Yuan Y, Zhao G, Lu J, Wang L, Shi Y, Zhang J. Enhancing the Thermostability of Bacillus licheniformis Alkaline Protease 2709 by Computation-Based Rational Design. Molecules. 2025; 30(5):1160. https://doi.org/10.3390/molecules30051160
Chicago/Turabian StyleYuan, Yuan, Guowei Zhao, Jing Lu, Lei Wang, Yawei Shi, and Jian Zhang. 2025. "Enhancing the Thermostability of Bacillus licheniformis Alkaline Protease 2709 by Computation-Based Rational Design" Molecules 30, no. 5: 1160. https://doi.org/10.3390/molecules30051160
APA StyleYuan, Y., Zhao, G., Lu, J., Wang, L., Shi, Y., & Zhang, J. (2025). Enhancing the Thermostability of Bacillus licheniformis Alkaline Protease 2709 by Computation-Based Rational Design. Molecules, 30(5), 1160. https://doi.org/10.3390/molecules30051160