
DFT Study on Fused N-Heteroaromatic Frameworks: Stability, Aromaticity, and Energetic Insights from Five-Membered Fused Six-Membered N-Heteroaromatic Skeletons



Abstract

1. Introduction
2. Result and Discussion
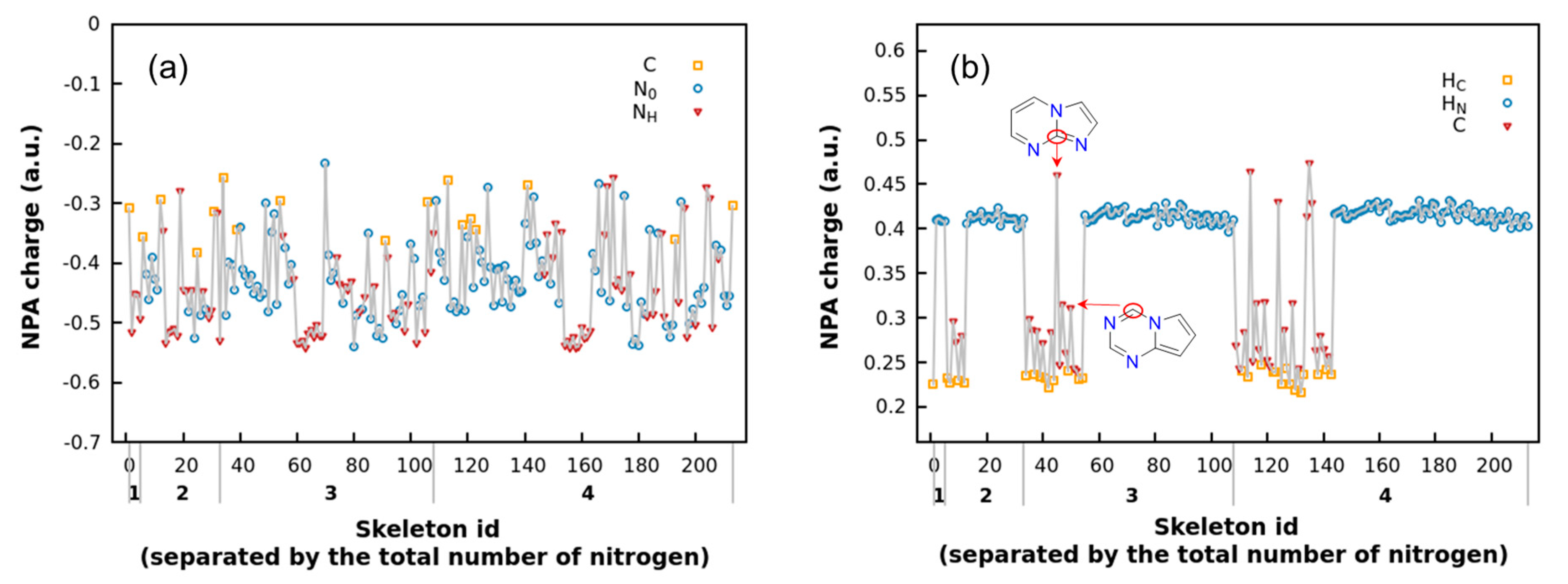
2.1. NPA Charges Analysis
- (1)
- Electronegativity trend: N > C > H.
- (2)
- NH tends to carry more negative charge due to the electron-donating effect of the H atom.
- (3)
- Catenated nitrogen substructures (N3 or N4) reduce the negative charge on nitrogen atoms compared to isolated nitrogen atoms. Nitrogen atoms at the ends of catenated nitrogen substructures carry more negative charge, whereas those in the middle exhibit less.
- (4)
- Both shared nitrogen (NS) and carbon (CS) atoms exhibit less negative charge compared to their isolated counterparts of the same type.
- (5)
- For the same type of atom, those in five-membered rings carry more negative charge than those in six-membered rings.
- (6)
- Charges tend to alternate in distribution; that is, if an adjacent atom carries a higher positive charge, the atom in question is likely to carry more negative charge.
2.2. Laplacian Bond Order Analysis
2.3. LOL-π Analysis
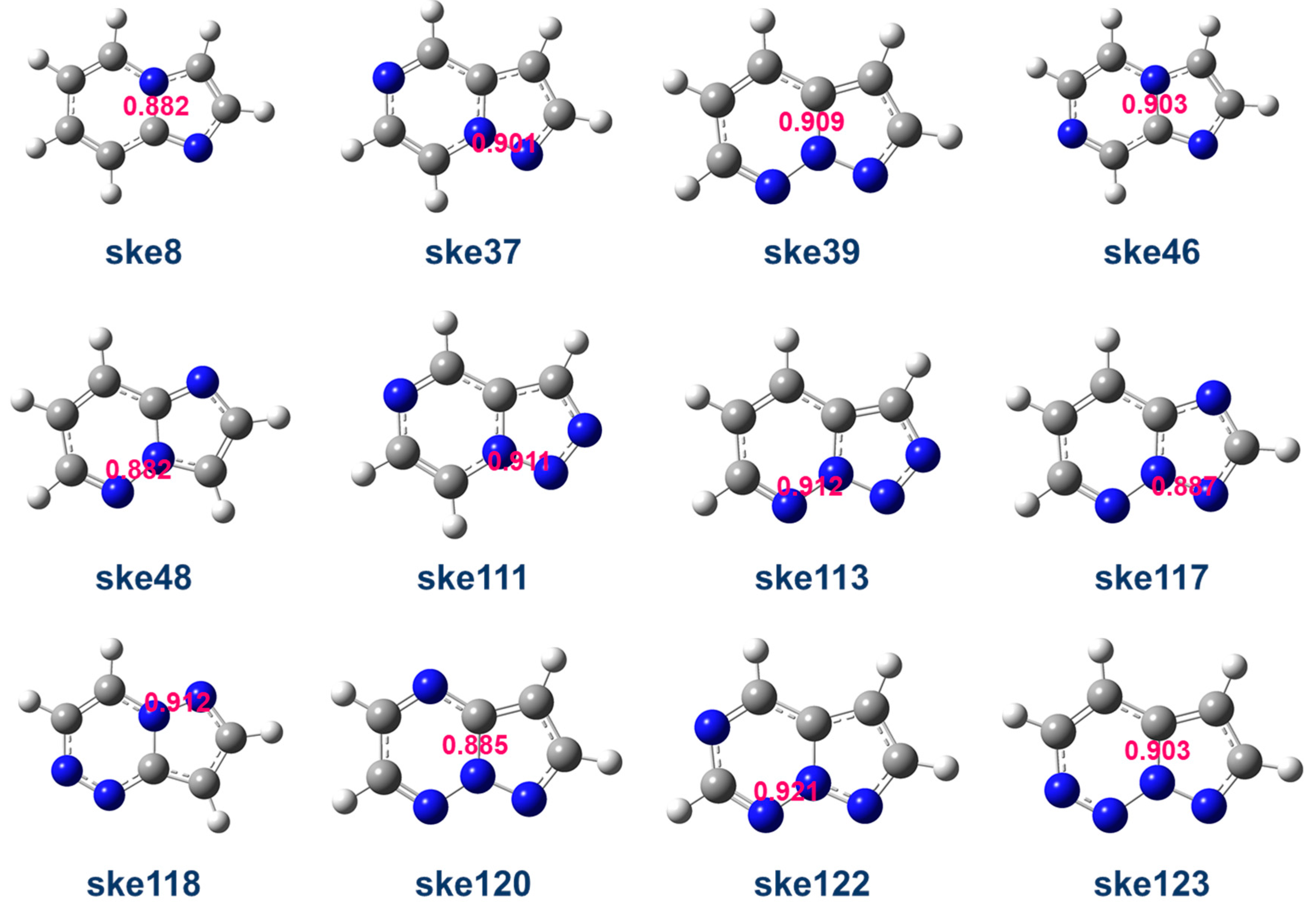
2.4. Multicenter Bond Order Analysis
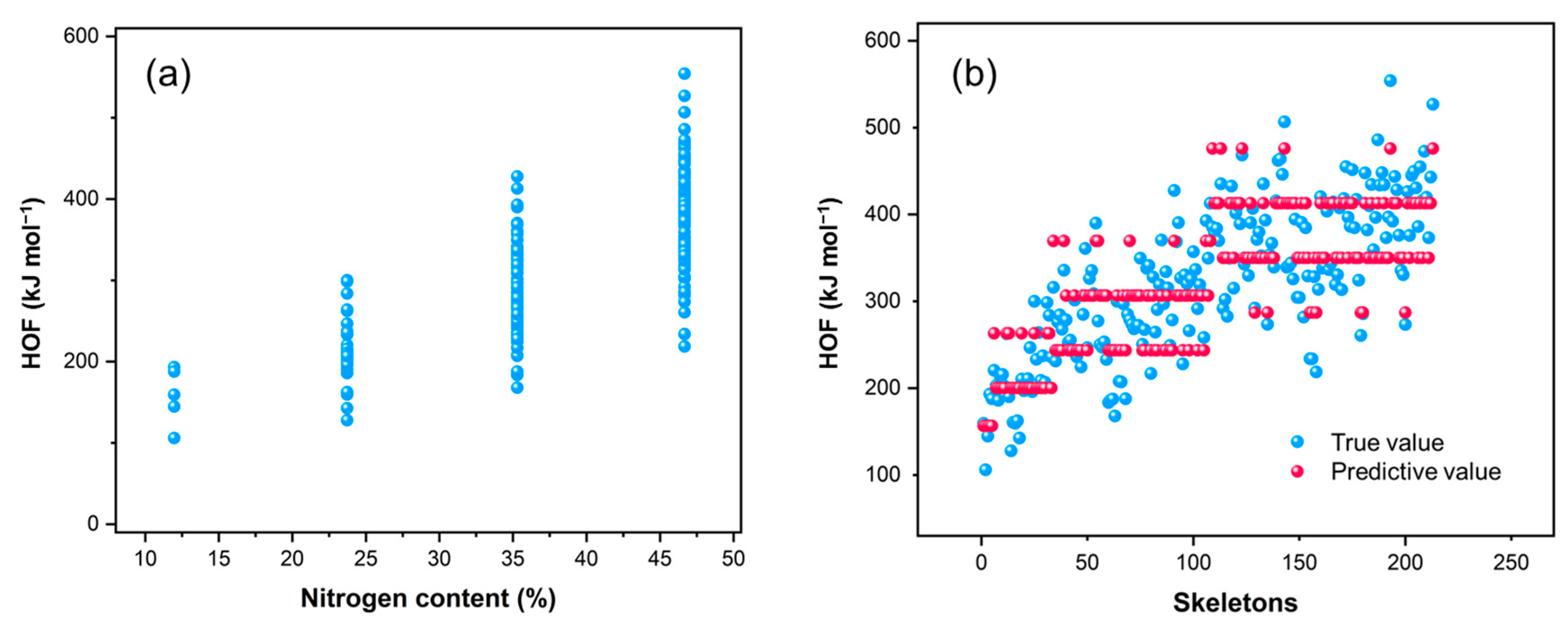
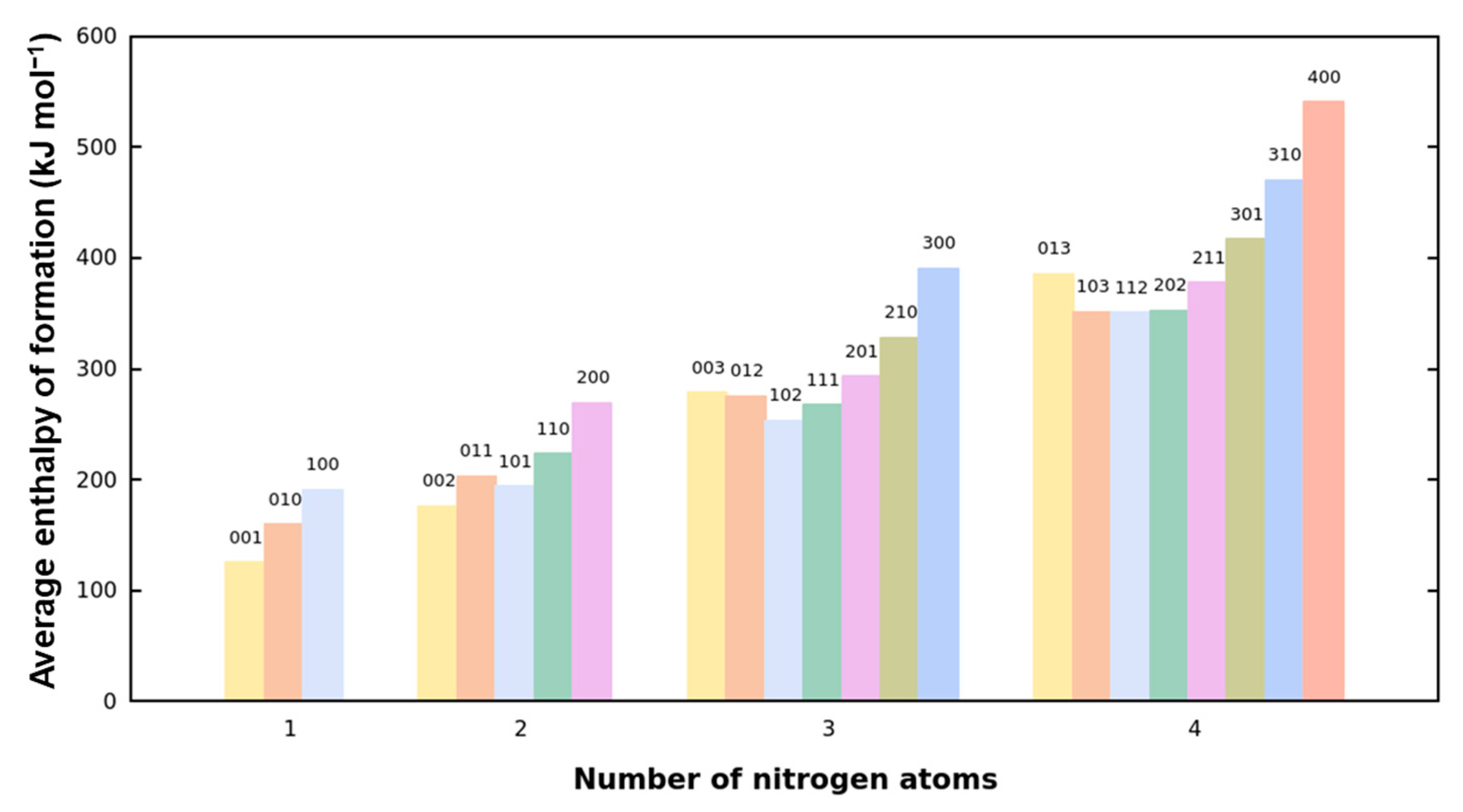
2.5. Enthalpy of Formation Analysis
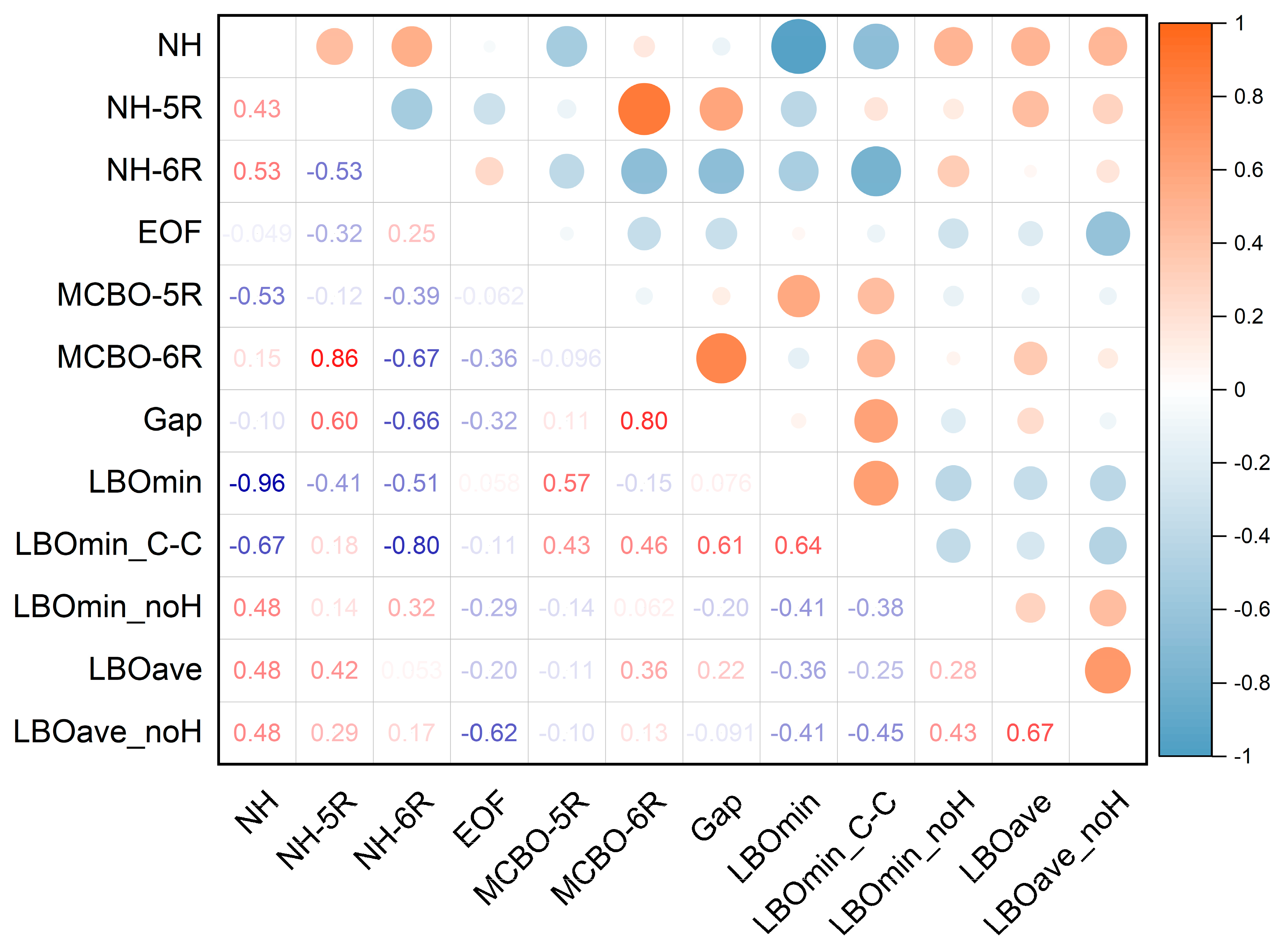
2.6. Correlation Analysis
3. Computing Method
4. Summary and Recommendations
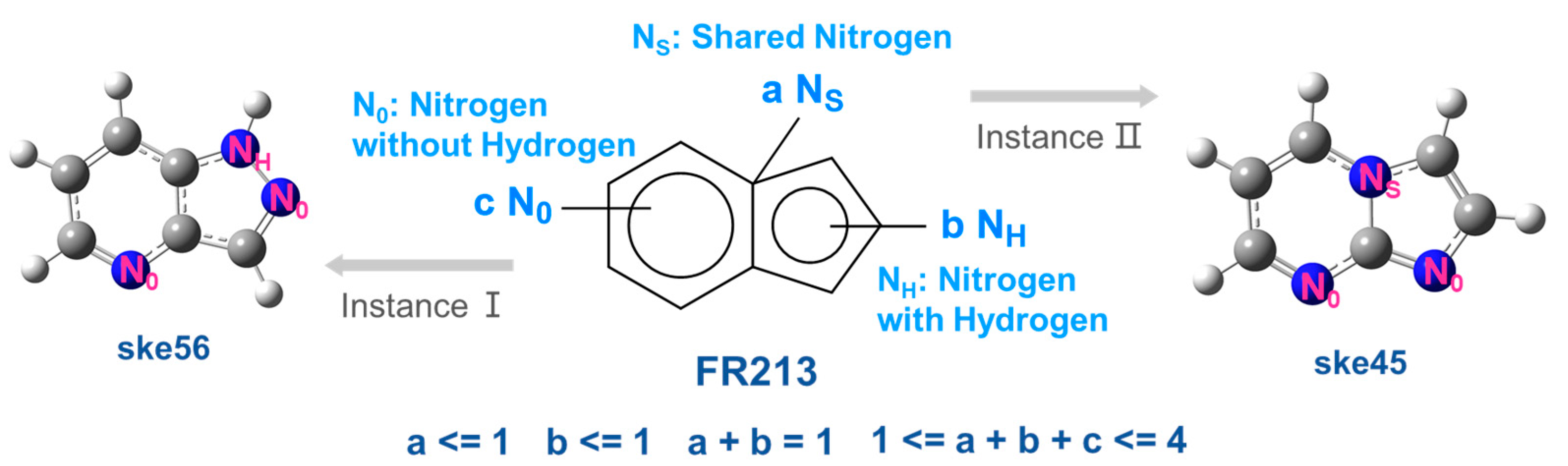
- (1)
- The nitrogen atoms in FR213 can be classified into three types, NS, NH, and N0, as described in the main text. They exhibit completely different conjugative and inductive properties. Different atomic types and their relative positions can have subtle effects on the NPA charge population of the skeletons.
- (2)
- In the context of LBO analysis, the LBOmin decreases as the number of nitrogen atoms increases, suggesting a reduction in the stability of the framework with higher nitrogen content. This trend is particularly pronounced when nitrogen atoms are incorporated into six-membered rings. N-H bonds are unequivocally the weakest bonds in the framework, yet their presence enhances the strength of other bonds in the framework. The LBO values of the bonds in the framework rings generally follow the trend C-C > C-N ≈ N=N > N-N. Among the C-N bonds, C-NS is the weakest. Besides, the N3 substructure does not have a significant effect on bond strength, but the N4 substructure does result in relatively weaker N-N bonds.
- (3)
- All molecules in FR213 have unique Lewis structures, and parameters such as bond length, LBO, and LOL-π paths can be associated with these structures. For example, regions corresponding to double bonds in the structure will exhibit shorter bond lengths, higher LBO values, and greater π electron density. Therefore, the strength of bonds can be qualitatively discussed based on the Lewis structures of the skeleton molecules.
- (4)
- In the context of MCBO analysis, the presence and position of N-H have a significant impact on the aromaticity of the framework molecules. N-H on the five-membered ring enhances the aromaticity of the six-membered ring, while N-H on the six-membered ring weakens the aromaticity of both the five-membered and six-membered rings. The MCBO of the six-membered ring shows a clear positive correlation with the HOMO-LUMO gap.
- (5)
- The EOF of FR213 is strongly correlated with the composition of its chemical bonds. By fitting the EOF to the quantities of C-C, C-N, and N-N bonds, we obtain a fitting equation with R2 = 0.98521 and RMSE = 40.4 kJ mol−1. The coefficients of this equation reflect the relative contributions of each bond type to the EOF. Specifically, the coefficient for the C-N bond is three times that of C-C, and the coefficient for the N-N bond is ten times that of C-C, indicating a significant contribution of nitrogen-containing bonds to the EOF. Furthermore, for a given bond composition, longer nitrogen chains do not result in a higher EOF. The presence or absence of N-H bonds has minimal impact on the EOF of the framework. Additionally, for the contribution to EOF: N6 > NS > N5.
- (1)
- In the synthesis of elemental explosives, skeletons containing N-H bonds are undesirable. Although N-H bonds can strengthen the framework, the N-H bond itself is the most unstable component, potentially leading to instability in explosive molecules, manifested as acidity or high sensitivity.
- (2)
- Achieving a balance between energy and stability depends on the distribution of nitrogen atom positions. While there is a general trend of decreasing stability with increasing nitrogen content, as shown in Figure 4, many tri-nitrogen and tetra-nitrogen frameworks can still maintain good structural stability. Specifically, placing more than two nitrogen atoms on six-membered rings should be avoided, as nitrogen atoms on these rings have a more detrimental effect on the framework’s stability. Moreover, continuous nitrogen chain structures do not confer energetic benefits and should be avoided in the framework to mitigate their negative impact on stability.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, H.; Shreeve, J.M. Azole-Based Energetic Salts. Chem. Rev. 2011, 111, 7377–7436. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Wang, B.; Qiu, L.; Xu, R.; Sheremetev, A.B. Recent Synthetic Efforts towards High Energy Density Materials: How to Design High-Performance Energetic Structures? FirePhysChem 2022, 2, 83–139. [Google Scholar] [CrossRef]
- Becuwe, A.; Delclos, A. Low-Sensitivity Explosive Compounds for Low Vulnerability Warheads. Propellants Explos. Pyrotech. 1993, 18, 1–10. [Google Scholar] [CrossRef]
- Hervé, G.; Roussel, C.; Graindorge, H. Selective Preparation of 3,4,5-Trinitro-1 H-Pyrazole: A Stable All-Carbon-Nitrated Arene. Angew. Chem. Int. Ed. 2010, 49, 3177–3181. [Google Scholar] [CrossRef] [PubMed]
- Pagoria, P.; Mitchell, A.; Schmidt, R.; Fried, L. Munitions Technology Development Program; UCRL-ID-103483-99; 1999; p. II-5. [Google Scholar]
- Wang, Y.; Liu, Y.; Song, S.; Yang, Z.; Qi, X.; Wang, K.; Liu, Y.; Zhang, Q.; Tian, Y. Accelerating the Discovery of Insensitive High-Energy-Density Materials by a Materials Genome Approach. Nat. Commun. 2018, 9, 2444. [Google Scholar] [CrossRef]
- Li, C.; Lei, C.; Tang, J.; Zhu, T.; Cheng, G.; Yang, H. C–C bonded bis-5,6 fused triazole–triazine compound: An advanced heat-resistant explosive with high energy and low sensitivity. Dalton Trans. 2022, 51, 15292–15299. [Google Scholar] [CrossRef]
- Zheng, X.; Xue, Y.; Dai, C.; Yang, H.; Cheng, G. The skeleton of 5,7-fused bicyclic imidazole-diazepine for heat-resistant energetic materials. Def. Technol. 2023, 27, 193–199. [Google Scholar] [CrossRef]
- Tang, Y.; He, C.; Imler, G.H.; Parrish, D.A.; Shreeve, J.M. Aminonitro Groups Surrounding a Fused Pyrazolotriazine Ring: A Superior Thermally Stable and Insensitive Energetic Material. ACS Appl. Energy Mater. 2019, 2, 2263–2267. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Ma, W.; Fei, T.; He, C.; Pang, S. Tri-Explosophoric Groups Driven Fused Energetic Heterocycles Featuring Superior Energetic and Safety Performances Outperforms HMX. Nat. Commun. 2022, 13, 5697. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Bond Order Analysis Based on the Laplacian of Electron Density in Fuzzy Overlap Space. J. Phys. Chem. A 2013, 117, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q.; Liu, Z. A Thorough Theoretical Exploration of Intriguing Characteristics of Cyclo [18]carbon: Geometry, Bonding Nature, Aromaticity, Weak Interaction, Reactivity, Excited States, Vibrations, Molecular Dynamics and Various Molecular Properties. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Schmider, H.L.; Becke, A.D. Chemical content of the kinetic energy density. J. Mol. Struct. THEOCHEM 2000, 527, 51–61. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure. Theor. Chem. Acc. 2020, 139, 25. [Google Scholar] [CrossRef]
- Giambiagi, M.; De Giambiagi, M.S.; Mundim, K.C. Definition of a multicenter bond index. Struct. Chem. 1990, 1, 423–427. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Gaussian 09 Revision D.01; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIREs Comput Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Skeleton Types | Six-Membered Rings | Average NMCBO-5R | Average NMCBO-6R |
|---|---|---|---|
| NH-5R | benzene | 0.51567 | 0.57977 |
| pyridine | 0.51199 | 0.58495 | |
| pyrazine | 0.50957 | 0.58365 | |
| pyridazine | 0.50939 | 0.58466 | |
| pyrimidine | 0.50873 | 0.58265 | |
| triazine | 0.50859 | 0.58706 | |
| Summary Average | 0.51087 | 0.58411 | |
| NH-6R | pyridine | 0.52345 | 0.51568 |
| pyridazine | 0.51281 | 0.51010 | |
| pyrimidine | 0.50961 | 0.50912 | |
| pyrazine | 0.49399 | 0.51652 | |
| triazine | 0.48808 | 0.50332 | |
| tetrazine | 0.46313 | 0.49413 | |
| Summary Average | 0.50449 | 0.50922 | |
| NS | pyrazine | 0.54020 | 0.53373 |
| pyridazine | 0.53737 | 0.52897 | |
| pyridine | 0.53629 | 0.53414 | |
| triazine | 0.52938 | 0.52845 | |
| pyrimidine | 0.52827 | 0.53232 | |
| tetrazine | 0.52249 | 0.52450 | |
| Summary Average | 0.53163 | 0.53039 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Z.; Li, C.; Li, S.; Yu, Q.; Zhang, J. DFT Study on Fused N-Heteroaromatic Frameworks: Stability, Aromaticity, and Energetic Insights from Five-Membered Fused Six-Membered N-Heteroaromatic Skeletons. Molecules 2025, 30, 1101. https://doi.org/10.3390/molecules30051101
Lu Z, Li C, Li S, Yu Q, Zhang J. DFT Study on Fused N-Heteroaromatic Frameworks: Stability, Aromaticity, and Energetic Insights from Five-Membered Fused Six-Membered N-Heteroaromatic Skeletons. Molecules. 2025; 30(5):1101. https://doi.org/10.3390/molecules30051101
Chicago/Turabian StyleLu, Zujia, Cong Li, Shaoqun Li, Qiyao Yu, and Jianguo Zhang. 2025. "DFT Study on Fused N-Heteroaromatic Frameworks: Stability, Aromaticity, and Energetic Insights from Five-Membered Fused Six-Membered N-Heteroaromatic Skeletons" Molecules 30, no. 5: 1101. https://doi.org/10.3390/molecules30051101
APA StyleLu, Z., Li, C., Li, S., Yu, Q., & Zhang, J. (2025). DFT Study on Fused N-Heteroaromatic Frameworks: Stability, Aromaticity, and Energetic Insights from Five-Membered Fused Six-Membered N-Heteroaromatic Skeletons. Molecules, 30(5), 1101. https://doi.org/10.3390/molecules30051101