
Anti-Butterfly Effect in Ribavirin Studied by Combined Experiment (PXRD/1H-14N NQR Cross-Relaxation Spectroscopy), Quantum Chemical Calculations, Molecular Docking, Molecular Dynamics Simulations, and Novel Structure-Binding Strength and Quadrupolar Indices

 , , ,
, , ,
Abstract

1. Introduction
2. Results and Discussion
2.1. Experimental Investigation
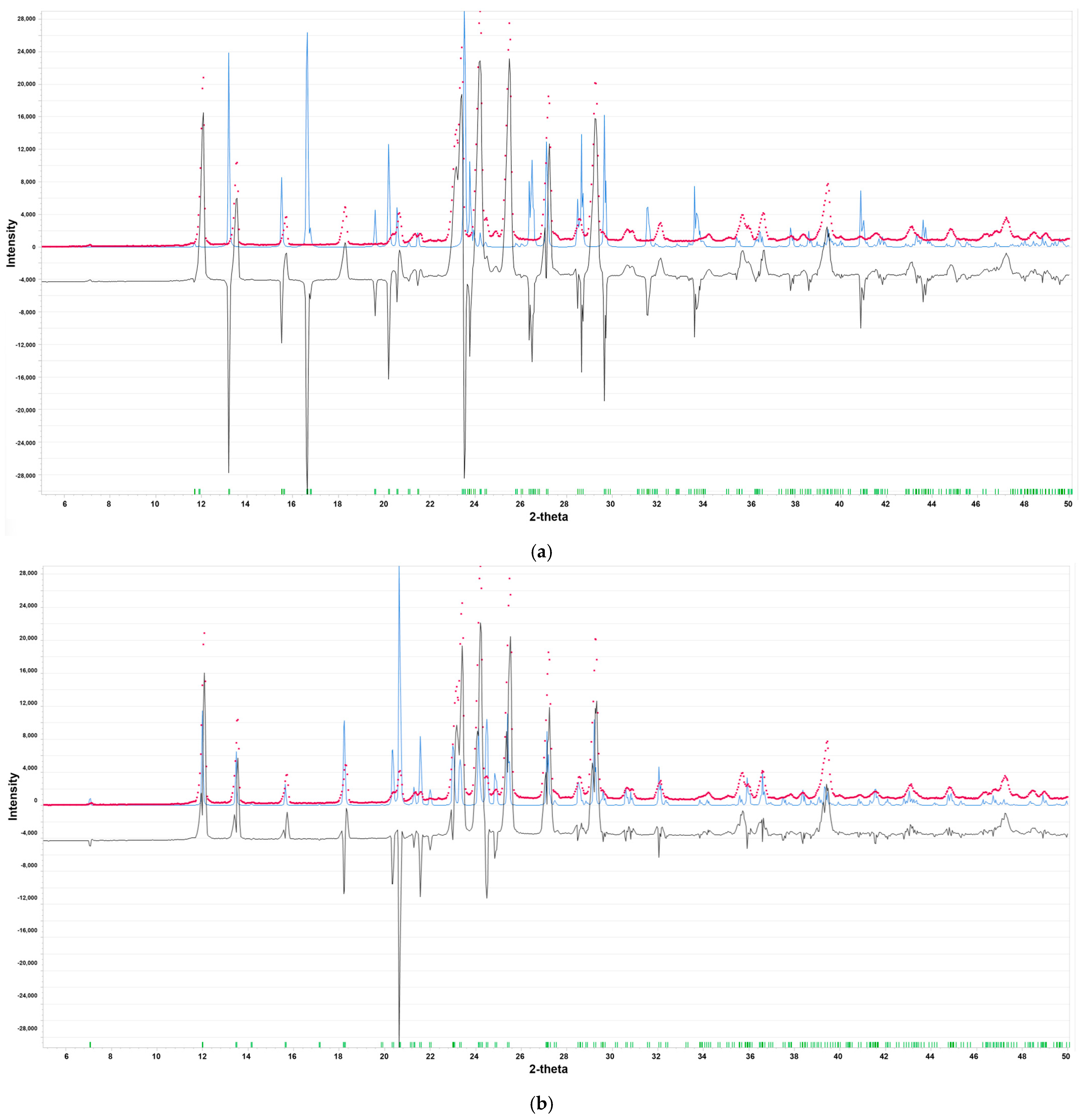
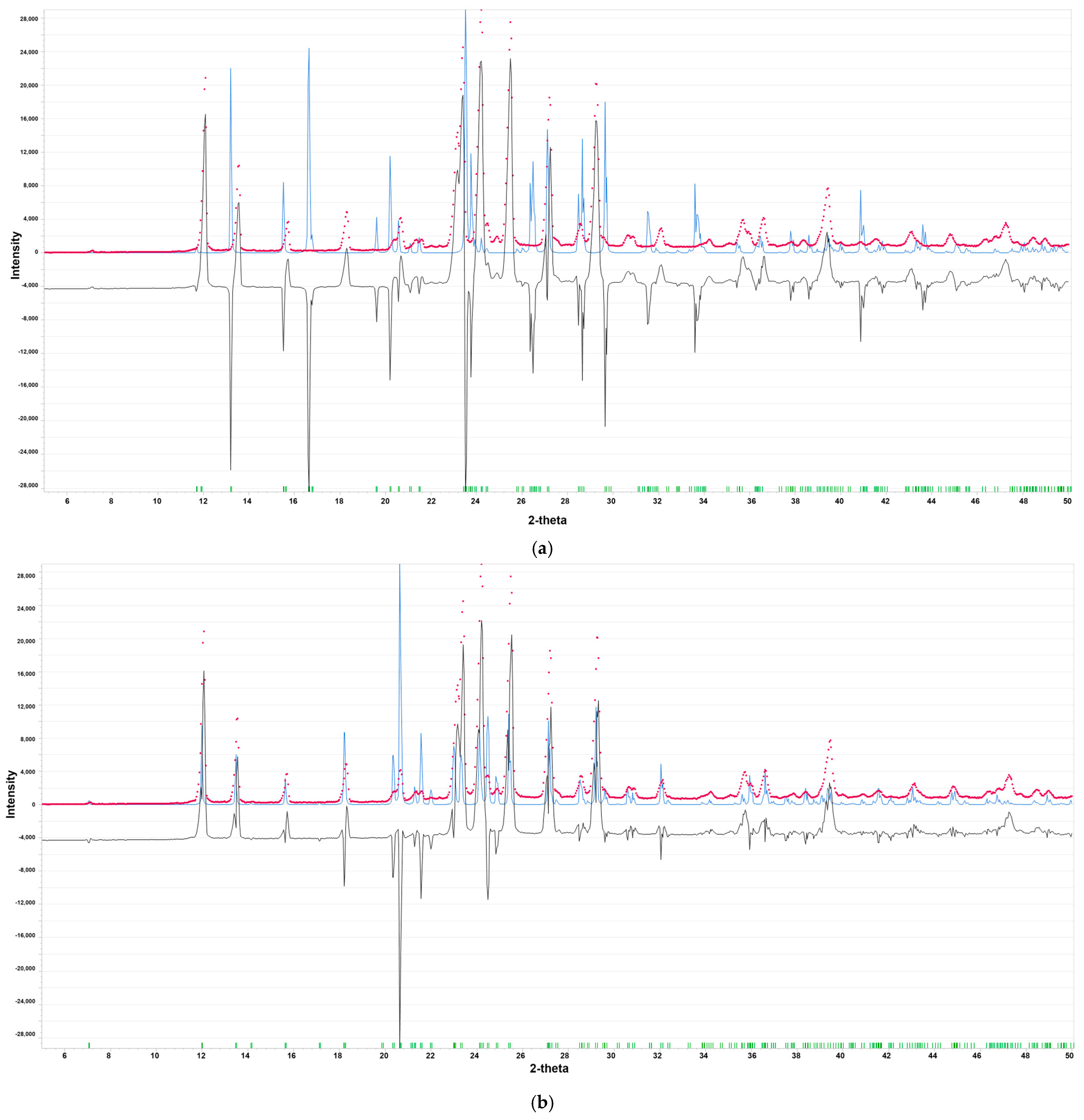
2.1.1. Polymorph Evaluation by PXRD
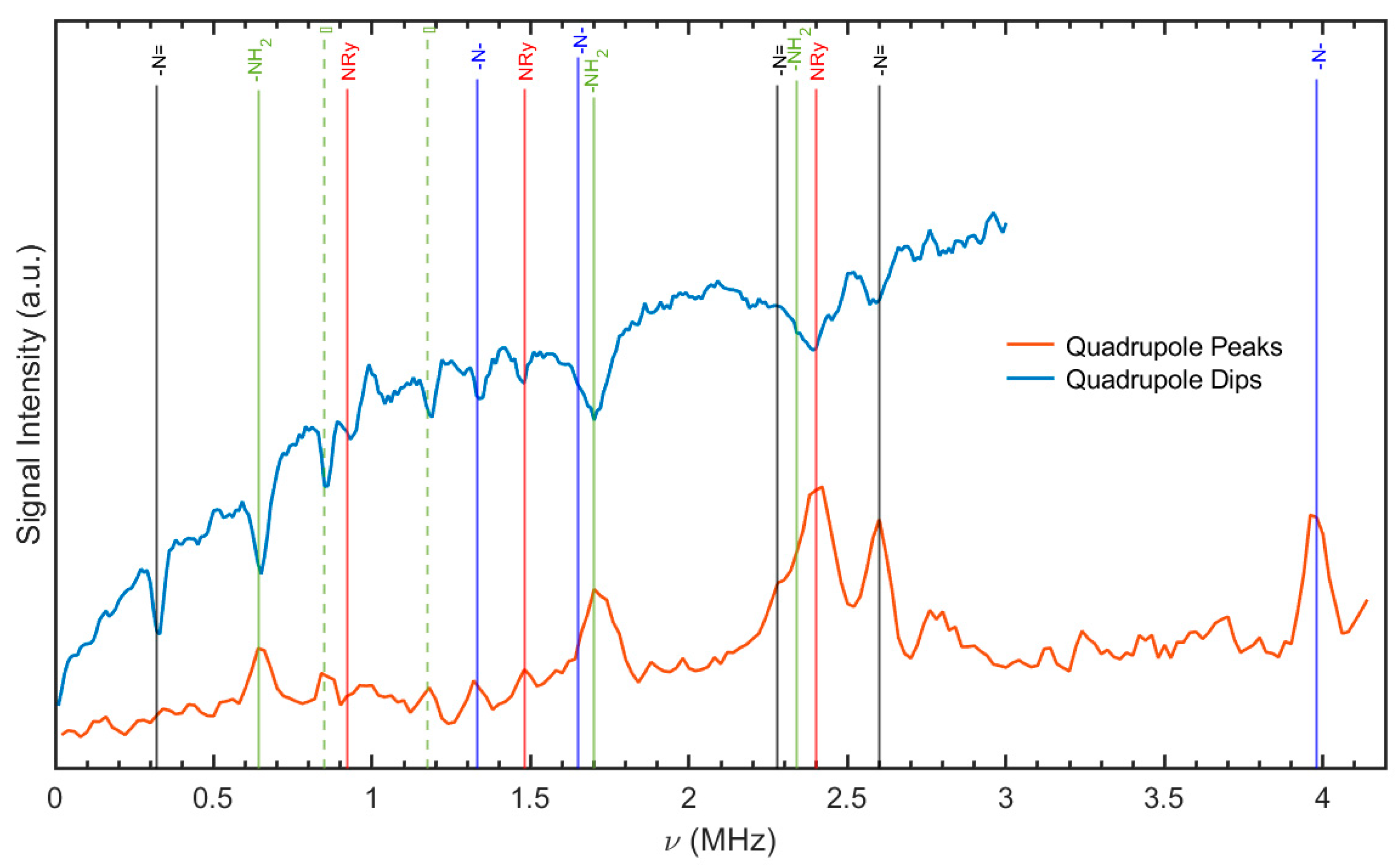
2.1.2. 1H-14N NQR Cross-Relaxation Spectroscopy
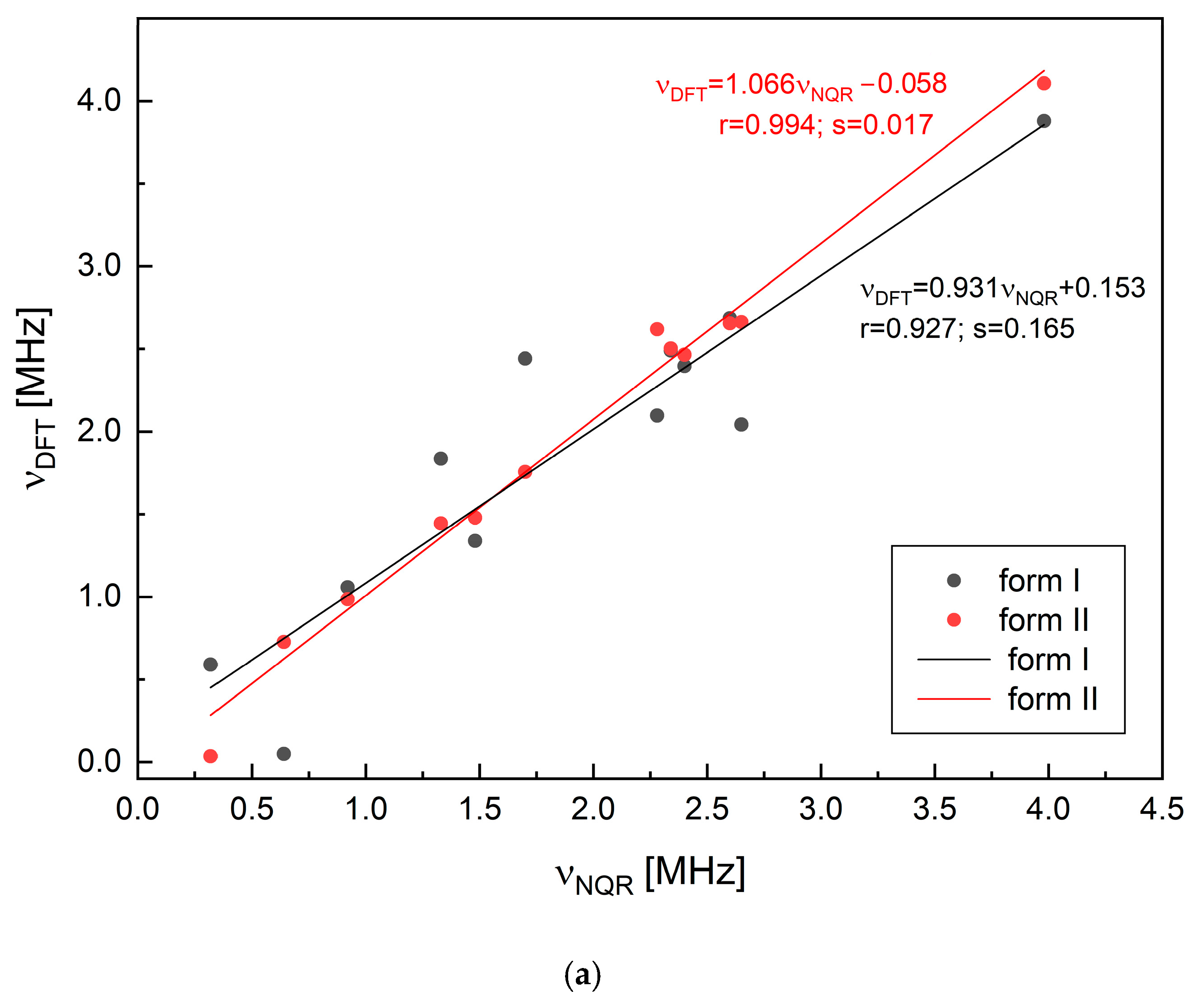
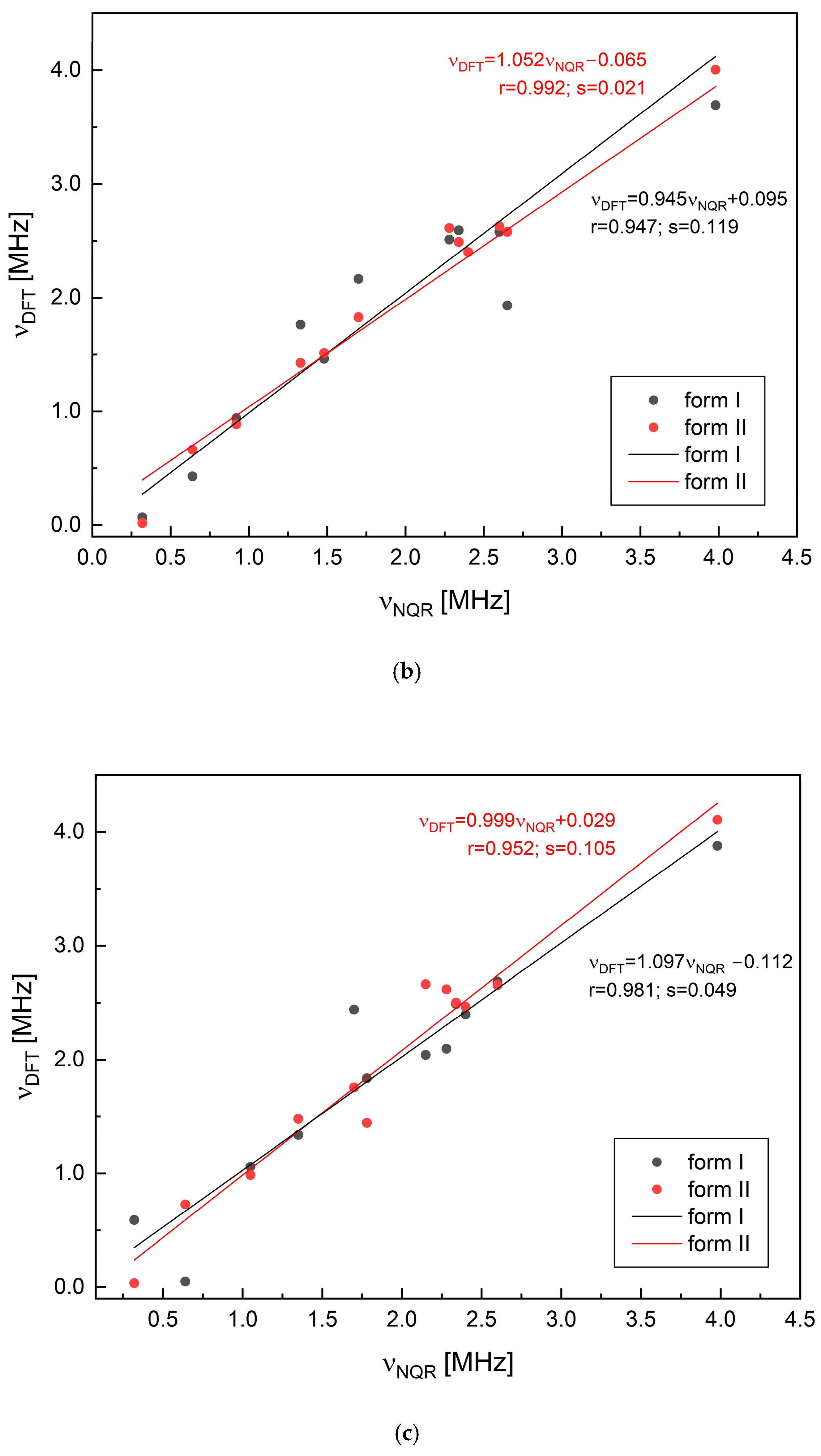
2.2. Solid-State DFT Calculations
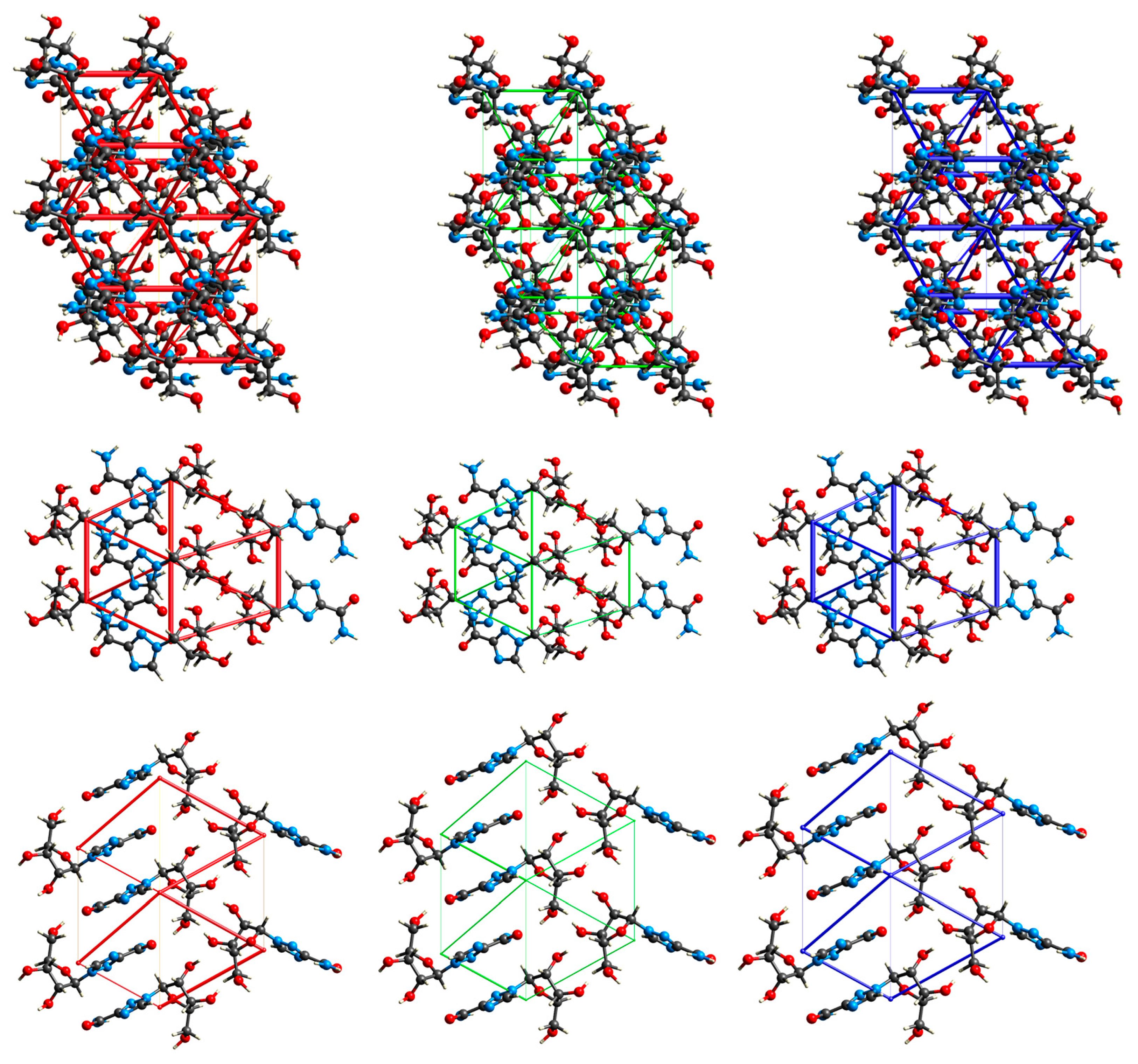
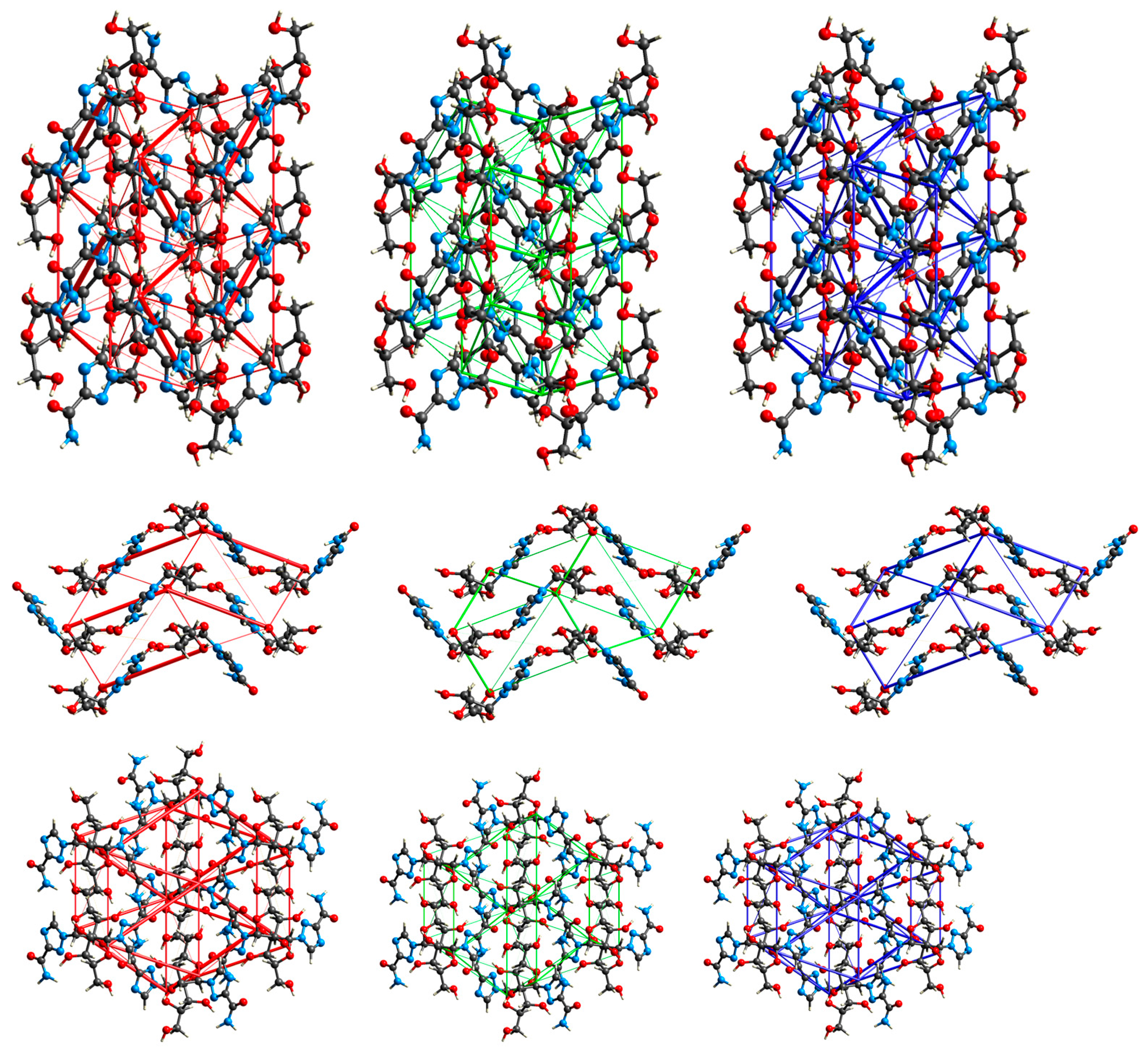
2.3. Binding Motifs in the Solid State
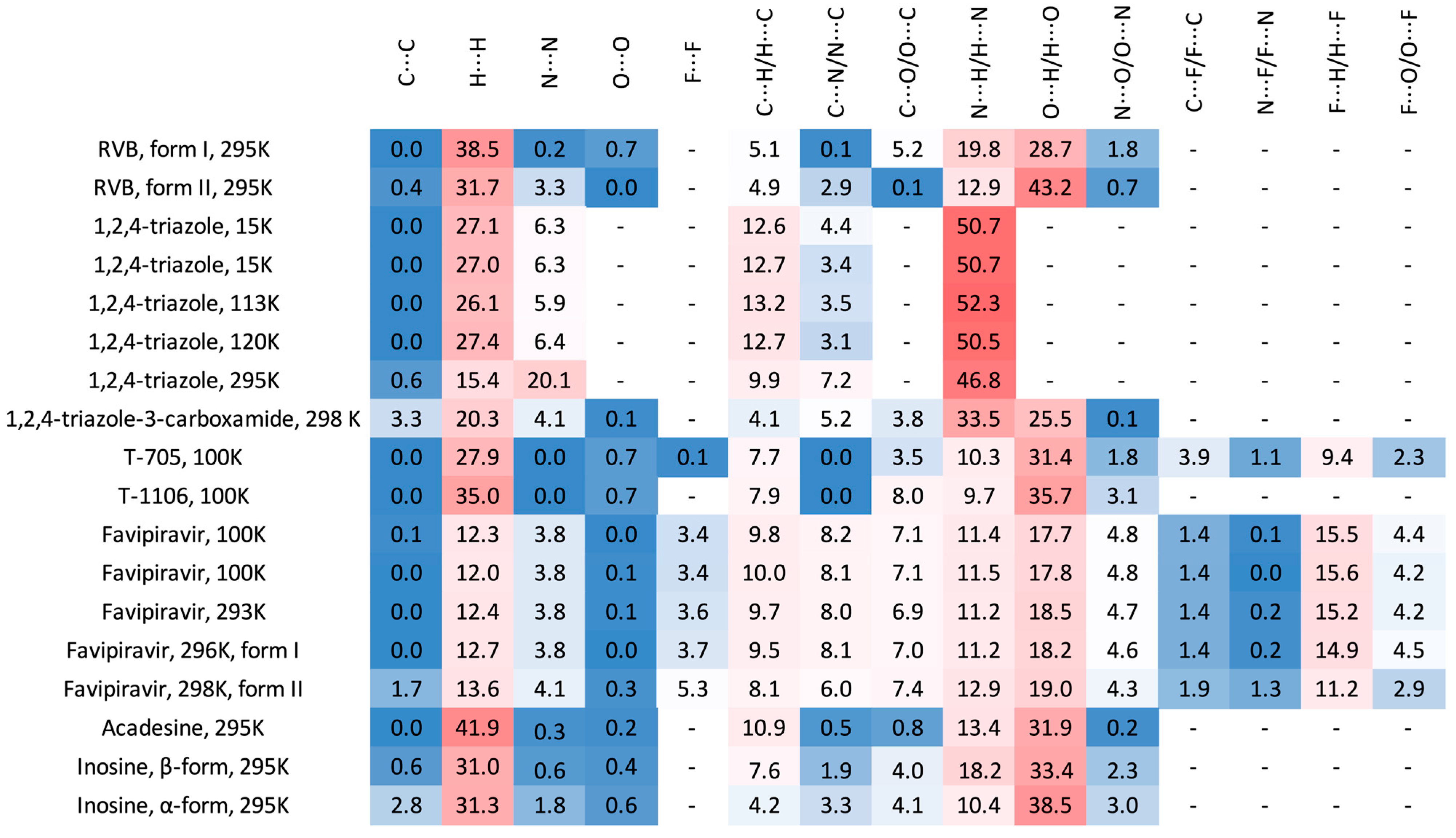
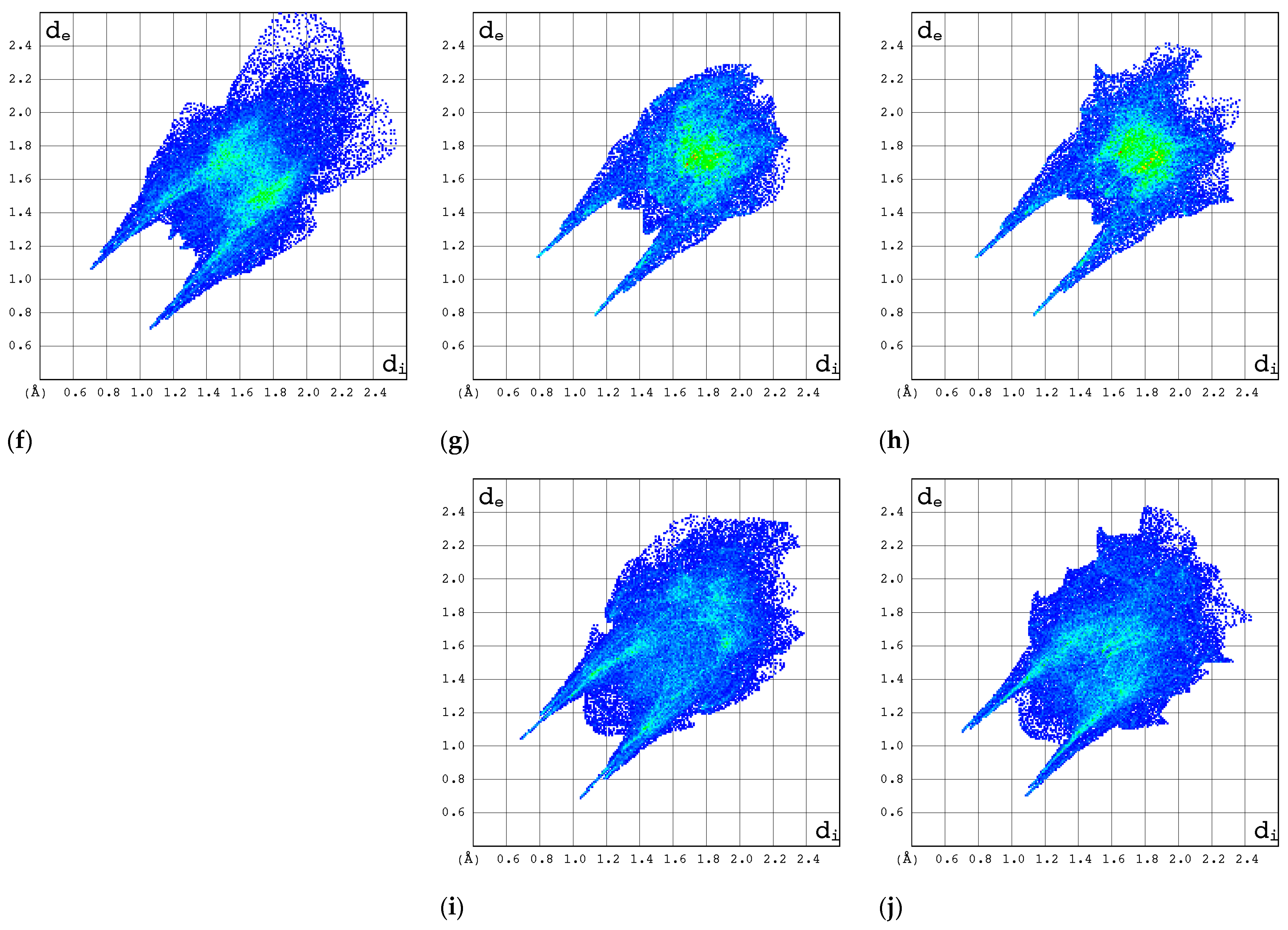
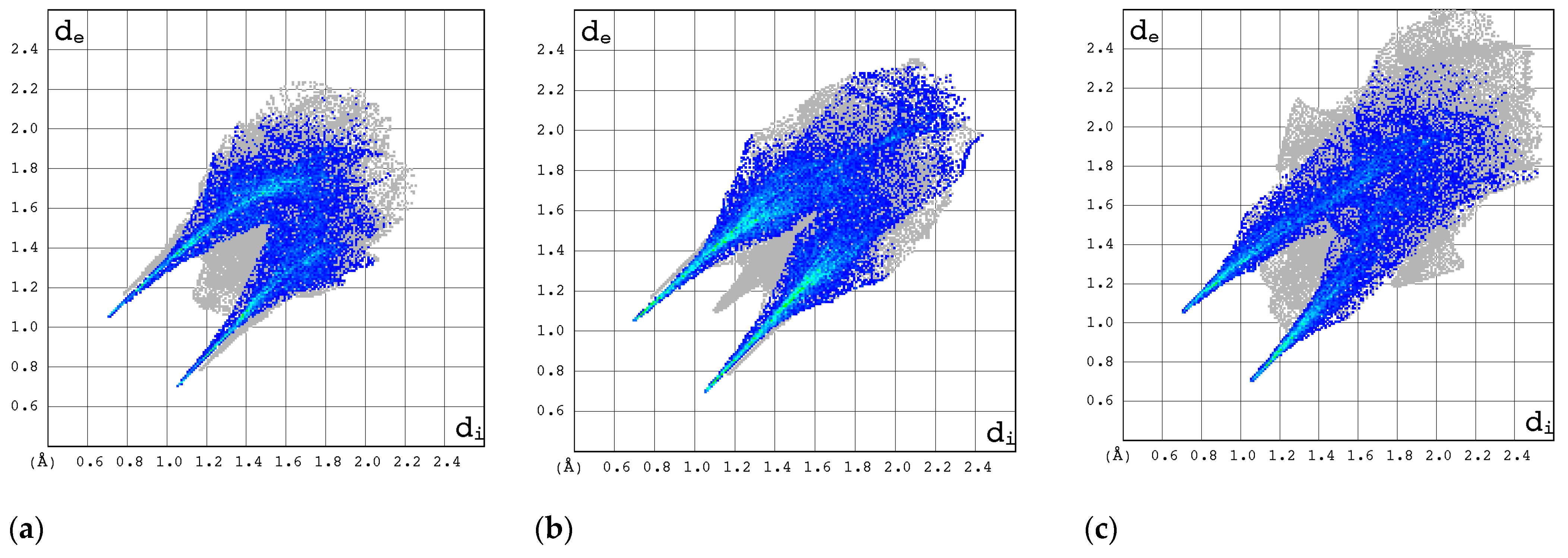
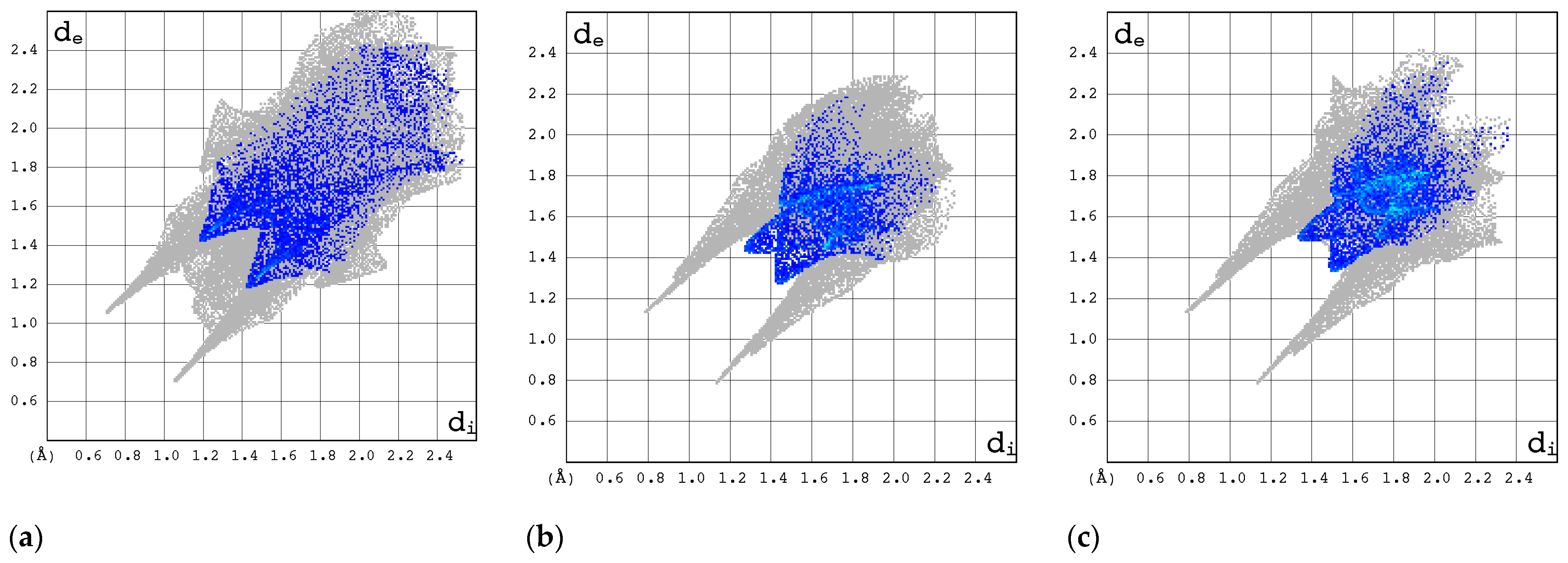
2.3.1. 3D Hirshfeld Surfaces
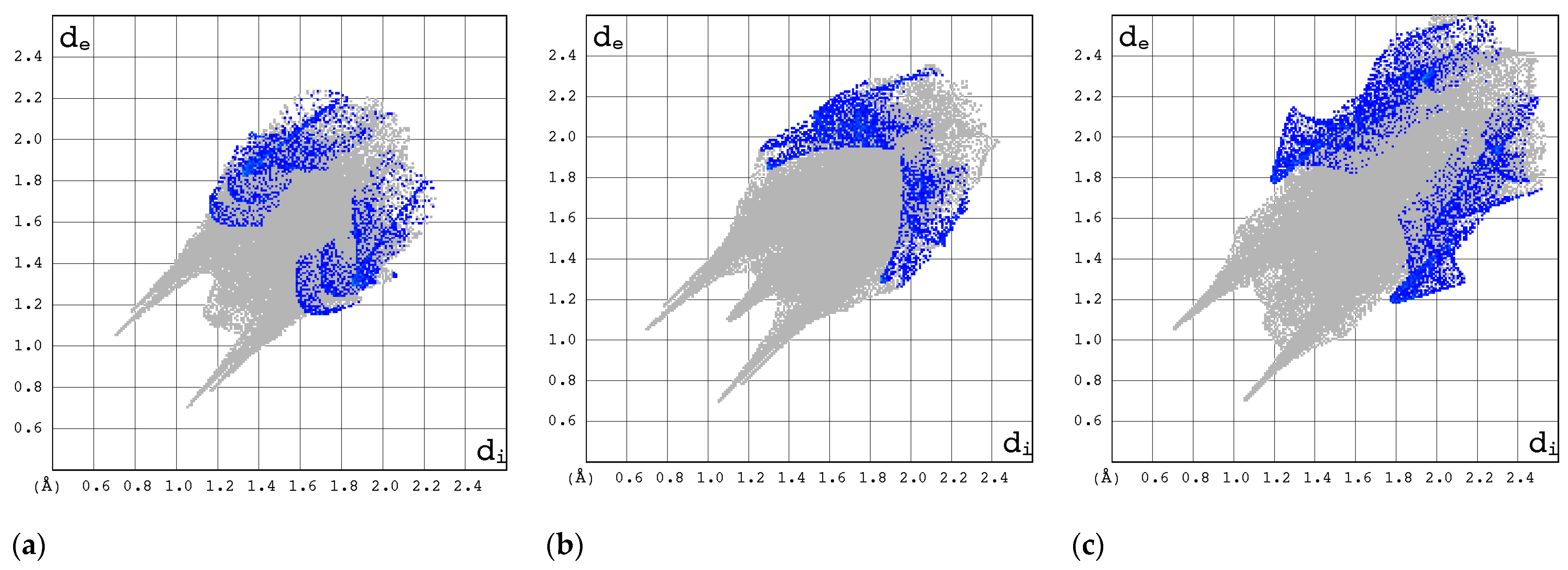
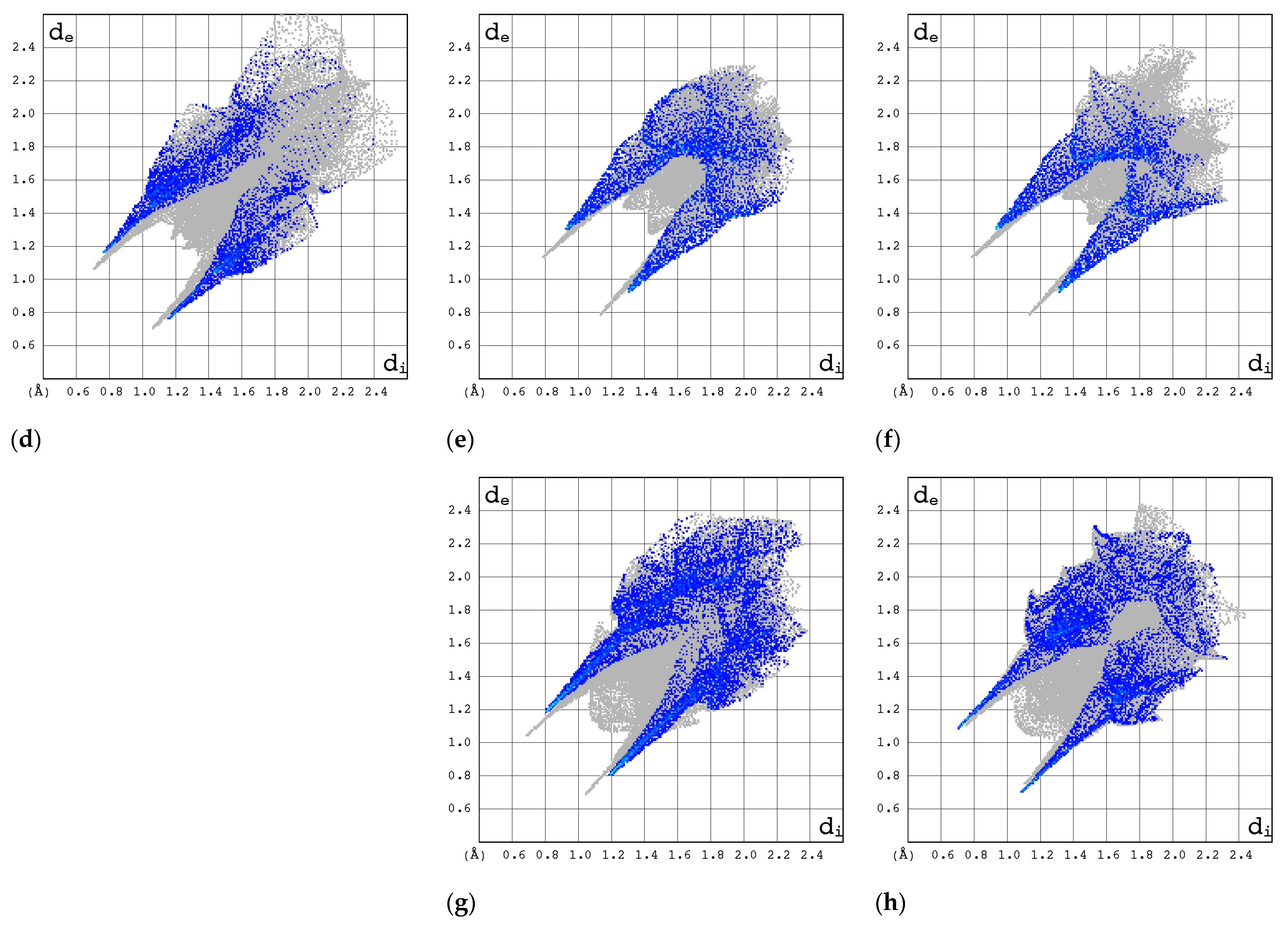
2.3.2. Enrichment Ratios
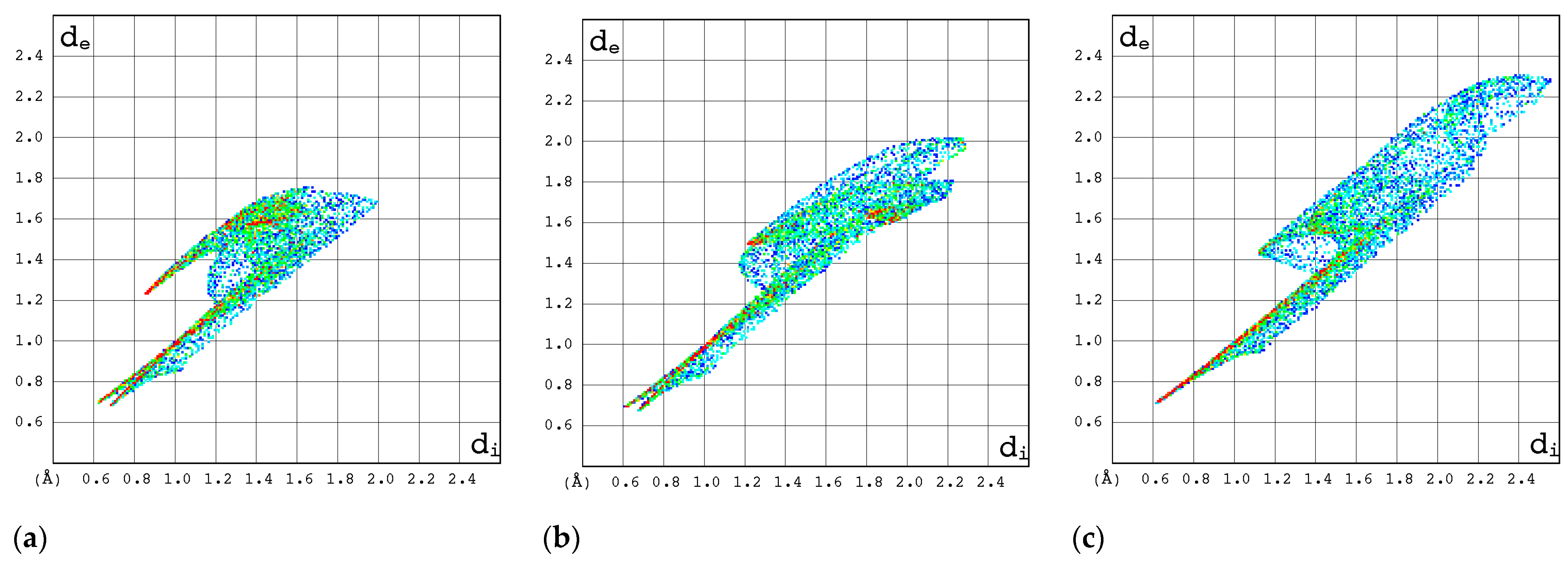
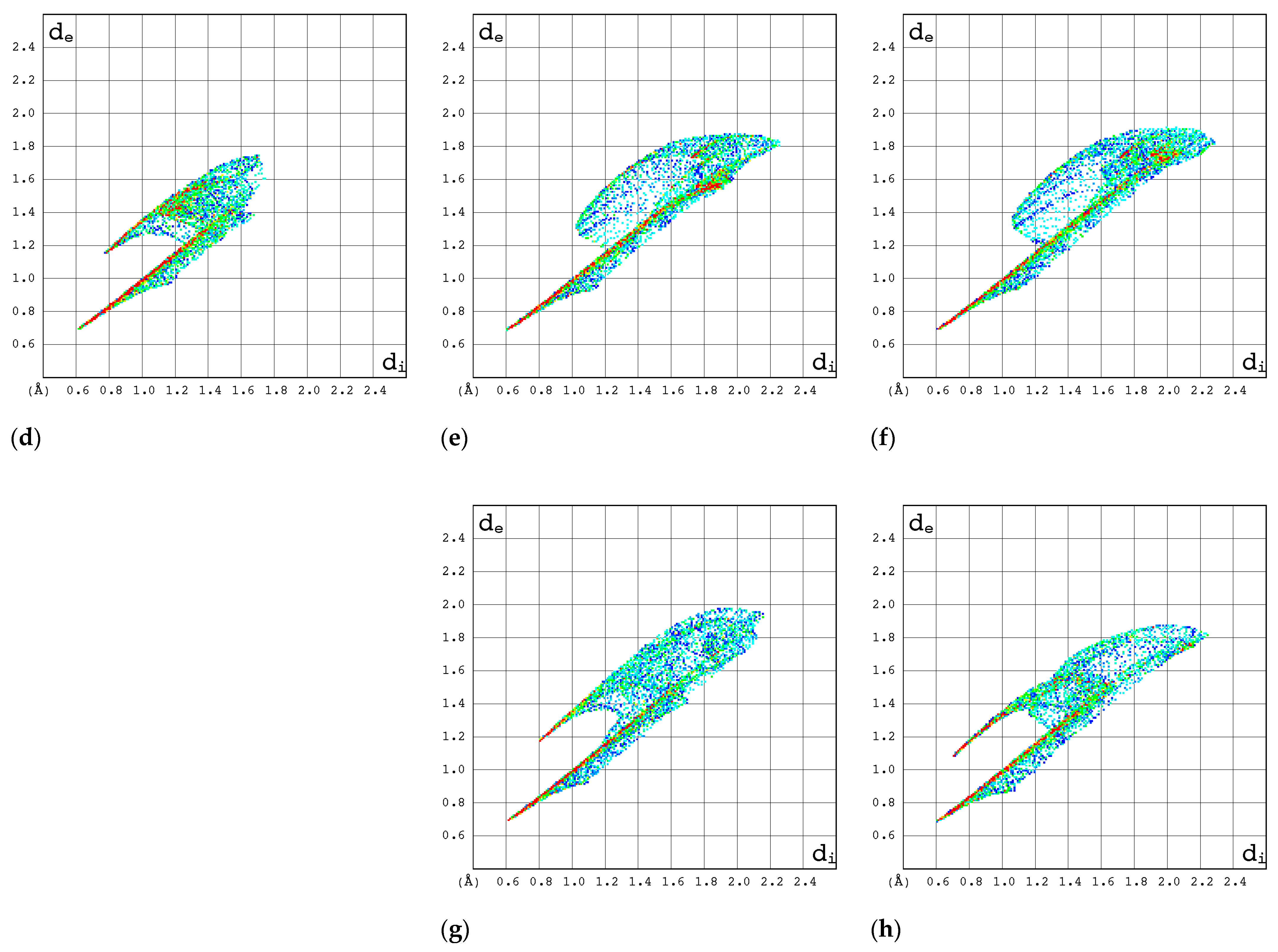
2.3.3. 2D Molecular Fingerprints
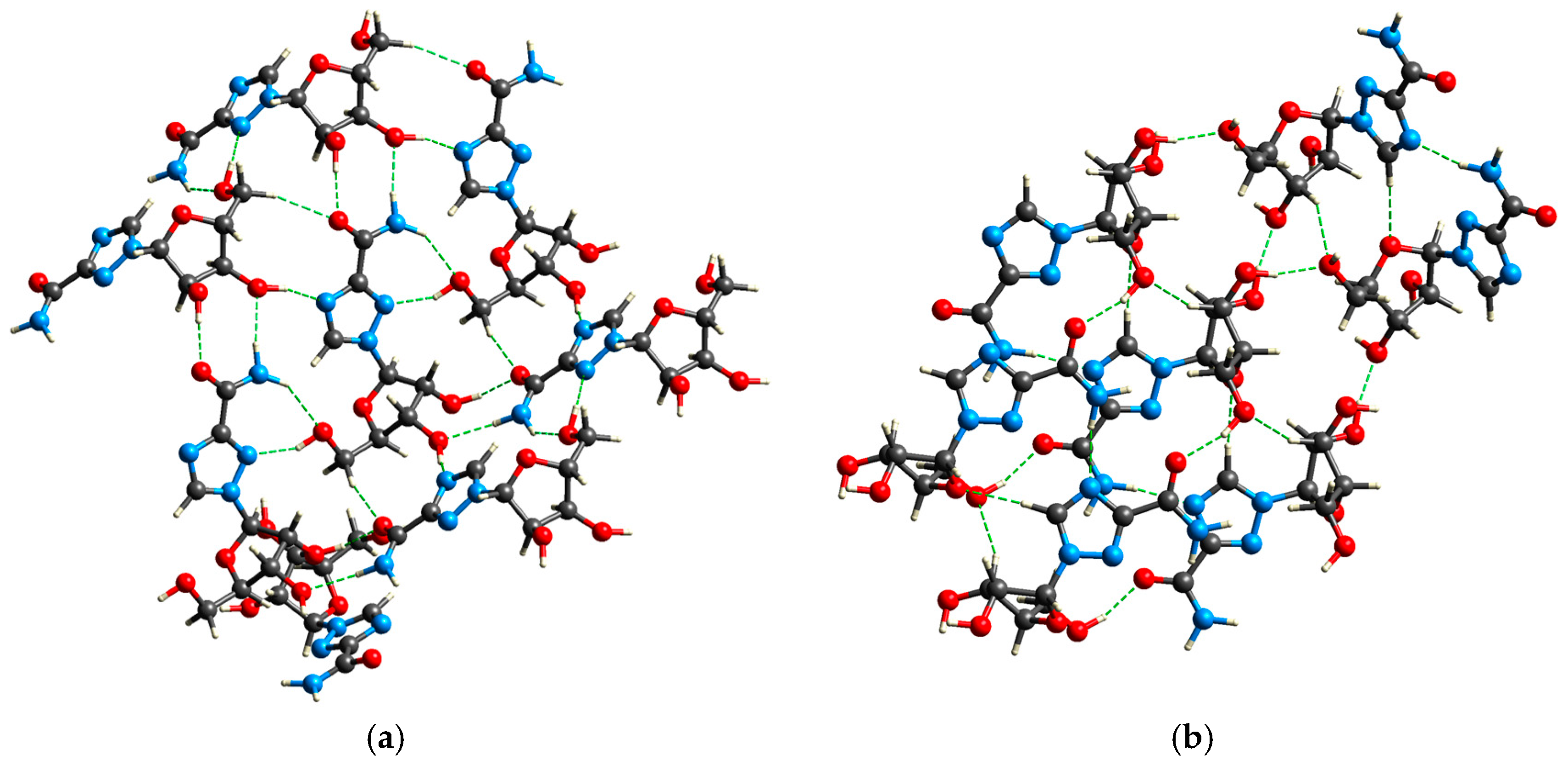
2.3.4. Intermolecular Interactions in the Solid State
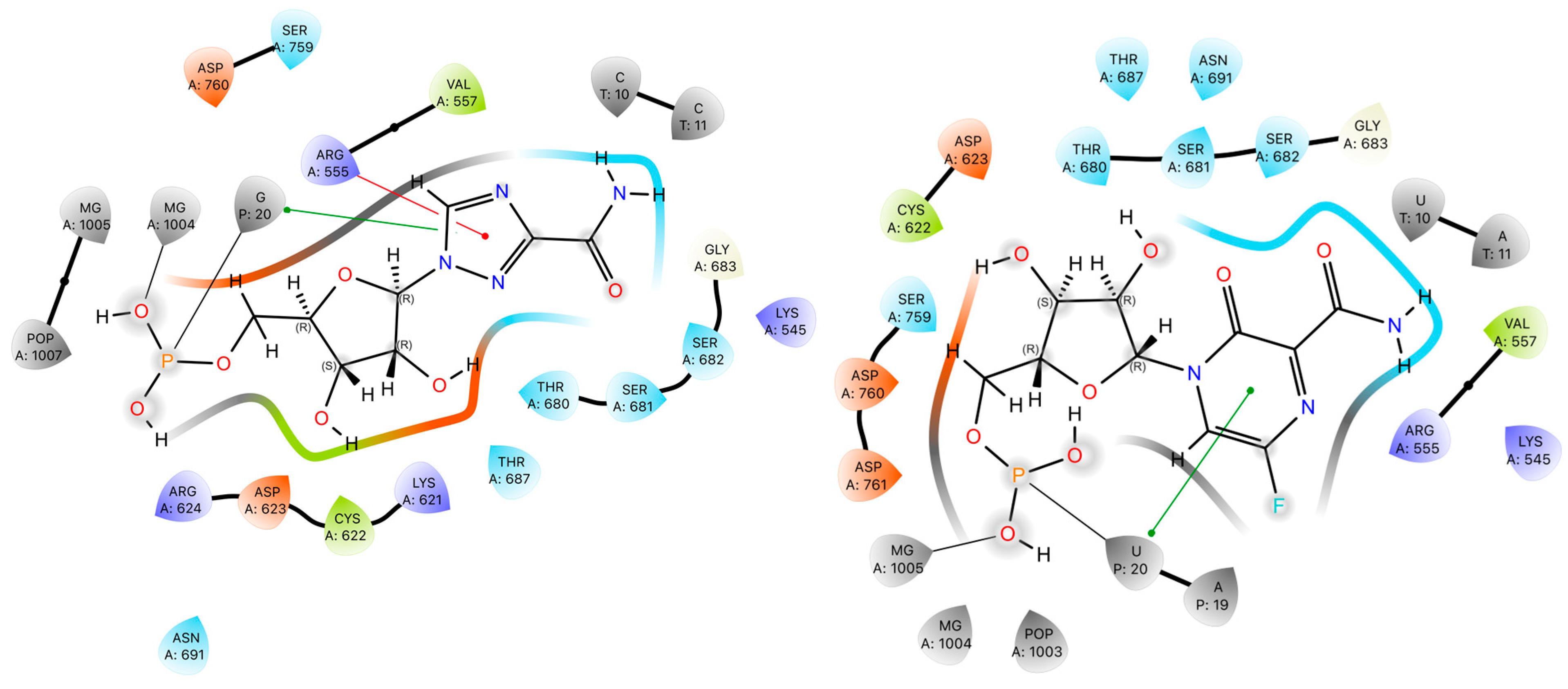
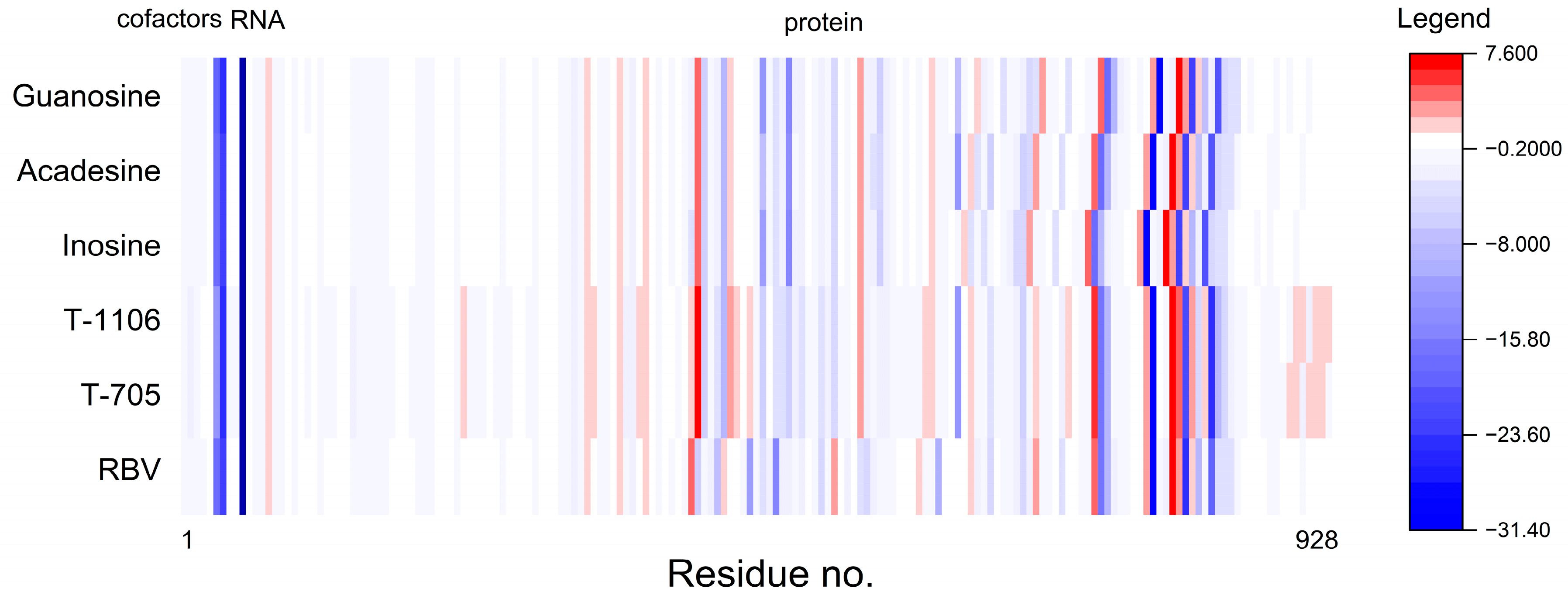
2.4. Binding Motifs in the Protein–Ligand Complex
2.4.1. The Role of Conformational Selection in Ligand Binding
2.4.2. The Role of Structural Similarity in the Binding of the Ligands
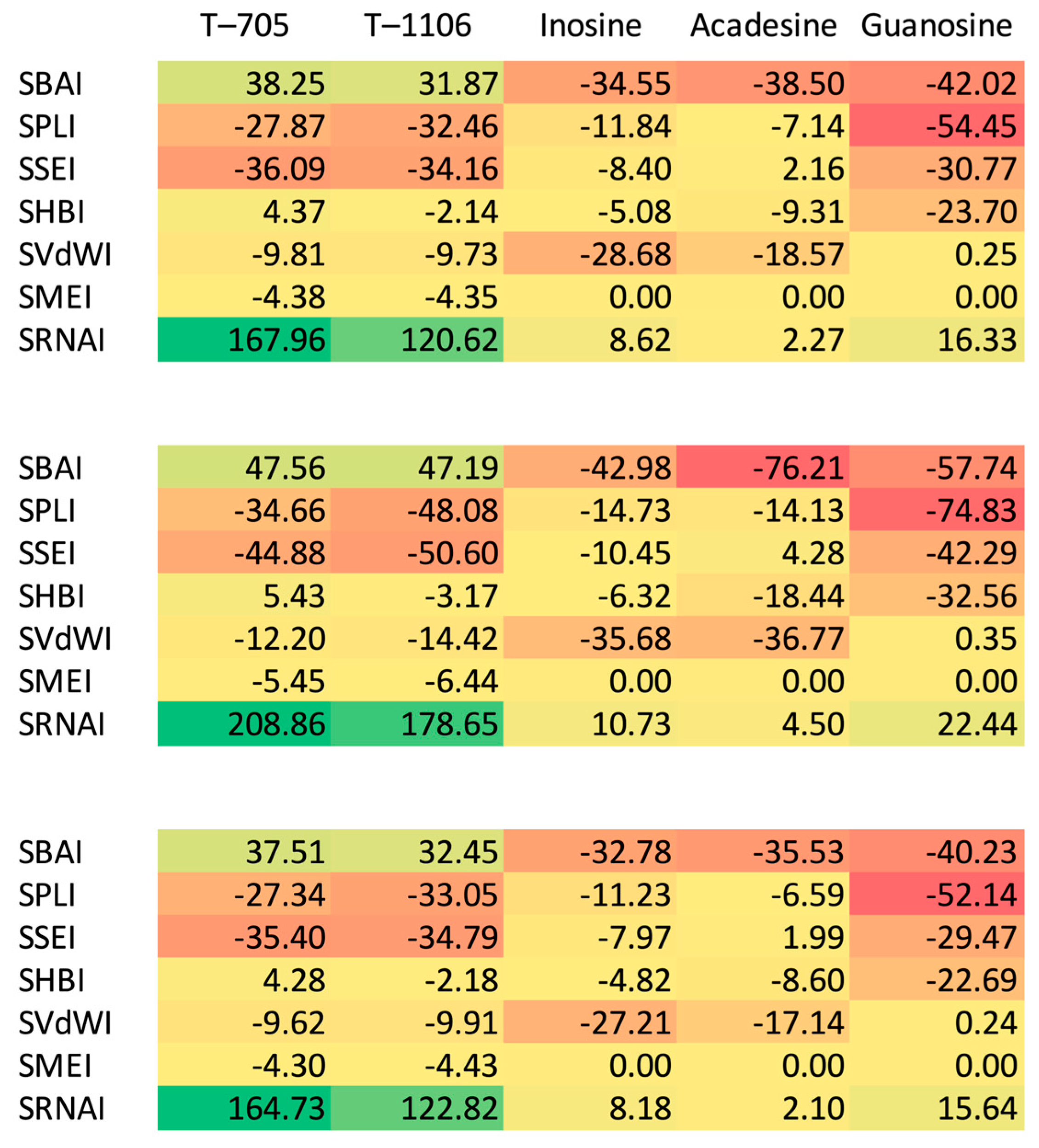
2.5. The Novel Quadrupolar Indices (QI)
2.5.1. The Quantification of the Solid State and Protein–Ligand Complexation Effects
2.5.2. The Quantification of the Similarity Between Binding Motifs in the Solid State and Protein–Ligand Complex
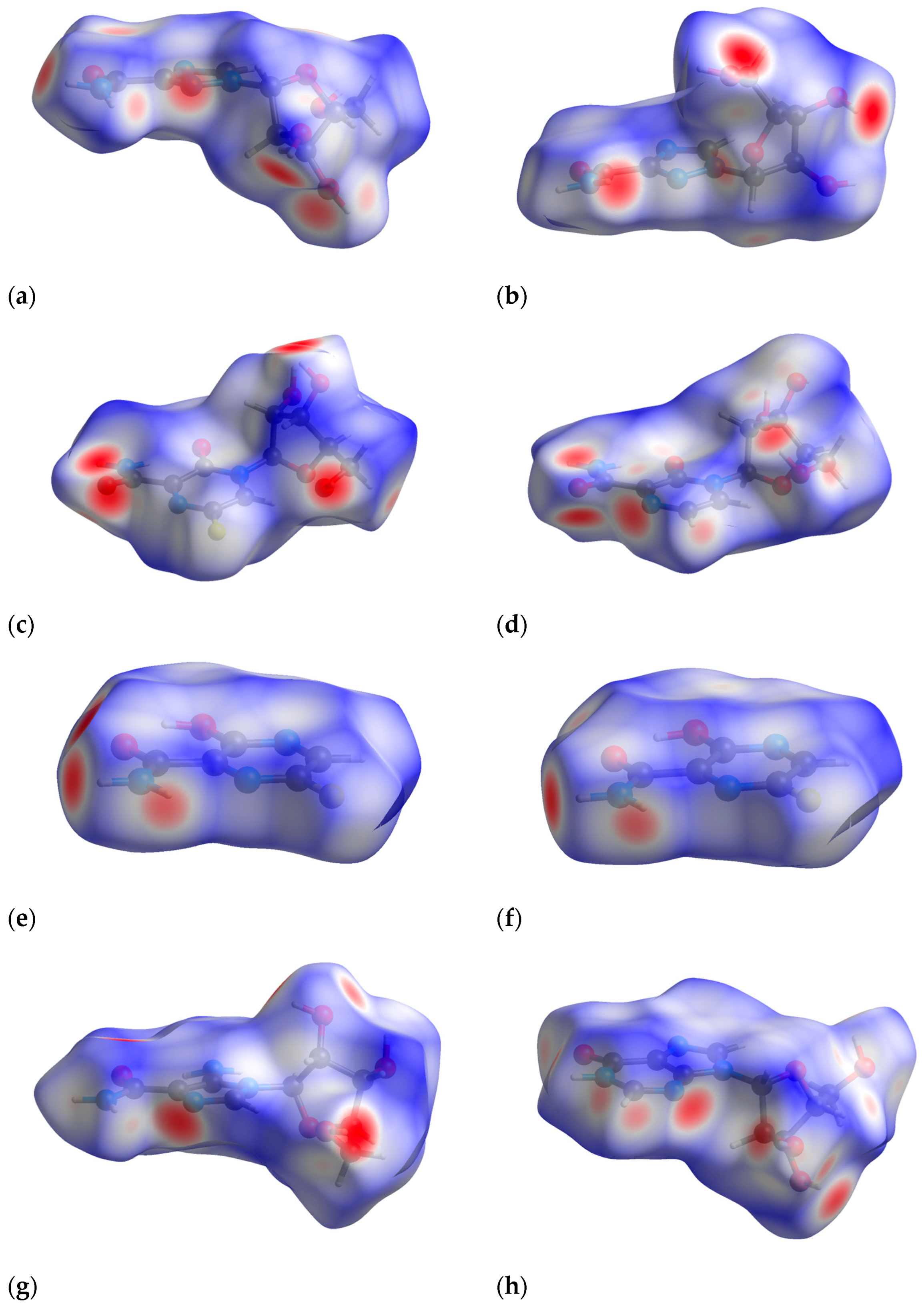
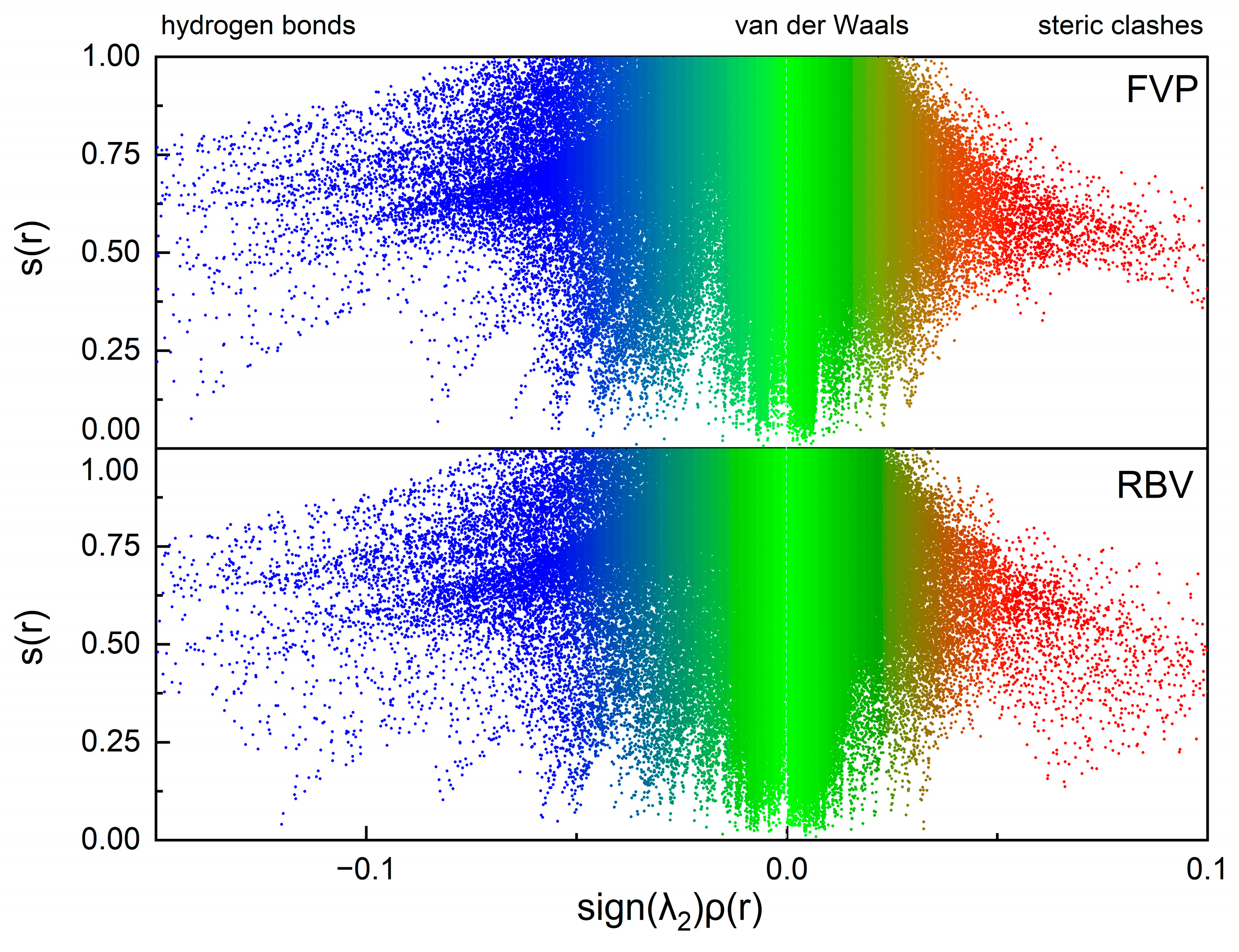
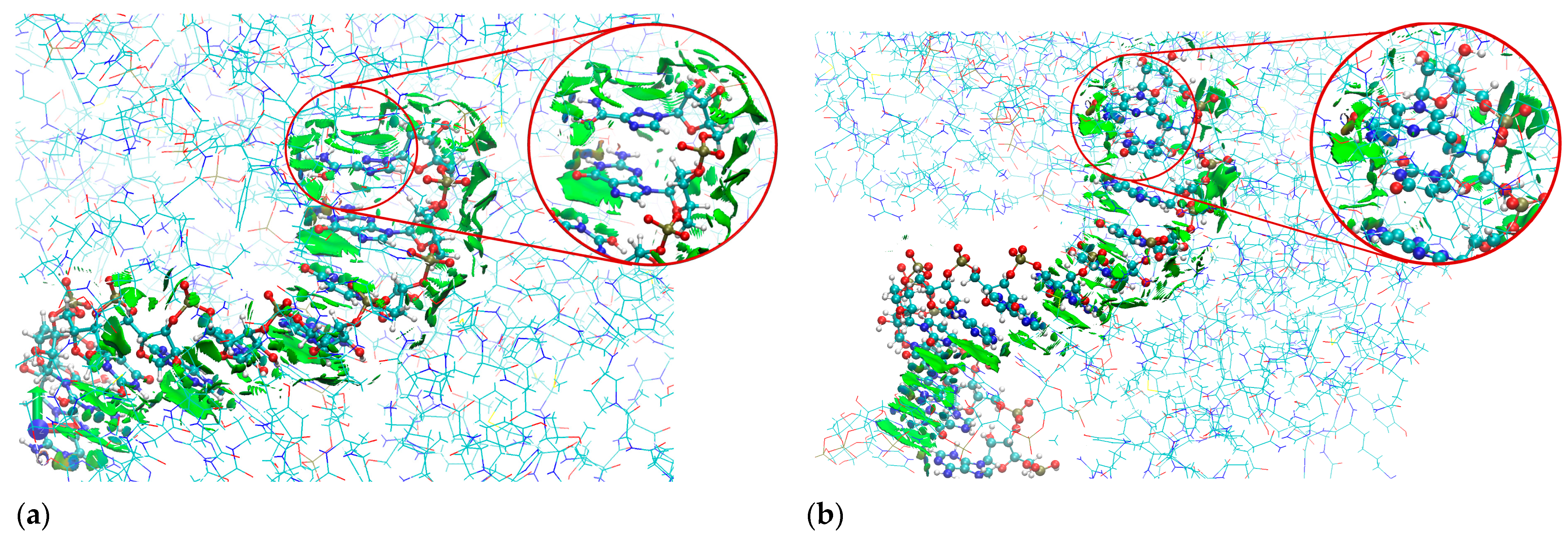
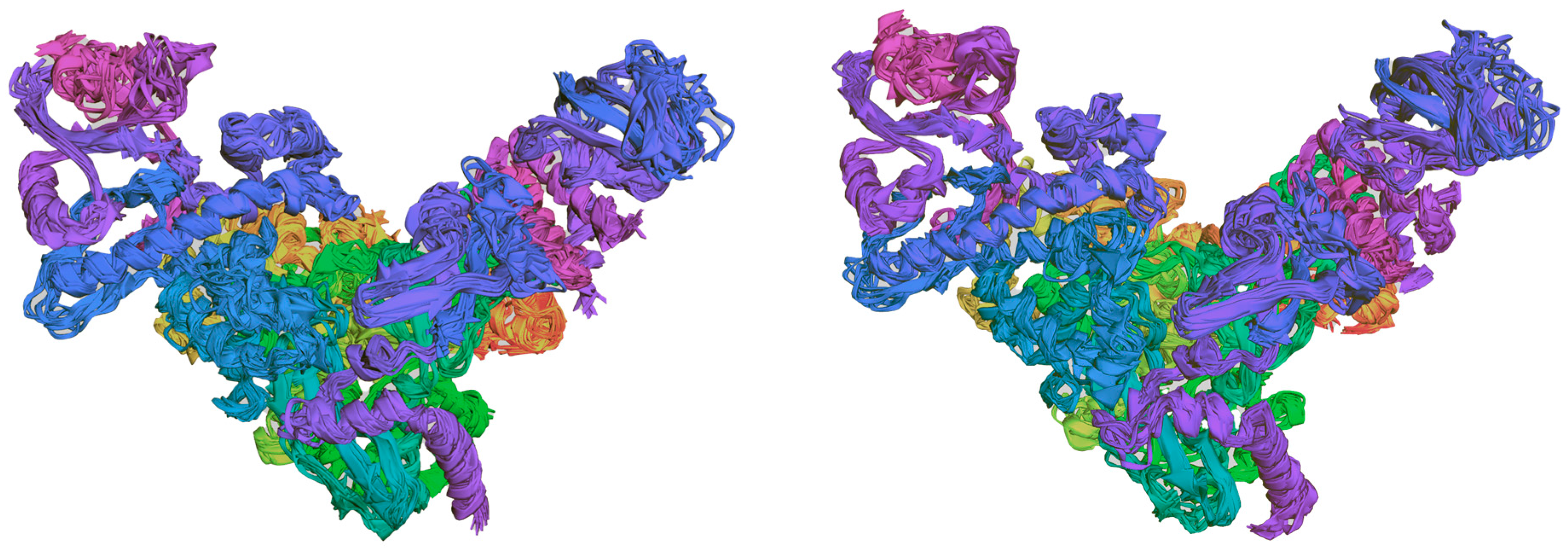
2.6. The Binding Mode Visualization
2.7. Molecular Dynamics Simulations
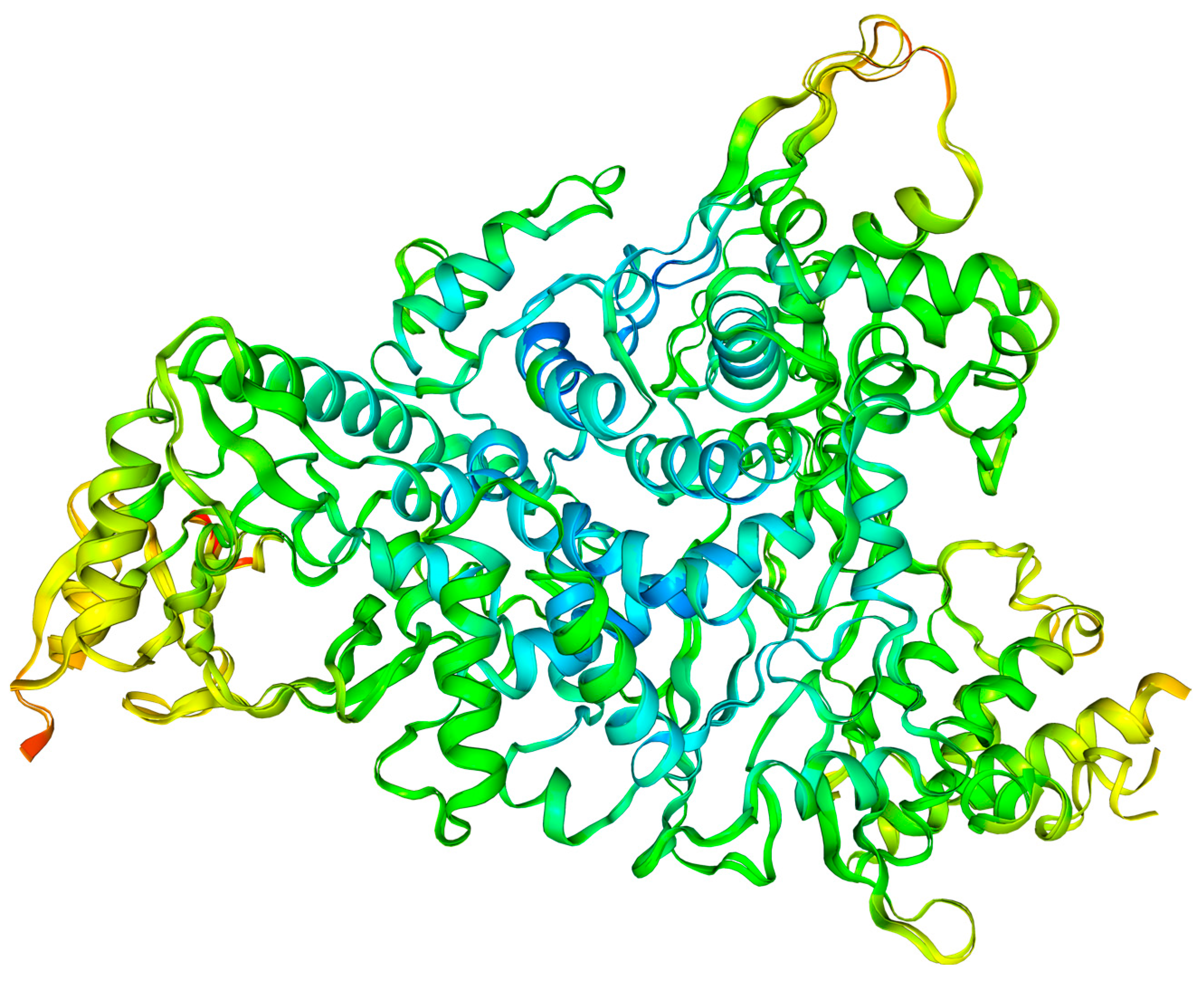
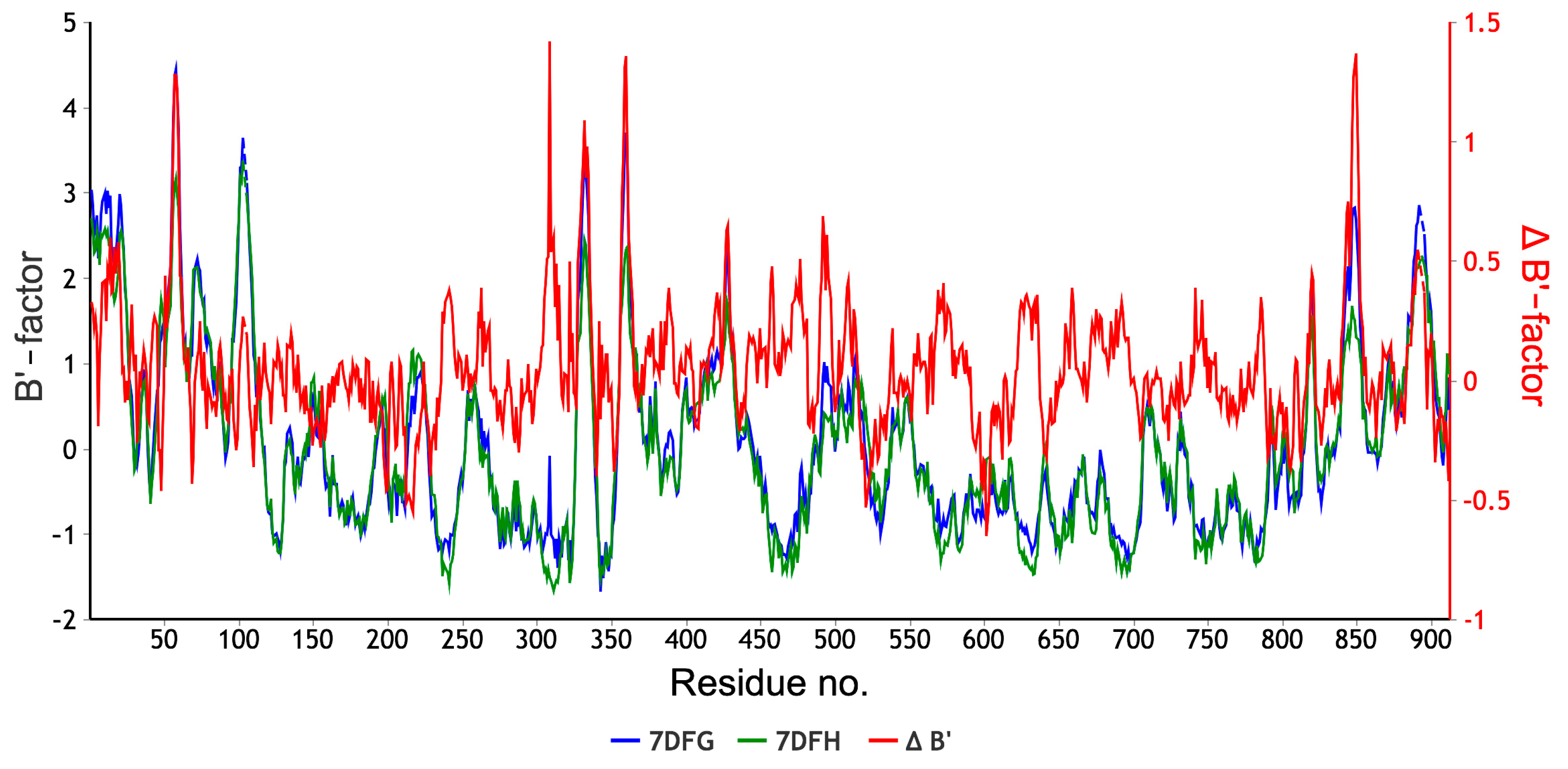
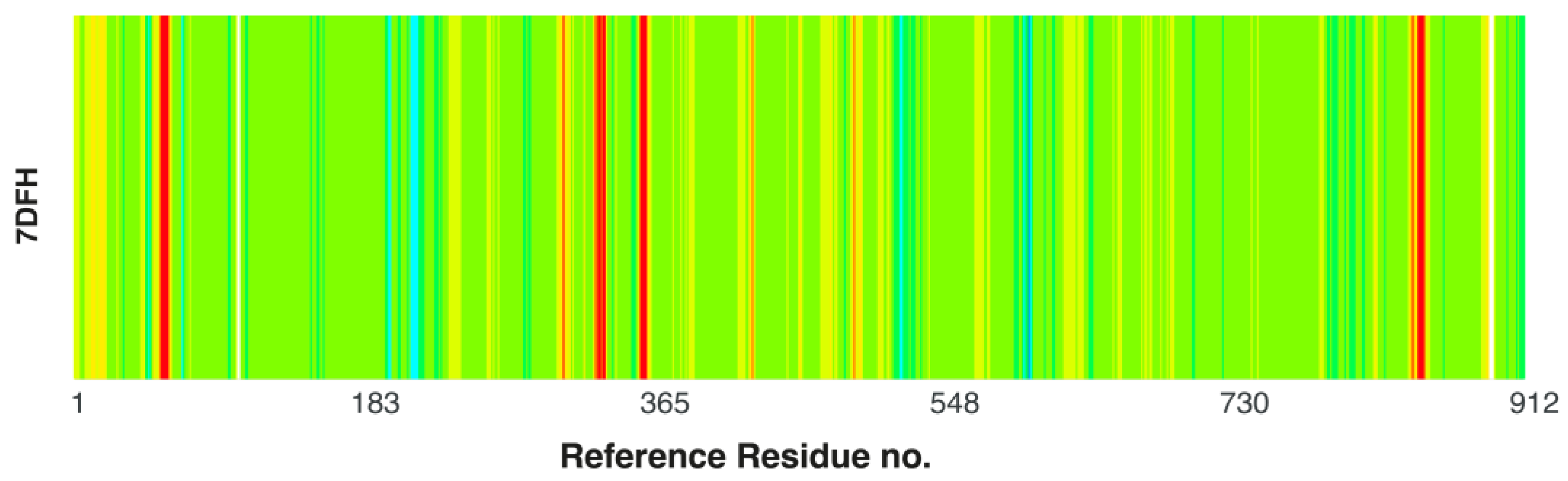
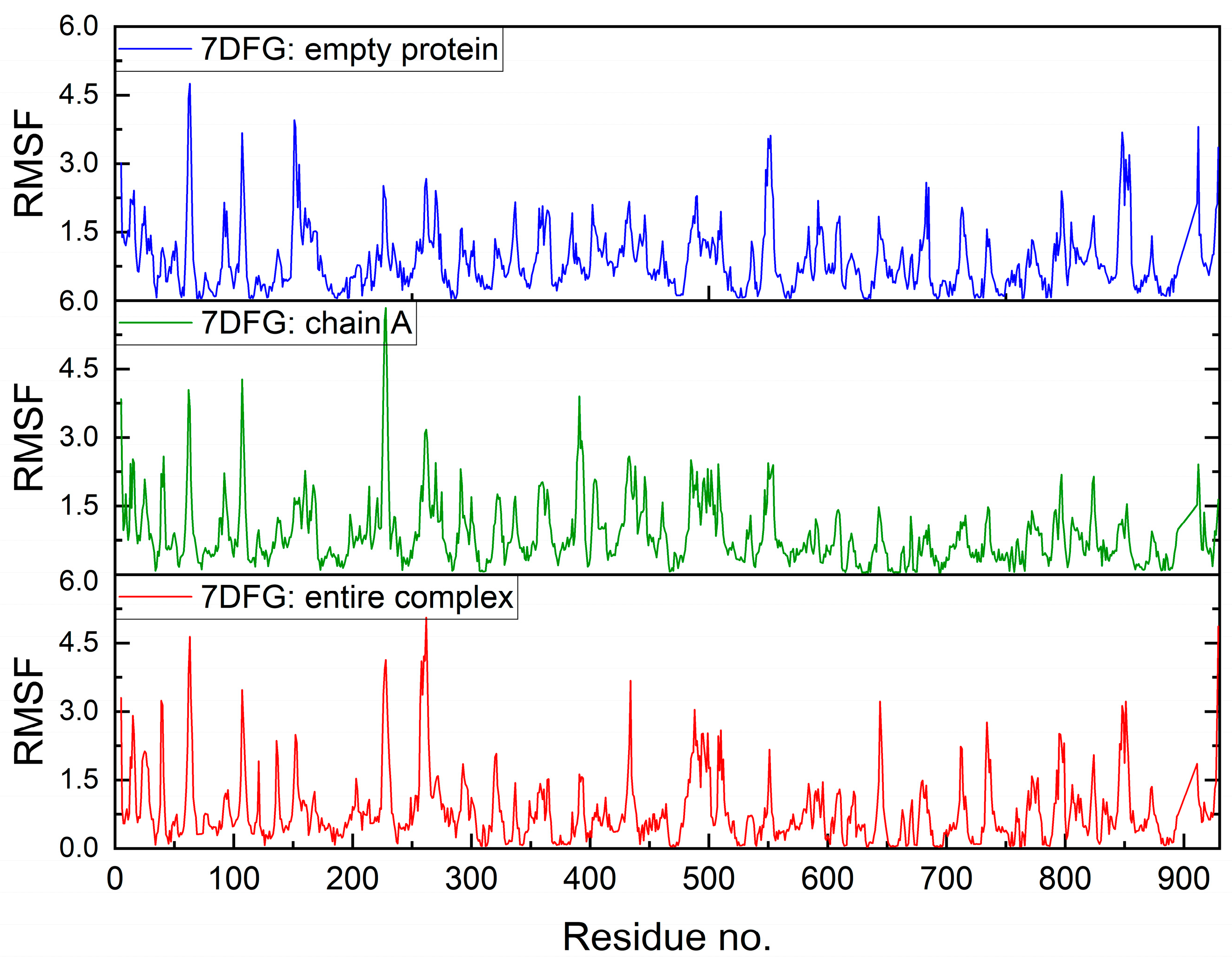
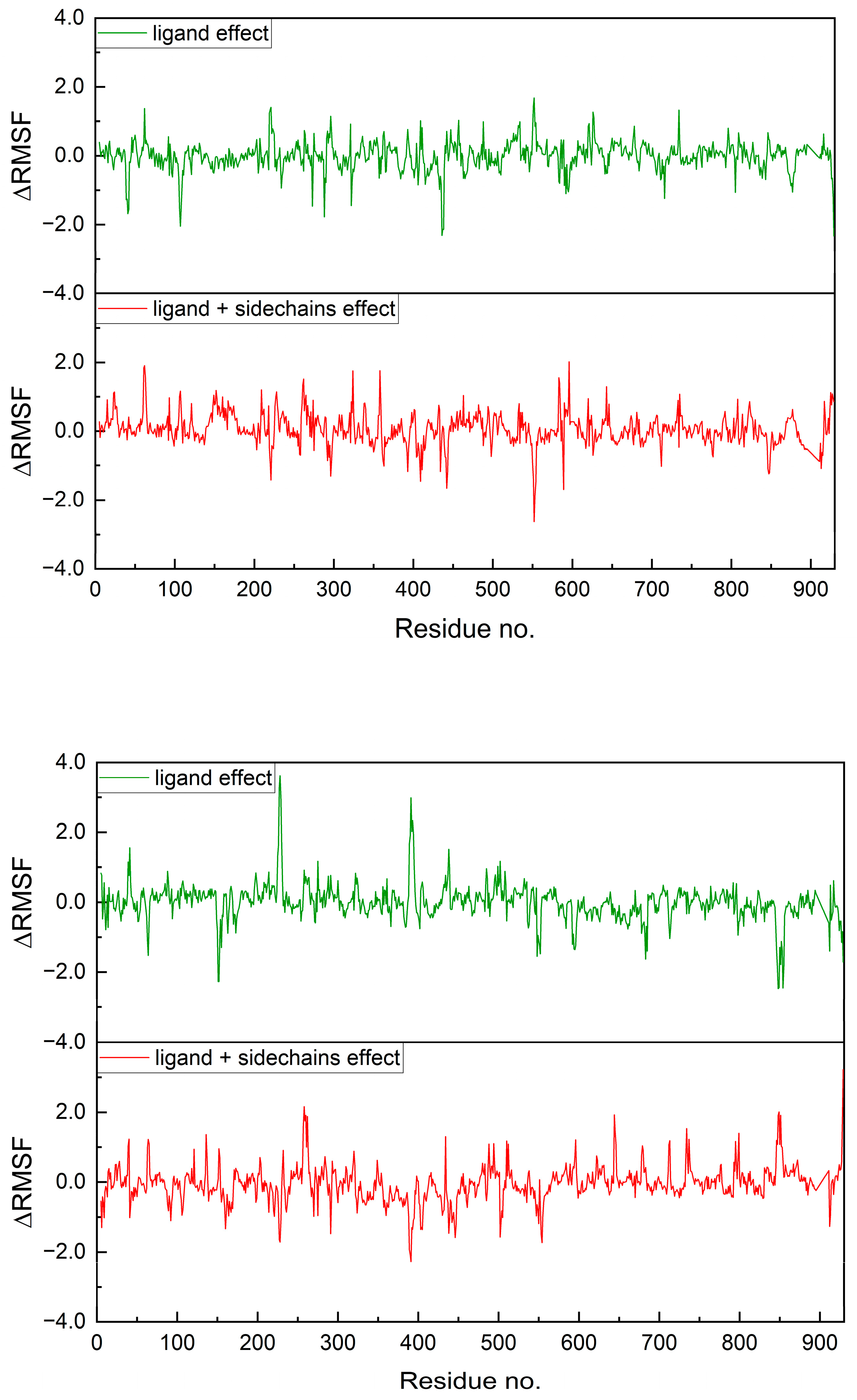
2.7.1. Protein Flexibility
2.7.2. Molecular Dynamics Simulation
3. Material
4. Methods
4.1. 14N Nuclear Quadrupole Resonance (NQR)
4.2. Density Functional Theory
4.3. 3D Hirshfeld Surfaces (3D HS)
4.4. Molecular Docking
4.5. Root Mean Square Deviation of the Binding Modes
4.6. Structure-Binding Strength Indices
- (a)
- Structure—Binding Affinity Index, SBAI:which measures the difference in the protein–ligand binding affinity between the candidate and reference ligands (numerator) with respect to their structural similarity (denominator);
- (b)
- Structure—Hydrogen Bonds Index, SHBI:which measure difference in the energy of hydrogen bonds (connecting the protein with the ligand) between the complex with the selected ligand and the reference compound (numerator) with respect to their structural dissimilarity, 1 − s, (denominator);
- (c)
- Structure—Steric Effect Index, SSEI:which measures difference in the energy of steric effects between the complex with the selected ligand and the reference (numerator) with respect to their structural dissimilarity, 1 − s, (denominator);
- (d)
- Structure—Protein-Ligand Index, SPLI:which measures the difference in the strength of the protein–ligand interactions between the ligand and reference (numerator) with respect to their structural dissimilarity, 1 − s, (denominator);
- (e)
- Structure—van der Waals Index, SVdWI:which measures the difference in the van der Waals term in the presence of the ligand and reference (numerator) with respect to the structural dissimilarity, 1 − s, (denominator);
- (f)
- Structure—RNA index, SRNAI:which measures the difference in the strength of the interactions between the RNA template and ligand, and the RNA template and reference (numerator) with respect to their structural dissimilarity, 1 − s, (denominator);
- (g)
- Structure—Metal Index, SMEI:which measures the difference in the strength of the interactions between the RNA template and ligand, and the RNA template and reference (numerator) with respect to their structural dissimilarity, 1 − s, (denominator);
4.7. Molecular Dynamics Simulation (MSD)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D HS | 3D Hirshfeld surfaces |
| 2D FP | 2D molecular fingerprints |
| CAS | Chemical abstracts service |
| CR | Cross-relaxation |
| DFT | Density functional theory |
| ED | Euclidean distance |
| EM | Electron microscopy |
| EXY | Enrichment ratios |
| FFC | Fast field-cycling |
| FVP | Favipiravir |
| GGA | Generalized gradient approximation |
| IMP | Inosine monophosphate |
| IMPDH | Inosine-5′-monophosphate dehydrogenase |
| M06L, M062X | Meta-hybrid Minnesota functionals |
| MD | Molecular docking |
| MDS | Molecular dynamic simulations |
| mRNA | Messenger RNA |
| NAD+ | Nicotinamide adenine dinucleotide |
| NMR | Nuclear magnetic resonance |
| NQR | Nuclear quadrupole resonance |
| PXRD | Powder X-ray diffraction |
| QTAIM | Quantum theory of atoms in molecules |
| QI | Quadrupolar indices |
| RBV | Ribavirin |
| RdRp | RNA-dependent RNA polymerase |
| RDS | Reduced density gradient |
| RMDS | Root mean square distance |
| RMSD | BM root mean square deviation of the binding modes |
| RMP | Ribavirin 5-monophosphate |
| RMSF | Root mean square fluctuation |
| RNA | Ribonucleic acid |
| RPBE | Revised Perdew–Burke–Ernzerhof |
| RTP | Ribavirin 5-triphosphate |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SBAI | Structure—binding affinity index |
| SBSI | Structure—binding strength indices |
| SHBI | Structure—hydrogen bonds index |
| SMEI | Structure—metal index |
| SSEI | Structure—steric effect index |
| SPLI | Structure—protein–ligand index |
| SRNAI | Structure—RNA index |
| SVdWI | Structure—van der Waals index |
| T-705 | 3,4-dihydro-6-fluoro-3-oxo-4-β-D-ribofuranosyl-2-pyrazine carboxamide |
| T-1106 | 3,4-dihydro-3-oxo-4-β-D-ribofuranosyl-2-pyrazine carboxamide |
| TK | Tkatchenko–Scheffler method |
| XMP | Xanthosine monophosphate (XMP) |
References
- Latosińska, J.N.; Latosińska, M.; Seliger, J.; Žagar, V.; Apih, T. Butterfly Effect in Cytarabine: Combined NMR-NQR Experiment, Solid-State Computational Modeling, Quantitative Structure-Property Relationships and Molecular Docking Study. Pharmaceuticals 2024, 17, 445. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Huffman, J.H.; Lois, G.P.K.; Allen, B.; Roland, J.T.W.; Robins, K. Broad-Spectrum Antiviral Activity of Virazole: 1-β-D-Ribofuranosyl-1,2,4-Triazole-3-Carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, J.T.; Robins, R.K.; Sidwell, R.W.; Simon, L.N. Design, Synthesis, and Broad Spectrum Antiviral Activity of 1-.Beta.-D-Ribofuranosyl-1,2,4-Triazole-3-Carboxamide and Related Nucleosides. J. Med. Chem. 1972, 15, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Web Annex A. World Health Organization Model List of Essential Medicines—23rd List, 2023. In The Selection and Use of Essential Medicines 2023: Executive Summary of the Report of the24th WHO Expert Committee on the Selection and Use of Essential Medicines; World Health Organization: Geneva, Switzerland, 2023; Available online: https://iris.who.int/bitstream/handle/10665/371090/WHO-MHP-HPS-EML-2023.02-eng.pdf?sequence=1 (accessed on 30 January 2025).
- Martin, P.; Jensen, D.M. Ribavirin in the Treatment of Chronic Hepatitis C. J. Gastro. Hepatol. 2008, 23, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Ganellin, C.R. (Eds.) Analogue-Based Drug Discovery, 1st ed.; Wiley: Hoboken, NJ, USA, 2006; ISBN 978-3-527-31257-3. [Google Scholar]
- De Clercq, E. The Race for Interferon-free HCV Therapies: A Snapshot by the Spring of 2012. Rev. Med. Virol. 2012, 22, 392–411. [Google Scholar] [CrossRef]
- Patki, S.A.; Gupta, P. Evaluation of Ribavirin in the Treatment of Acute Hepatitis. Chemotherapy 1982, 28, 298–303. [Google Scholar] [CrossRef]
- Gerolami, R.; Borentain, P.; Raissouni, F.; Motte, A.; Solas, C.; Colson, P. Treatment of Severe Acute Hepatitis E by Ribavirin. J. Clin. Virol. 2011, 52, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.; Banks, G.; Smith, R. Ribavirin: A Clinical Overview. Eur. J. Epidemiol. 1986, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tam, R.C.; Lau, J.Y.; Hong, Z. Mechanisms of Action of Ribavirin in Antiviral Therapies. Antivir. Chem. Chemother. 2001, 12, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.L.B.; Culjkovic-Kraljacic, B. Ribavirin as an Anti-Cancer Therapy: Acute Myeloid Leukemia and Beyond? Leuk. Lymphoma 2010, 51, 1805–1815. [Google Scholar] [CrossRef]
- Wambecke, A.; Laurent-Issartel, C.; Leroy-Dudal, J.; Giffard, F.; Cosson, F.; Lubin-Germain, N.; Uziel, J.; Kellouche, S.; Carreiras, F. Evaluation of the Potential of a New Ribavirin Analog Impairing the Dissemination of Ovarian Cancer Cells. PLoS ONE 2019, 14, e0225860. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Ribavirin Suppresses eIF4E-Mediated Oncogenic Transformation by Physical Mimicry of the 7-Methyl Guanosine mRNA Cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef]
- Kentsis, A.; Volpon, L.; Topisirovic, I.; Soll, C.E.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Further Evidence That Ribavirin Interacts with eIF4E. RNA 2005, 11, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, W.; Yang, R.; Zhang, Q.; Hao, L.; Bian, E.; Yang, Y.; Huang, X.; Wu, Y.; Zhang, B. Ribavirin Inhibits the Growth and Ascites Formation of Hepatocellular Carcinoma through Downregulation of Type I CARM1 and Type II PRMT5. Toxicol. Appl. Pharmacol. 2022, 435, 115829. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.P.; Kigwana, L.J.; Streeter, D.G.; Robins, R.K.; Simon, L.N.; Roboz, J. The Relationship between the Metabolism of Ribavirin and Its Proposed Mechanism of Action. Ann. N. Y. Acad. Sci. 1977, 284, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Nyström, K.; Waldenström, J.; Tang, K.-W.; Lagging, M. Ribavirin: Pharmacology, Multiple Modes of Action and Possible Future Perspectives. Future Virol. 2019, 14, 153–160. [Google Scholar] [CrossRef]
- Willis, R.C.; Carson, D.A.; Seegmiller, J.E. Adenosine Kinase Initiates the Major Route of Ribavirin Activation in a Cultured Human Cell Line. Proc. Natl. Acad. Sci. USA 1978, 75, 3042–3044. [Google Scholar] [CrossRef]
- Parker, W.B. Metabolism and Antiviral Activity of Ribavirin. Virus Res. 2005, 107, 165–171. [Google Scholar] [CrossRef]
- Russmann, S.; Grattagliano, I.; Portincasa, P.; Palmieri, V.; Palasciano, G. Ribavirin-Induced Anemia: Mechanisms, Risk Factors and Related Targets for Future Research. Curr. Med. Chem. 2006, 13, 3351–3357. [Google Scholar] [CrossRef] [PubMed]
- Garden, B.B. Handbook on Mulches; World’s Best Illustrated Garden and Horticultural Handbooks; Brooklyn Botanic Garden: Brooklyn, NY, USA, 1980; ISBN B000H46LKW. [Google Scholar]
- Latosińska, J.N.; Latosińska, M.; Seliger, J.; Žagar, V.; Apih, T.; Grieb, P. Elucidating the Role of Noncovalent Interactions in Favipiravir, a Drug Active against Various Human RNA Viruses; a 1H–14N NQDR/Periodic DFT/QTAIM/RDS/3D Hirshfeld Surfaces Combined Study. Molecules 2023, 28, 3308. [Google Scholar] [CrossRef]
- Latosińska, M.; Latosińska, J.N. Favipiravir Analogues as Inhibitors of SARS-CoV-2 RNA-Dependent RNA Polymerase, Combined Quantum Chemical Modeling, Quantitative Structure–Property Relationship, and Molecular Docking Study. Molecules 2024, 29, 441. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.; Smith, J.A.S. Nitrogen-14 Quadrupole Cross-Relaxation Spectroscopy. Proc. R. Soc. Lond. A 1988, 416, 149–178. [Google Scholar] [CrossRef]
- Gregorovič, A.; Apih, T.; Seliger, J. 1H–14N Cross-Relaxation Spectrum Analysis in Sildenafil and Sildenafil Citrate. Solid State Nucl. Magn. Reson. 2016, 78, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kidd, W.C.; Varlashkin, P.; Li, C. The Applicability of Powder X-Ray Diffraction to the Quantification of Drug Substance Polymorphs Using a Model Organic System. Powder Diffr. 1993, 8, 180–187. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; The International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 2003; ISBN 978-0-19-855865-1. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Seliger, J.; Žagar, V.; Apih, T.; Gregorovič, A.; Latosińska, M.; Olejniczak, G.A.; Latosińska, J.N. Polymorphism and Disorder in Natural Active Ingredients. Low and High-Temperature Phases of Anhydrous Caffeine: Spectroscopic (1H–14N NMR–NQR/14N NQR) and Solid-State Computational Modelling (DFT/QTAIM/RDS) Study. Eur. J. Pharm. Sci. 2016, 85, 18–30. [Google Scholar] [CrossRef]
- Latosińska, J.N.; Latosińska, M.; Olejniczak, G.A.; Seliger, J.; Žagar, V. Topology of the Interactions Pattern in Pharmaceutically Relevant Polymorphs of Methylxanthines (Caffeine, Theobromine, and Theophiline): Combined Experimental (1H–14N Nuclear Quadrupole Double Resonance) and Computational (DFT and Hirshfeld-Based) Study. J. Chem. Inf. Model. 2014, 54, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Latosinńska, J.N. Applications of Nuclear Quadrupole Resonance Spectroscopy in Drug Development. Expert Opin. Drug Discov. 2007, 2, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Blinc, R.; Seliger, J.; Zidansek, A.; Žagar, V.; Milia, F.; Robert, H. 14N Nuclear Quadrupole Resonance of Some Sulfa Drugs. Solid State Nucl. Magn. Reson. 2006, 30, 61–68. [Google Scholar] [CrossRef]
- Latosińska, J.N. Nuclear Quadrupole Resonance Spectroscopy in Studies of Biologically Active Molecular Systems—A Review. J. Pharm. Biomed. Anal. 2005, 38, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Trontelj, Z.; Pirnat, J.; Jazbinšek, V.; Lužnik, J.; Srčič, S.; Lavrič, Z.; Beguš, S.; Apih, T.; Žagar, V.; Seliger, J. Nuclear Quadrupole Resonance (NQR)—A Useful Spectroscopic Tool in Pharmacy for the Study of Polymorphism. Crystals 2020, 10, 450. [Google Scholar] [CrossRef]
- Latosińska, J.N. NQR Parameters: Electric Field Gradient Tensor and Asymmetry Parameter Studied in Terms of Density Functional Theory. Int. J. Quantum Chem. 2003, 91, 284–296. [Google Scholar] [CrossRef]
- Li, F.; Chen, S.; Hu, H.; Liang, C.; Sun, S.; Jin, C.; Chen, F. Crystallization Selectivity of Ribavirin Solution and Amorphous Phase. Molecules 2023, 28, 6320. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, P.; Sundaralingam, M. The Crystal and Molecular Structures of Two Polymorphic Crystalline Forms of Virazole (1-β-D-Ribofuranosyl-1,2,4-Triazole-3-Carboxamide). A New Synthetic Broad Sprectrum Antiviral Agent. Acta Crystallogr. B Struct. Sci. 1976, 32, 419–426. [Google Scholar] [CrossRef]
- Seliger, J.; Osredkar, R.; Mali, M.; Blinc, R. 14N Quadrupole Resonance of Some Liquid Crystalline Compounds in the Solid. J. Chem. Phys. 1976, 65, 2887–2891. [Google Scholar] [CrossRef]
- Seliger, J.; Blinc, R.; Arend, H.; Kind, R. Proton-14N Double Resonance Study of the Structural Phase Transitions in the Perovskite Type Layer Compound (CH3NH3)2CdCl4. Z. Phys. B 1976, 25, 189–195. [Google Scholar] [CrossRef]
- Palmer, M.H.; Stephenson, D.; Smith, J.A.S. 14N Quadrupole Coupling Tensors in Solid Pyrazole, 1H-1,2,4-Triazole, and 1H-Tetrazole: Theory and Experiment. Chem. Phys. 1985, 97, 103–111. [Google Scholar] [CrossRef]
- Schempp, E.; Bray, P.J. Nuclear Quadrupole Resonance in 5-Membered Heterocycles: Pyrazole, Imidazole, 1,2,4-Triazole. Phys. Lett. A 1967, 25, 414–415. [Google Scholar] [CrossRef]
- Guibé, L.; Lucken, E.A.C. Pure Quadrupole Resonance of 14N in Some Five-Membered Heterocyclic Compounds. Mol. Phys. 1968, 14, 73–77. [Google Scholar] [CrossRef]
- Latosińska, J.N.; Latosińska, M.; Koput, J. The Tautomeric Equilibria of Cytosine Studied by NQR Spectroscopy and HF, MP2 and DFT Calculations. J. Mol. Struct. 2003, 648, 9–18. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C. Hydrogen Bonds as Design Elements in Organic Chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Huchting, J.; Winkler, M.; Nasser, H.; Meier, C. Synthesis of T-705-Ribonucleoside and T-705-Ribonucleotide and Studies of Chemical Stability. ChemMedChem 2017, 12, 652–659. [Google Scholar] [CrossRef]
- Adamiak, D.A.; Saenger, W. Precursors in the Biosynthesis of Purine Nucleotides. The Crystal Structures of 5-Amino-1-β-D-Riboluranosylimidazole-4-Carboxamide (AICAR) and Its 5’-(Dihydrogen Phosphate). Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 1979, 35, 924–928. [Google Scholar] [CrossRef]
- Subramanian, E. Inosine. Cryst. Struct. Commun. 1979, 8, 777. [Google Scholar]
- Munns, A.R.I.; Tollin, P. The Crystal and Molecular Structure of Inosine. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 1101–1113. [Google Scholar] [CrossRef]
- Sugawara, Y.; Iimura, Y.; Iwasaki, H.; Urabe, H.; Saito, H. Reversible Crystal Transition of Guanosine between the Dihydrate and Anhydrous States Coupled with Adsorption-Desorption Process. J. Biomol. Struct. Dyn. 1994, 11, 721–729. [Google Scholar] [CrossRef]
- Xie, Q.-R. 1H-1,2,4-Triazole-3-Carboxamide. Acta Crystallogr. E Struct. Rep. Online 2008, 64, o1110. [Google Scholar] [CrossRef] [PubMed]
- Goloveshkin, A.S.; Korlyukov, A.A.; Vologzhanina, A.V. Novel Polymorph of Favipiravir—An Antiviral Medication. Pharmaceutics 2021, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Li, Z.; Kong, L.; Xie, Y.; Zhang, T.; Xu, W. Synthesis and Crystal Structure of 6-Fluoro-3-Hydroxypyrazine-2-Carboxamide. Drug Discov. Ther. 2014, 8, 117–120. [Google Scholar] [CrossRef]
- Ghosh, P.; Panda, S.J.; Purohit, C.S. A new short synthesis route for favipiravir and its analogue: Their tautomerization behaviour. New J. Chem. 2022, 46, 18824–18831. [Google Scholar] [CrossRef]
- Tiyasakulchai, T.; Charoensetakul, N.; Khamkhenshorngphanuch, T.; Thongpanchang, C.; Srikun, O.; Yuthavong, Y.; Srimongkolpithak, N. Scalable Synthesis of Favipiravir via Conventional and Continuous Flow Chemistry. RSC Adv. 2021, 11, 38691–38693. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, P.; Koritsanszky, T.; Luger, P. Experimental Electron Density Study of 1,2,4-Triazole at 15 K. A Comparison with Ab Initio-Calculations. Z. Für Krist. Cryst. Mater. 1997, 212, 213–220. [Google Scholar] [CrossRef]
- Goldstein, P.; Ladell, J.; Abowitz, G. Refinement of the Crystal and Molecular Structure of 1,2,4-Triazole (C2H3N3) at Low Temperature. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 1969, 25, 135–143. [Google Scholar] [CrossRef]
- Deuschl, H. Die Röntgenstrukturanalyse von 1,2,4-Triazol. Ber. Bunsenges Phys. Chem. 1965, 69, 550–557. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Ruble, J.R.; Yates, J.H. Neutron Diffraction at 15 and 120 K and Ab Initio Molecular-Orbital Studies of the Molecular Structure of 1,2,4-Triazole. Acta Crystallogr. B Struct. Sci. 1983, 39, 388–394. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.F.; Perry, J.K.; Olinares, P.D.B.; Lee, H.W.; Chen, J.; Appleby, T.C.; Feng, J.Y.; Bilello, J.P.; Ng, H.; Sotiris, J.; et al. Structural Basis for Substrate Selection by the SARS-CoV-2 Replicase. Nature 2023, 614, 781–787. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428.e13. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Z.; Yu, Z. Structure of COVID-19 RNA-Dependent RNA Polymerase Bound to Favipiravir; Worldwide Protein Data Bank Foundation: Piscataway, NJ, USA, 2021. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Z.; Yu, Z. Structure of COVID-19 RNA-Dependent RNA Polymerase Bound to Ribavirin; Worldwide Protein Data Bank Foundation: Piscataway, NJ, USA, 2021. [Google Scholar] [CrossRef]
- Wang, S.; Xie, J.; Pei, J.; Lai, L. CavityPlus 2022 Update: An Integrated Platform for Comprehensive Protein Cavity Detection and Property Analyses with User-Friendly Tools and Cavity Databases. J. Mol. Biol. 2023, 435, 168141. [Google Scholar] [CrossRef]
- Miles, D.W.; Robins, R.K. Circular Dichroism Studies on Azole Nucleosides. The Solution Conformation of 5-Amino-7-.Beta.-D-Ribofuranosylimidazole-4-Carboxamide, Bredinin, Pyrazofurin, Ribavirin, and Related Nucleosides. J. Phys. Chem. 1983, 87, 2444–2450. [Google Scholar] [CrossRef]
- Cao, L.; Wang, X. Structure of Tomato Spotted Wilt Virus L Protein Binding to Ribavirin; Worldwide Protein Data Bank Foundation: Piscataway, NJ, USA, 2024. [Google Scholar] [CrossRef]
- Alam, I.; Lee, J.-H.; Cho, K.J.; Han, K.R.; Yang, J.M.; Chung, M.S.; Kim, K.H. Crystal Structures of Murine Norovirus-1 RNA-Dependent RNA Polymerase in Complex with 2-Thiouridine or Ribavirin. Virology 2012, 426, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Prosise, G.L.; Wu, J.Z.; Luecke, H. Crystal Structure of Tritrichomonas Foetus Inosine Monophosphate Dehydrogenase in Complex with the Inhibitor Ribavirin Monophosphate Reveals a Catalysis-Dependent Ion-Binding Site. J. Biol. Chem. 2002, 277, 50654–50659. [Google Scholar] [CrossRef] [PubMed]
- Risal, D.; Strickler, M.D.; Goldstein, B.M. Crystal Structure of Human Inosine Monophosphate Dehydrogenase Type II Complexed with the MPA/NAD Analog C2-MAD; Worldwide Protein Data Bank Foundation: Piscataway, NJ, USA, 2004. [Google Scholar] [CrossRef]
- Kusakabe, Y.; Ishihara, M.; Tanaka, N. Crystal Structure of Mouse SAHH Complexed with Ribavirin; Worldwide Protein Data Bank Foundation: Piscataway, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Benarroch, D.; Egloff, M.-P.; Mulard, L.; Guerreiro, C.; Romette, J.-L.; Canard, B. A Structural Basis for the Inhibition of the NS5 Dengue Virus mRNA 2′-O-Methyltransferase Domain by Ribavirin 5′-Triphosphate. J. Biol. Chem. 2004, 279, 35638–35643. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Orta, C.; Arias, A.; Pérez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. Sequential Structures Provide Insights into the Fidelity of RNA Replication. Proc. Natl. Acad. Sci. USA 2007, 104, 9463–9468. [Google Scholar] [CrossRef] [PubMed]
- Waller, I. Zur Frage der Einwirkung der Wärmebewegung auf die Interferenz von Röntgenstrahlen. Z. Phys. 1923, 17, 398–408. [Google Scholar] [CrossRef]
- Debye, P. Interferenz von Röntgenstrahlen Und Wärmebewegung. Annal. Phys. 1913, 348, 49–92. [Google Scholar] [CrossRef]
- Latosińska, J.N.; Latosińska, M.; Seliger, J.; Žagar, V. Exploring Partial Structural Disorder in Anhydrous Paraxanthine through Combined Experiment, Solid-State Computational Modelling, and Molecular Docking. Processes 2023, 11, 2740. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Account. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Kristallogr. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Packwood, D.; Kermode, J.; Mones, L.; Bernstein, N.; Woolley, J.; Gould, N.; Ortner, C.; Csányi, G. A Universal Preconditioner for Simulating Condensed Phase Materials. J. Chem. Phys. 2016, 144, 164109. [Google Scholar] [CrossRef]
- Pyykkö, P. Year-2008 Nuclear Quadrupole Moments. Mol. Phys. 2008, 106, 1965–1974. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Jelsch, C.; Ejsmont, K.; Huder, L. The Enrichment Ratio of Atomic Contacts in Crystals, an Indicator Derived from the Hirshfeld Surface Analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, C. GEMDOCK: A Generic Evolutionary Method for Molecular Docking. Proteins 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Gehlhaar, D.K.; Bouzida, D.; Rejto, P.A. Fully Automated and Rapid Flexible Docking of Inhibitors Covalently Bound to Serine Proteases. In Evolutionary Programming VII; Porto, V.W., Saravanan, N., Waagen, D., Eiben, A.E., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 1998; Volume 1447, pp. 449–461. ISBN 978-3-540-64891-8. [Google Scholar]
- Vangone, A.; Schaarschmidt, J.; Koukos, P.; Geng, C.; Citro, N.; Trellet, M.E.; Xue, L.C.; Bonvin, A.M.J.J. Large-Scale Prediction of Binding Affinity in Protein–Small Ligand Complexes: The PRODIGY-LIG Web Server. Bioinformatics 2019, 35, 1585–1587. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Latosińska, M.; Latosińska, J.N. The Chameleon Strategy—A Recipe for Effective Ligand Screening for Viral Targets Based on Four Novel Structure–Binding Strength Indices. Viruses 2024, 16, 1073. [Google Scholar] [CrossRef] [PubMed]
- Latosińska, M.; Latosińska, J.N. Serine/Threonine Protein Kinases as Attractive Targets for Anti-Cancer Drugs—An Innovative Approach to Ligand Tuning Using Combined Quantum Chemical Calculations, Molecular Docking, Molecular Dynamic Simulations, and Network-like Similarity Graphs. Molecules 2024, 29, 3199. [Google Scholar] [CrossRef]
- Kurkcuoglu, Z.; Koukos, P.I.; Citro, N.; Trellet, M.E.; Rodrigues, J.P.G.L.M.; Moreira, I.S.; Roel-Touris, J.; Melquiond, A.S.J.; Geng, C.; Schaarschmidt, J.; et al. Performance of HADDOCK and a Simple Contact-Based Protein–Ligand Binding Affinity Predictor in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018, 32, 175–185. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
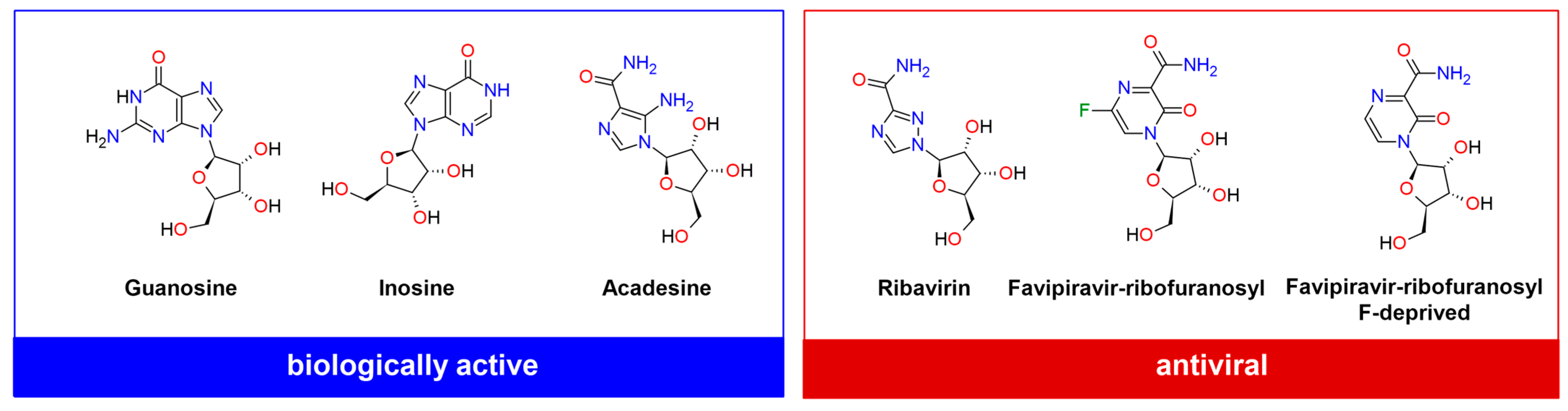
| Compound | CAS | Biological Activity Profile | Similarity | ||
|---|---|---|---|---|---|
| Tanimoto * | Atom Pair Tanimoto ** | Maximum Common Substructure *** | |||
| Ribavirin, 1-(β-D-Ribofuranosyl)-1H-1,2,4-triazole-3-carboxamide, RBV | 36791-04-5 | broad spectrum anti-viral, potential anti-cancer | 1.0 | 1.0 | 1.0 |
| Guanosine, 9-(β-D-Ribofuranosyl)guaninę, G | 118-00-3 | required for an RNA splicing reaction in mRNA; neuroprotective agent reducing neuroinflammation, oxidative stress, and excitotoxicity | 0.4476 | 0.598 | 0.5417 |
| Inosine, 9-(β-D-Ribofuranosyl) hypoxanthine, I | 58-63-9 | regulation of RNA editing, metabolic enzyme activity, and signaling pathways; promotion of T cell proliferation and differentiation and enhancement of the response to immune-checkpoint inhibitors therapy; anti-inflammatory, antinociceptive, immunomodulatory, and neuroprotective | 0.4526 | 0.560 | 0.5652 |
| Acadesine, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside, AICA-riboside/AICAR | 2627-69-2 | hypoglycemic, cardioprotective, and anti-neoplastic agent; regulates lipid and glucose metabolism, pro-inflammatory responses, cytokine production, cell proliferation, and apoptosis; improve ischemia or reperfusion injury and kidney fibrosis in rats; protection against acute tubular necrosis; potential anti-cancer | 0.4675 | 0.731 | 0.8421 |
| Favipiravir -ribofuranosyl, 6-fluoro-3-hydroxy-2-pyrazinecarboxamide-ribofuranosyl, Favipiravir -Ry, FVP-R | 740790-94-7 | broad spectrum anti-viral, potential anti-cancer | 0.4342 | 0.545 | 0.4231 |
| Favipiravir -ribofuranosyl (F-deprived), 4-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-3-oxopyrazine-2-carboxamide | 356782-84-8 | yellow fever virus anti-viral | 0.4298 | 0.615 | 0.4400 |
| Nitrogen Site | ν+ (MHz) | ν− (MHz) | ν0 (MHz) | |e2qQ/h| (MHz) | η | Assignment |
|---|---|---|---|---|---|---|
| N(1) | 2.340 | 1.700 | 0.640 | 2.693 | 0.475 | –NH2 |
| N(2) | 3.980 | 2.650 | 1.330 | 4.420 | 0.602 | –N= ring position N(2) |
| N(3) | 2.600 | 2.280 | 0.320 | 3.253 | 0.197 | –N= ring position N(4) |
| N(4) | 2.400 | 1.480 | 0.920 | 2.587 | 0.711 | >N-sugar |
| Nitrogen Site | ν+ (MHz) | ν− (MHz) | ν0 (MHz) | |e2qQ/h| (MHz) | η | Assignment |
|---|---|---|---|---|---|---|
| 1H-1,2,4-triazole | 3.793 (3.877) | 2.405 (2.470) | 1.384 (1.407) | 4.132 (4.231) | 0.672 (0.665) | –N= ring position N(2) |
| 2.503 (2.551) | 2.257 (2.279) | 0.246 (0.272) | 3.173 (3.220) | 0.155 (0.169) | –N= ring position N(4) | |
| 2.114 | 1.442 | 0.672 | 2.371 | 0.567 | >NH |
| Polymorph | Structure Geometry | ν+ (MHz) | ν− (MHz) | ν0 (MHz) | |e2qQ/h| (MHz) | η | Assignment |
|---|---|---|---|---|---|---|---|
| RBV form II | hydrogen positions optimized | 2.503 | 1.755 | 0.727 | 2.629 | 0.750 | –NH2 |
| 4.107 | 2.663 | 1.444 | 4.513 | 0.640 | –N= ring position N(2) | ||
| 2.655 | 2.619 | 0.035 | 3.516 | 0.020 | –N= ring position N(4) | ||
| 2.465 | 1.479 | 0.986 | 2.852 | 0.511 | >N-sugar | ||
| RBV form II | hydrogen positions optimized | 2.491 | 2.441 | 0.049 | 3.288 | 0.030 | -NH2 |
| 3.878 | 2.043 | 1.835 | 3.947 | 0.930 | –N= ring position N(2) | ||
| 2.686 | 2.096 | 0.590 | 3.188 | 0.370 | –N= ring position N(4) | ||
| 2.397 | 1.338 | 1.058 | 2.490 | 0.851 | >N-sugar | ||
| RBV form I | full geometry optimization | 2.490 | 1.828 | 0.662 | 2.879 | 0.460 | –NH2 |
| 4.004 | 2.578 | 1.426 | 4.388 | 0.650 | –N= ring position N(2) | ||
| 2.629 | 2.612 | 0.017 | 3.494 | 0.010 | –N= ring position N(4) | ||
| 2.401 | 1.514 | 0.887 | 2.610 | 0.680 | >N-sugar | ||
| RBV form I | full geometry optimization | 2.593 | 2.165 | 0.428 | 3.172 | 0.270 | –NH2 |
| 3.694 | 1.931 | 1.763 | 3.750 | 0.940 | –N= ring position N(2) | ||
| 2.579 | 2.512 | 0.068 | 3.394 | 0.040 | –N= ring position N(4) | ||
| 2.402 | 1.462 | 0.940 | 2.576 | 0.730 | >N-sugar |
| Structure | <C(5)N(1)CO |
|---|---|
| Ribavirin, form I | 10.41 |
| Ribavirin, form II | 119.00 |
| Favipiravir -ribofuranosyl, Favipiravir—ribofuranosyl, 3,4-dihydro-6-fluoro-3-oxo-4-β-D-ribofuranosyl-2-pyrazine carboxamide, T-705 | 15.40 |
| 3,4-dihydro-3-oxo-4-β-D-ribofuranosyl-2-pyrazine carboxamide, T-1106 | 7.62 |
| Acadesine | 62.77 |
| Inosine, α-form | 12.44 |
| Inosine, β-form | 139.87 |
| Guanosine.H2O | 122.49 |
| Structure | ED | RMSD |
|---|---|---|
| Ribavirin, form I, 295 K * | 18.7 | 4.83 |
| 1H-1,2,4-triazole, 15 K ** | 58.2 | 15.03 |
| 1H-1,2,4-triazole, 113 K | 59.4 | 15.33 |
| 1H-1,2,4-triazole, 120 K | 58.0 | 14.99 |
| 1H-1,2,4-triazole, 295 K | 60.1 | 15.51 |
| 1H-1,2,4-triazole-3-carboxamide, 298 K | 29.9 | 7.73 |
| Favipiravir-ribofuranosyl, 3,4-dihydro-6-fluoro-3-oxo-4-β-D-ribofuranosyl-2-pyrazine carboxamide, T-705, 100 K | 17.6 | 4.55 |
| 3,4-dihydro-3-oxo-4-β-D-ribofuranosyl-2-pyrazine carboxamide, T-1106, 100 K | 13.2 | 3.41 |
| Favipiravir, 100 K **, form I | 37.8 | 9.75 |
| Favipiravir, 293 K, form I | 36.9 | 9.52 |
| Favipiravir, 296 K, form I | 36.9 | 9.52 |
| Favipiravir, 298 K, form II | 34.2 | 8.83 |
| Acadesine | 55.4 | 14.31 |
| Inosine, α-form | 49.9 | 12.89 |
| Inosine, β-form | 51.3 | 13.26 |
| Structure | Atom | C | H | N | O | F |
|---|---|---|---|---|---|---|
| Ribavirin, form I | Surface % | 5.15 | 65.3 | 11.00 | 18.55 | - |
| C | 2.16 | 0.00 | - | - | - | |
| H | 0.92 | 0.76 | 0.90 | - | - | |
| N | 2.84 | 0.00 | 1.38 | 0.17 | - | |
| O | 0.05 | 2.72 | 1.18 | 0.44 | 0.20 | |
| Ribavirin, form II (stable) | Surface % | 4.30 | 62.25 | 11.45 | 22.00 | - |
| C | 2.16 | - | - | - | - | |
| H | 0.92 | 0.82 | - | - | - | |
| N | 2.84 | 0.90 | 2.52 | - | - | |
| O | 0.05 | 1.58 | 0.12 | 0.00 | - | |
| T-705 | Surface % | 7.5 | 57.3 | 6.6 | 20.25 | 8.45 |
| C | 0.00 | - | - | - | - | |
| H | 0.90 | 0.85 | - | - | - | |
| N | 0.00 | 1.36 | 0.00 | - | - | |
| O | 1.15 | 1.35 | 0.67 | 0.17 | - | |
| F | 3.00 | 0.97 | 0.99 | 0.70 | 0.03 | |
| T-1106 | Surface % | 7.95 | 61.6 | 6.4 | 24.05 | - |
| C | 0.00 | - | - | - | - | |
| H | 0.81 | 0.92 | - | - | - | |
| N | 0.00 | 1.23 | 0.00 | - | - | |
| O | 2.09 | 1.20 | 1.01 | 0.12 | - | |
| Favipiravir, form I | Surface % | 13.45 | 39.2 | 16.45 | 17.05 | 14.05 |
| C | 0.94 | - | - | - | - | |
| H | 0.77 | 0.89 | - | - | - | |
| N | 1.36 | 1.01 | 1.52 | - | - | |
| O | 1.61 | 1.41 | 0.77 | 0.10 | - | |
| F | 0.53 | 1.02 | 0.30 | 0.61 | 1.11 | |
| Favipiravir, form II | Surface % | 13.05 | 39.7 | 15.9 | 17.2 | 14.25 |
| C | 0.00 | - | - | - | - | |
| H | 0.92 | 0.81 | - | - | - | |
| N | 1.95 | 0.90 | 1.50 | - | - | |
| O | 1.56 | 1.34 | 0.84 | 0.00 | - | |
| F | 0.40 | 1.32 | 0.04 | 0.92 | 0.75 | |
| Surface % | 6.1 | 70.0 | 7.35 | 16.65 | - | |
| Acadesine | C | 0.00 | - | - | - | - |
| H | 1.28 | 0.86 | - | - | - | |
| N | 0.56 | 1.30 | 0.56 | - | - | |
| O | 0.39 | 1.37 | 0.08 | 0.07 | - | |
| Surface % | 7.35 | 60.6 | 11.75 | 20.2 | - | |
| C | 1.11 | - | - | - | - | |
| Inosine, α-form | H | 0.85 | 0.84 | - | - | - |
| N | 1.10 | 1.28 | 0.43 | - | - | |
| O | 1.35 | 1.36 | 0.46 | 0.10 | - | |
| Inosine, β-form (stable) | Surface % | 8.45 | 63.45 | 9.7 | 18.4 | - |
| C | 3.78 | - | - | - | - | |
| H | 0.40 | 1.04 | - | - | - | |
| N | 1.89 | 0.80 | 1.91 | - | - | |
| O | 1.32 | 1.24 | 0.81 | 0.12 | - |
| Structure | ED | |
|---|---|---|
| RBV Form II | RBV Form I | |
| RBV | 5.09 | 5.09 |
| T-705 | 5.68 | 3.74 |
| T-1106 | 4.93 | 0.88 |
| Favipiravir, form I | 3.26 | 3.02 |
| Favipiravir, form II | 3.56 | 3.27 |
| Acadesine | 3.76 | 2.52 |
| Inosine, α-form | 3.24 | 2.11 |
| Inosine, β-form | 2.54 | 4.85 |
| N···C | N···H | N···N | N···O | N···F | RMSD_N | |
|---|---|---|---|---|---|---|
| Ribavirin, form I | 38.1 | 60.3 | 0 | 1.6 | - | 4.86 |
| Ribavirin, form II | 43.6 | 51.5 | 3.2 | 1.8 | - | 0 * |
| T-705 | 45.3 | 51.4 | 0 | 1.7 | 1.6 | 1.77 |
| T-1106 | 45.3 | 52.7 | 0 | 2.0 | - | 1.71 |
| Favipiravir, form I | 43.2 | 48.2 | 3.1 | 3.0 | 2.4 | 1.91 |
| Favipiravir, form II | 44.5 | 47.1 | 3.1 | 3.2 | 2.1 | 2.30 |
| 1H-1,2,4-triazole | 39.5 | 39.2 | 21.3 | - | - | 9.96 |
| 1H-1,2,4-triazole-3-carboxamide | 33.2 | 63.7 | 3.2 | - | - | 7.17 |
| Inosine, α-form | 56.6 | 42.7 | 0.3 | 0.4 | - | 7.17 |
| Inosine, β-form | 59.4 | 33.3 | 0.5 | 6.8 | - | 11.07 |
| Acadesine | 39.4 | 59.7 | 0.2 | 0.7 | - | 4.36 |
| Guanosine.H2O | 50.8 | 48.2 | 0.7 | 0.3 | - | 3.77 |
| Structure | Eelectrostatic | Epolarization | Edispersion | Erepulsion | Etotal |
|---|---|---|---|---|---|
| Ribavirin, form I | −205.58 | −46.75 | −138.88 | 140.47 | −250.79 |
| Ribavirin, form II | −253.46 | −50.71 | −125.55 | 168.60 | −261.28 |
| T-705 | −196.01 | −42.99 | −117.32 | 140.83 | −215.56 |
| T-1106 | −166.51 | −40.04 | −133.96 | 129.24 | −211.35 |
| Favipiravir, form I | −79.49 | −14.81 | −107.71 | 62.57 | −139.47 |
| Favipiravir, form II | −61.76 | −11.85 | −67.63 | 48.19 | −93.08 |
| 1H-1,2,4-triazole | −93.36 | −17.12 | −51.01 | 63.23 | −98.28 |
| 1H-1,2,4-triazole-3-carboxamide | −164.93 | −37.25 | −87.89 | 106.25 | −183.91 |
| Inosine, β-form | −204.66 | −41.24 | −132.37 | 153.85 | −224.51 |
| Acadesine | −190.61 | −40.59 | −129.09 | 135.91 | −224.45 |
| Polymorph | Moiety | Type of Interactions | Hydrogen Bond | dDA [Å] | <DAH |
|---|---|---|---|---|---|
| RBV, Form I | aglycone | intramolecular | NH···N(2) | 2.793 | 81.40 |
| intermolecular | NH···O | 3.251 | 143.92 | ||
| NH···O | 2.976 | 156.63 | |||
| O···HO | 2.685 | 154.94 | |||
| O···HC | 3.464 | 162.63 | |||
| N(4) ···HO | 2.860 | 153.48 | |||
| N(2) ···HO | 2.899 | 138.27 | |||
| RBV, Form II | aglycone | intramolecular | NH···N(2) | 2.806 | 94.46 |
| intermolecular | NH···N(4) | 2.940 | 167.00 | ||
| NH…O | 3.242 | 110.98 | |||
| NH···HC | 4.152 | 153.72 | |||
| O···HO | 2.689 | 156.39 | |||
| C(5) ···HO | 3.283 | 176.19 | |||
| glycone | C···HO | 3.456 | 147.84 | ||
| O···HO | 2.684 | 159.04 | |||
| O···HO | 2.745 | 158.23 |
| Ligand | Interaction Type | Residue | H-A | D-A | <DAH | Donor | Acceptor |
|---|---|---|---|---|---|---|---|
| RBV | Hydrogen bond | Arg555 | 3.02 | 3.40 | 104.28 | N | O |
| Asp623 | 2.58 | 3.06 | 110.83 | O | N3 | ||
| Asp623 | 3.39 | 3.97 | 121.64 | O | O | ||
| Ser682 | 3.24 | 4.07 | 142.89 | –NH from amine | O3 | ||
| Ser682 | 3.12 | 3.47 | 102.94 | –N(2)= | O2 | ||
| Metal complexation | Mg2+ | - | 2.72 | - | RBV-MP | P | |
| FVP | Hydrogen bond | Lys545 | 2.86 | 131.57 | 131.57 | –NH2 | N3 |
| Lys545 | 3.09 | 3.64 | 114.95 | –N(3) | N2 | ||
| Asp623 | 3.50 | 3.97 | 111.96 | –NH from amine | O3 | ||
| Thr680 | 3.11 | 3.87 | 135.50 | O3 | O3 | ||
| Metal complexation | Mg2+ | - | 2.84 | - | FVP-RMP | P |
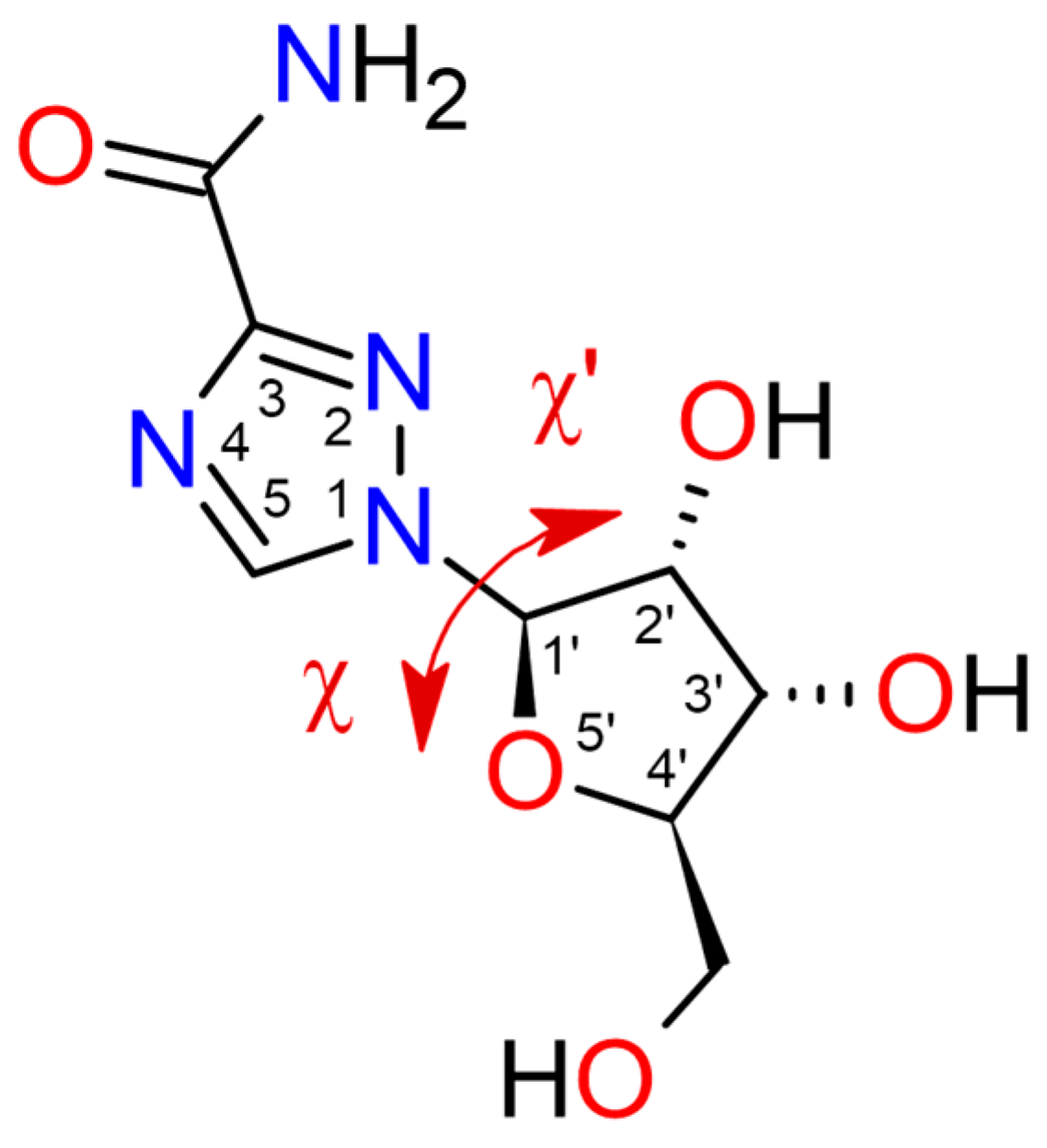
| Structure | Protein | Type | Ligand | χ [°] <C(5)NCO | χ’ [°] <N(2)NCO | θ [°] Ring Angle | Conformation | Helix Type | Binding Affinity [kJ/mol] |
|---|---|---|---|---|---|---|---|---|---|
| 7DFG [67] | SARS-CoV-2, RdRp | positive-single stranded RNA | FVP-RMP | 32.0 | −148.5 | 36.9 | anti | C3′-endo sugars (A-helix) | −71.98 * (−22.91 **) |
| 7DFH [68] | SARS-CoV-2, RdRp | positive-single stranded RNA | RBV-MP | 40.8 | −139.6 | 41.0 | anti | -50.34 (−19.11 **) | |
| 8KI6 [71] | Tomato spotted wilt virus L, RdRp | negative-stranded RNA | RBV | 75.5 | −104.5 | 78.9 | anti | −16.07 | |
| 3SFU [72] | Murine norovirus, RdRp | positive-single-stranded RNA | RBV | 97.0 | −84.6 | 70.5 | high-anti | −16.85 | |
| 1ME8 [73] | Inosine-5′-monophosphate dehydrogenase | enzyme, DNA/RNA synthesis | RBV-MP | 84.2 | −95.7 | 79.3 | anti | −26.07 | |
| 1NF7 [74] | Inosine-5′-monophosphate dehydrogenase | enzyme, DNA/RNA synthesis | RBV-MP | 81.0 | −99.2 | 78.1 | anti | −26.07 | |
| [41] | Solid | Form II | RBV | 119 | −62.2 | 78.2 | high-anti | - | |
| - | Solid, optimized *** | Form II | RBV | 122.5 | −61.7 | 78.0 | high-anti | - | |
| [41] | Solid | Form I | RBV | 10.4 | −170.6 | 77.7 | anti | - | |
| - | Solid optimized *** | Form I | RBV | 13.46 | −171.7 | 77.9 | anti | - | |
| [50] | Solid | - | FVP-R | 15.4 | −164.9 | 83.9 | anti | - | |
| 5AXD [75] | Mouse S-adenosyl homocysteine hydrolase, SAHH | DNA/RNA metabolism | RBV | 75.3 | −107.5 | 76.3 | anti | C3′-endo sugars (A-helix) | −24.83 |
| 1R6A [76] | Denge virus, 2′-o-methyltransferase | positive-single-stranded RNA | RBV-MP | 41.0 | −138.9 | 89.1 | anti | −22.62 | |
| 2E9R [77] | Foot-and-mouth disease (FMDV) virus, RdRp | positive-single-stranded RNA | RBV-TP | −11.9 | 167.5 | 71.0 | anti | −22.09 |
| Parameter | T-705 | T-1106 | RBV | Inosine | Acadesine | Guanosine |
|---|---|---|---|---|---|---|
| Total energy | −498.80 | −496.06 | −448.26 | −402.65 | −402.98 | −383.74 |
| Protein–ligand | −191.67 | −188.93 | −207.44 | −200.96 | −203.64 | −177.36 |
| Steric | −134.72 | −135.66 | −155.14 | −150.54 | −156.29 | −138.14 |
| Hydrogen bonds | −21.75 | −18.06 | −19.28 | −16.50 | −14.32 | −6.19 |
| VdW | −55.00 | −55.00 | −60.55 | −44.85 | −50.66 | −60.69 |
| Mg2+ Zn2+ | −40.47 | −40.47 | −42.95 | −42.95 | −42.95 | −42.95 |
| RNA template | −292.90 | −266.65 | −197.87 | −202.59 | −199.08 | −206.89 |
| Ligand | RMSD_BM (kJ/mol) | Manhattan Distance (kJ/mol) |
|---|---|---|
| T-705 | 5.742 | 276.32 |
| T-1106 | 5.747 | 278.89 |
| Inosine | 5.934 | 427.14 |
| Acadesine | 2.864 | 184.35 |
| Guanosine | 6.120 | 456.79 |
| Ligand | Binding Affinity (kJ/mol) | SBAI (kJ/mol) | ||
|---|---|---|---|---|
| Tanimoto Similarity | Atom Pair Tanimoto Similarity | Maximum Common Substructure Similarity | ||
| T-705 | −71.98 | 38.2 | 37.5 | 47.6 |
| T-1106 | −68.51 | 31.9 | 32.4 | 47.2 |
| RBV | −50.34 | 100.0 | 100.0 | 100.0 |
| Inosine | −31.43 | −34.5 | −43.5 | −43.0 |
| Acadesine | −29.84 | −38.5 | −129.8 | −76.2 |
| Guanosine | −27.13 | −42.0 | −50.6 | −57.7 |
| Form | Solid State | Single Molecule | Solid-State Effect | Assignment | |||
|---|---|---|---|---|---|---|---|
| |e2qQ/h| (MHz) | η | |e2qQ/h| (MHz) | η | Δs (MHz) | δs | ||
| RBV I | 3.288 | 0.030 | 3.946 | 0.110 | −0.658 | −0.08 | -NH2 |
| 3.947 | 0.930 | 4.142 | 0.680 | −0.195 | 0.25 | –N= ring position N(2) | |
| 3.188 | 0.370 | 3.926 | 0.090 | −0.738 | 0.28 | –N= ring position N(4) | |
| 2.490 | 0.851 | 2.649 | 0.730 | −0.159 | 0.121 | >N-sugar | |
| RBV II | 2.629 | 0.750 | 3.758 | 0.070 | −1.129 | 0.68 | –NH2 |
| 4.513 | 0.640 | 4.527 | 0.660 | −0.014 | −0.02 | –N= ring position N(2) | |
| 3.516 | 0.020 | 4.063 | 0.130 | −0.547 | −0.11 | –N= ring position N(4) | |
| 2.852 | 0.511 | 2.734 | 0.670 | 0.118 | −0.159 | >N-sugar | |
| FVP I | 2.336 | 0.81 | 2.968 | 0.51 | −0.632 | 0.3 | –NH2 |
| 4.56 | 0.24 | 4.829 | 0.32 | −0.269 | −0.08 | –N= ring position N(4) | |
| 4.739 | 0.35 | 4.564 | 0.33 | 0.175 | 0.02 | –N= ring position N(1) | |
| FVP II | 2.308 | 0.76 | 2.935 | 0.48 | −0.627 | 0.28 | –NH2 |
| 4.615 | 0.34 | 4.839 | 0.4 | −0.224 | −0.06 | –N= ring position N(4) | |
| 4.676 | 0.38 | 4.554 | 0.39 | 0.122 | −0.01 | –N= ring position N(1) | |
| Protein-Ligand Complex | Single Molecule | Complexation Effect | Assignment | ||||
|---|---|---|---|---|---|---|---|
| |e2qQ/h| (MHz) | η | |e2qQ/h| (MHz) | η | Δc (MHz) | δc | ||
| RBV | 2.742 | 0.610 | 4.212 | 0.230 | −1.47 | 0.38 | –NH2 |
| 4.810 | 0.630 | 4.662 | 0.650 | 0.148 | −0.02 | –N= ring position N(2) | |
| 4.519 | 0.090 | 4.525 | 0.070 | −0.006 | 0.02 | –N= ring position N(4) | |
| 2.842 | 0.511 | 2.850 | 0.640 | −0.008 | −0.129 | >N-sugar | |
| FVP-R | 2.445 | 0.300 | 3.810 | 0.120 | −1.365 | 0.18 | –NH2 |
| 4.637 | 0.420 | 4.784 | 0.400 | −0.147 | 0.02 | –N= ring position N(4) | |
| 1.989 | 0.180 | 2.283 | 0.400 | −0.294 | −0.22 | –N= ring position N(1) | |
| Form II | Form I | Assignment | |||
|---|---|---|---|---|---|
| ΔCS (MHz) | δcs | ΔCS (MHz) | δCS | ||
| RBV | 0.113 | −0.14 | −0.546 | 0.58 | –NH2 |
| 0.297 | −0.01 | 0.863 | −0.3 | –N= ring position N(2) | |
| 1.003 | 0.07 | 1.331 | −0.28 | –N= ring position N(4) | |
| −0.01 | 0 | 0.352 | −0.34 | >N-sugar | |
| FVP/FVP-R | −0.109 | 0.51 | −0.137 | 0.46 | –NH2 |
| −0.077 | −0.18 | −0.022 | −0.08 | –N= ring position N(4) | |
| 2.75 | 0.17 | 2.687 | 0.2 | –N= ring position N(1)/>N-sugar | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latosińska, J.N.; Latosińska, M.; Seliger, J.; Žagar, V.; Apih, T. Anti-Butterfly Effect in Ribavirin Studied by Combined Experiment (PXRD/1H-14N NQR Cross-Relaxation Spectroscopy), Quantum Chemical Calculations, Molecular Docking, Molecular Dynamics Simulations, and Novel Structure-Binding Strength and Quadrupolar Indices. Molecules 2025, 30, 1096. https://doi.org/10.3390/molecules30051096
Latosińska JN, Latosińska M, Seliger J, Žagar V, Apih T. Anti-Butterfly Effect in Ribavirin Studied by Combined Experiment (PXRD/1H-14N NQR Cross-Relaxation Spectroscopy), Quantum Chemical Calculations, Molecular Docking, Molecular Dynamics Simulations, and Novel Structure-Binding Strength and Quadrupolar Indices. Molecules. 2025; 30(5):1096. https://doi.org/10.3390/molecules30051096
Chicago/Turabian StyleLatosińska, Jolanta Natalia, Magdalena Latosińska, Janez Seliger, Veselko Žagar, and Tomaž Apih. 2025. "Anti-Butterfly Effect in Ribavirin Studied by Combined Experiment (PXRD/1H-14N NQR Cross-Relaxation Spectroscopy), Quantum Chemical Calculations, Molecular Docking, Molecular Dynamics Simulations, and Novel Structure-Binding Strength and Quadrupolar Indices" Molecules 30, no. 5: 1096. https://doi.org/10.3390/molecules30051096
APA StyleLatosińska, J. N., Latosińska, M., Seliger, J., Žagar, V., & Apih, T. (2025). Anti-Butterfly Effect in Ribavirin Studied by Combined Experiment (PXRD/1H-14N NQR Cross-Relaxation Spectroscopy), Quantum Chemical Calculations, Molecular Docking, Molecular Dynamics Simulations, and Novel Structure-Binding Strength and Quadrupolar Indices. Molecules, 30(5), 1096. https://doi.org/10.3390/molecules30051096