
Formation and Chemical Structure of Carbon-13 Tracer Lignin-Carbohydrate Complexes (LCCs) During Kraft Pulping



Abstract
1. Introduction
2. Results and Discussion
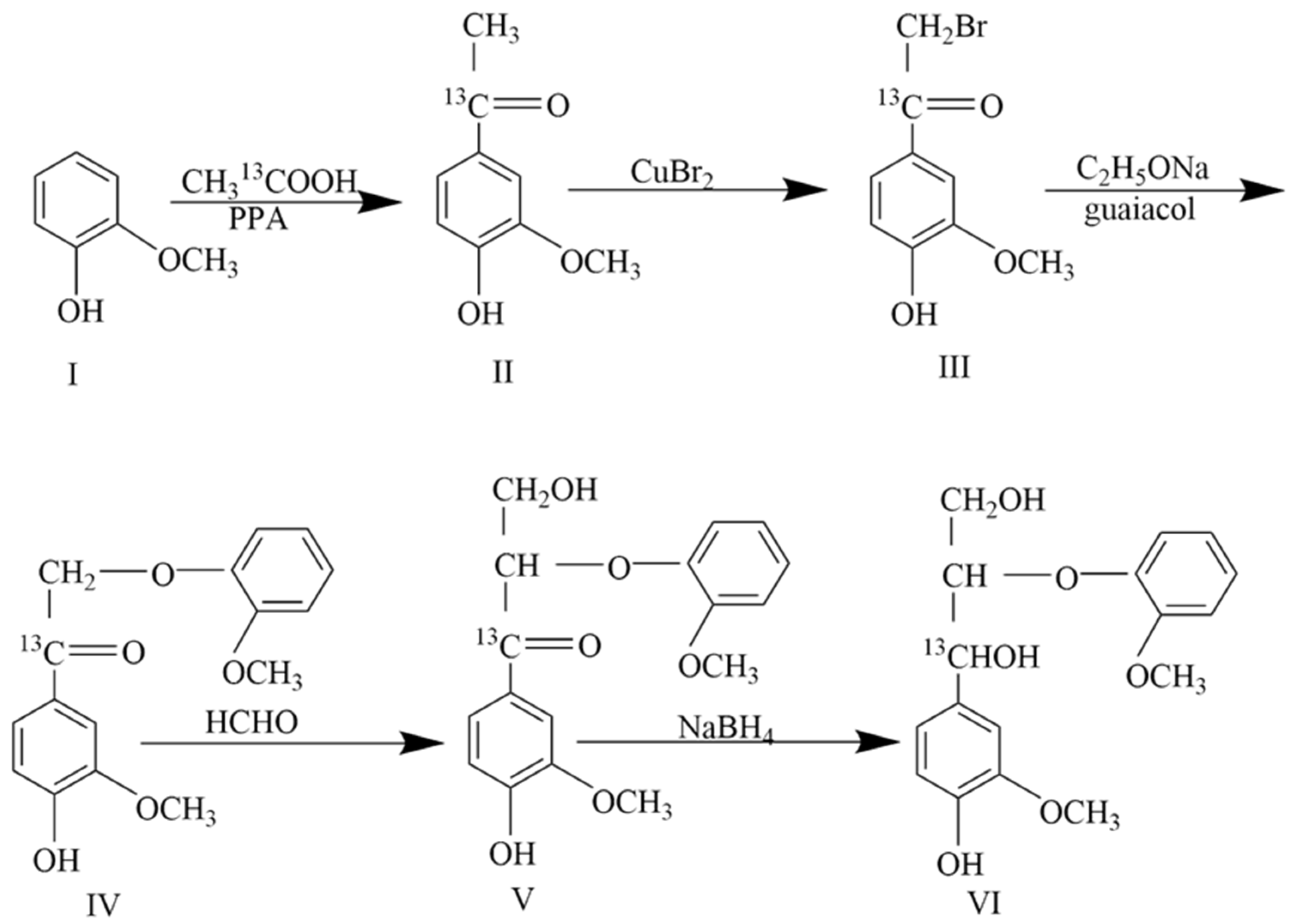
2.1. Modification of the Synthesis Method of Guaiacylglycerol-β-Guaiacyl Ether-[α-13C]
2.1.1. Improvement in Bromination Reaction Solvent
2.1.2. Optimization of Bromination Reaction Time
2.1.3. Purification of Bromination Product
2.1.4. Analysis of 4-(α-Bromoacetyl)-Guaiacol-[α-13C]
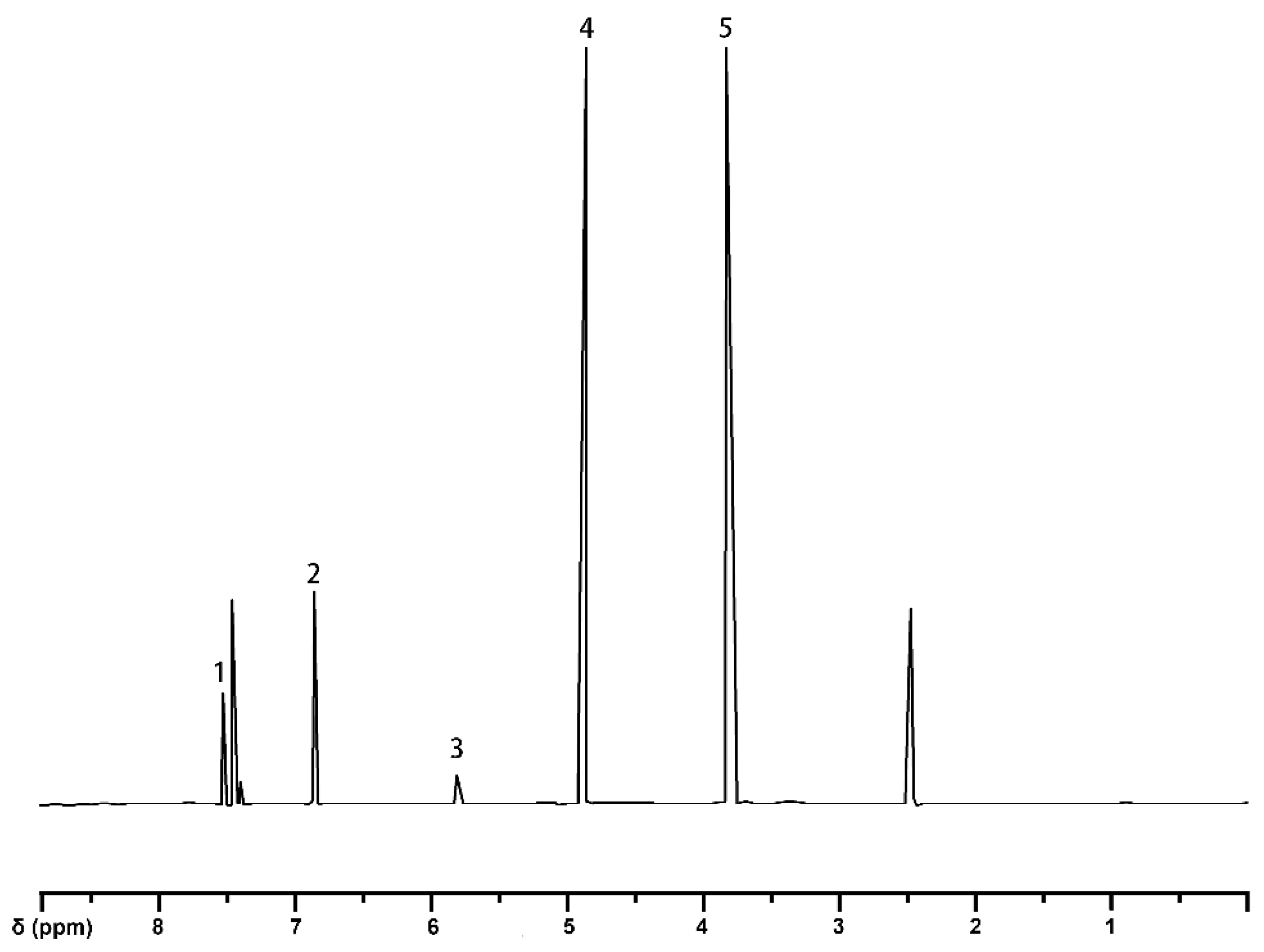
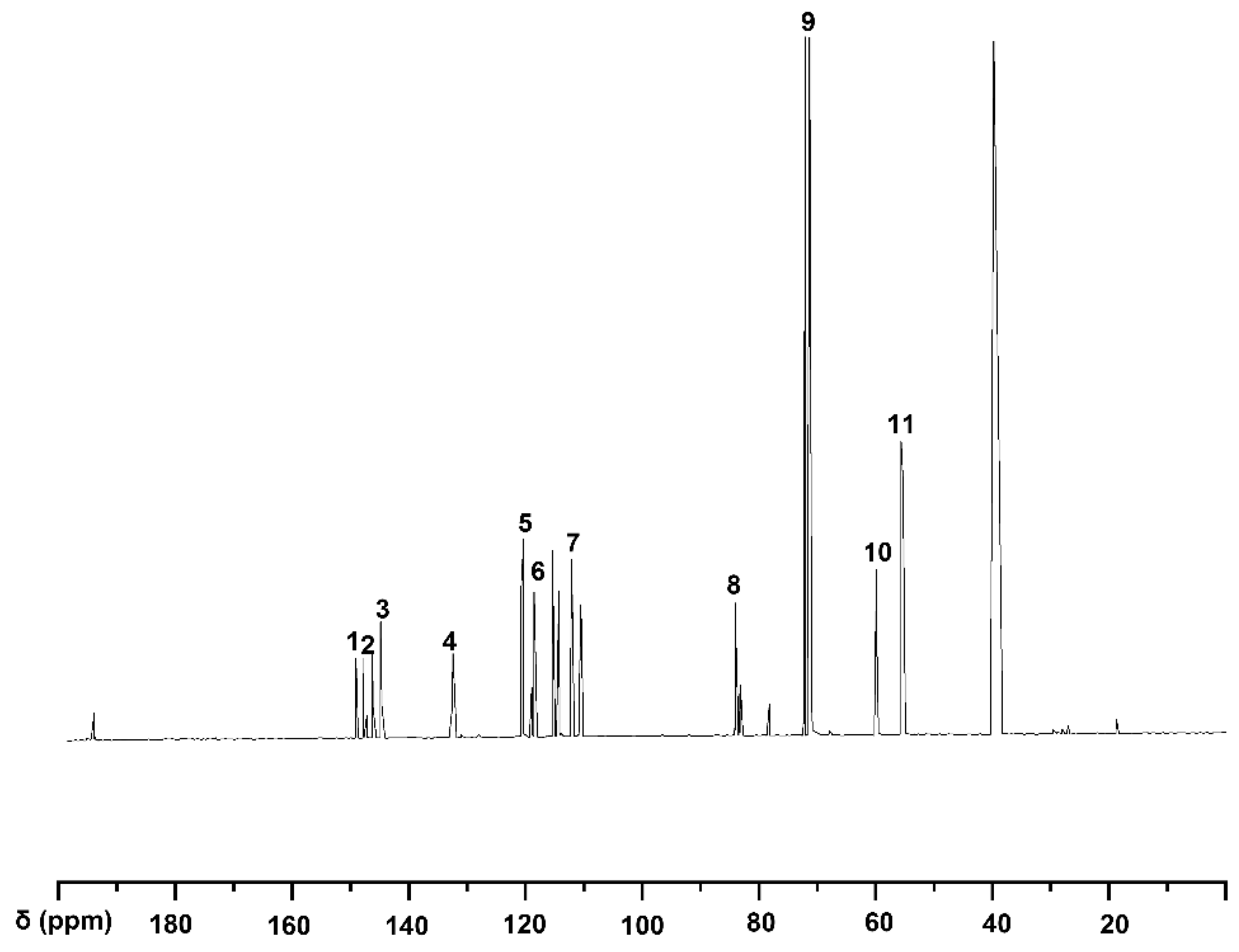
2.1.5. Analysis of Guaiacylglycerol-β-Guaiacyl Ether-[α-13C]
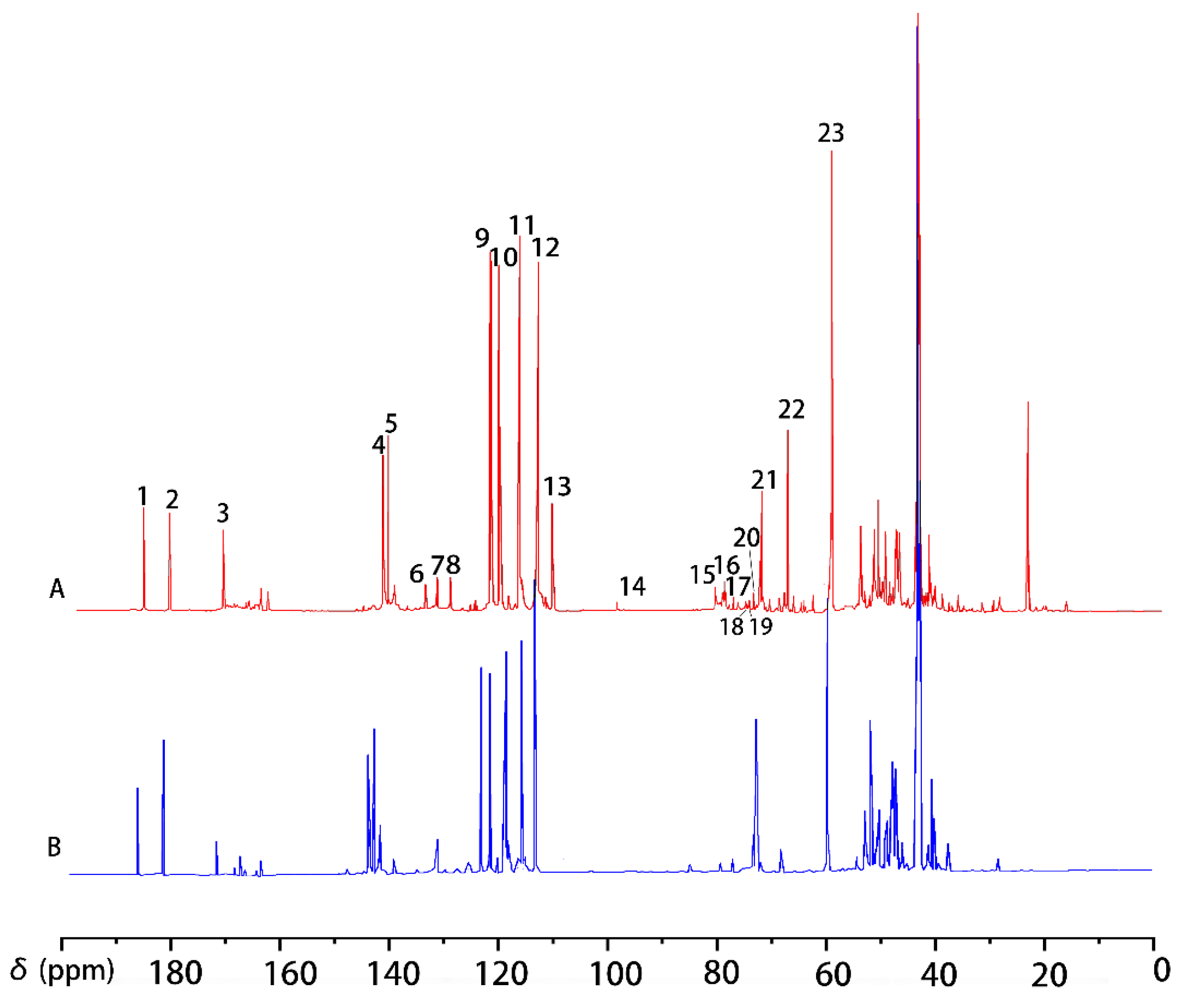
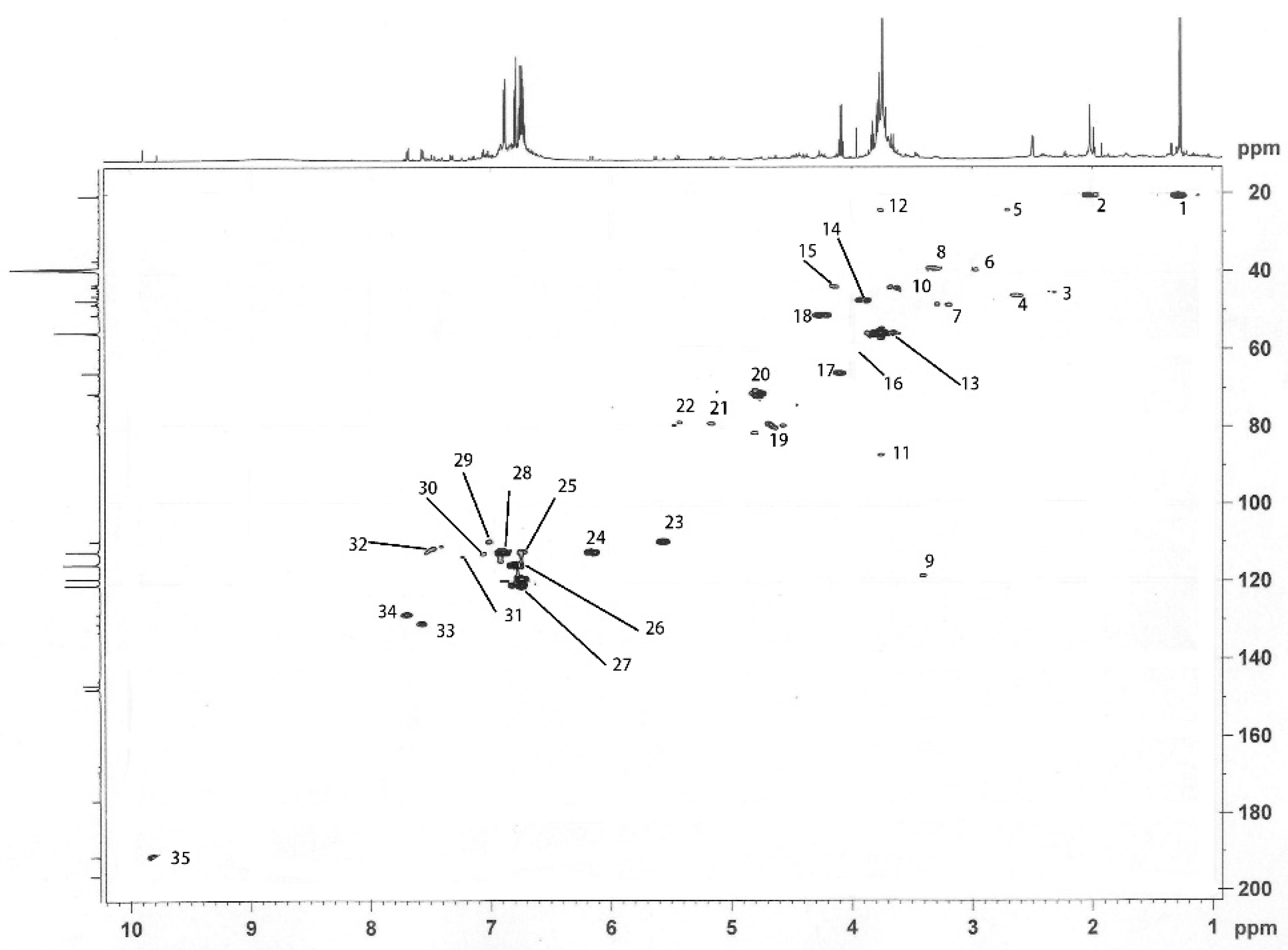
2.2. Co-Polymerized Products of Guaiacylglycerol-β-Guaiacyl Ether-[α-13C] with Xylose in Kraft Pulping Process
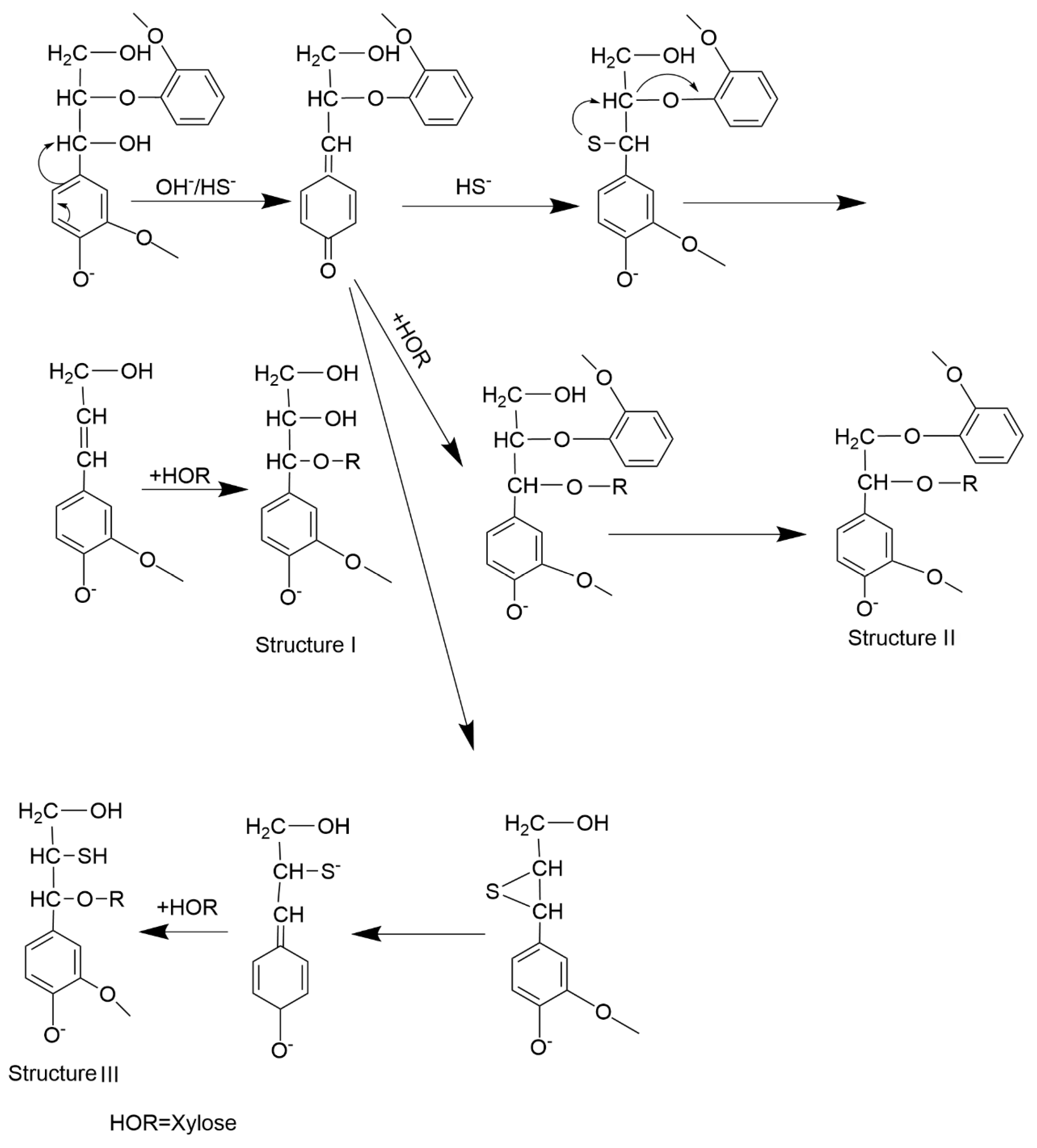
2.3. Formation Mechanism of LCC Structure During the Kraft Cooking Process
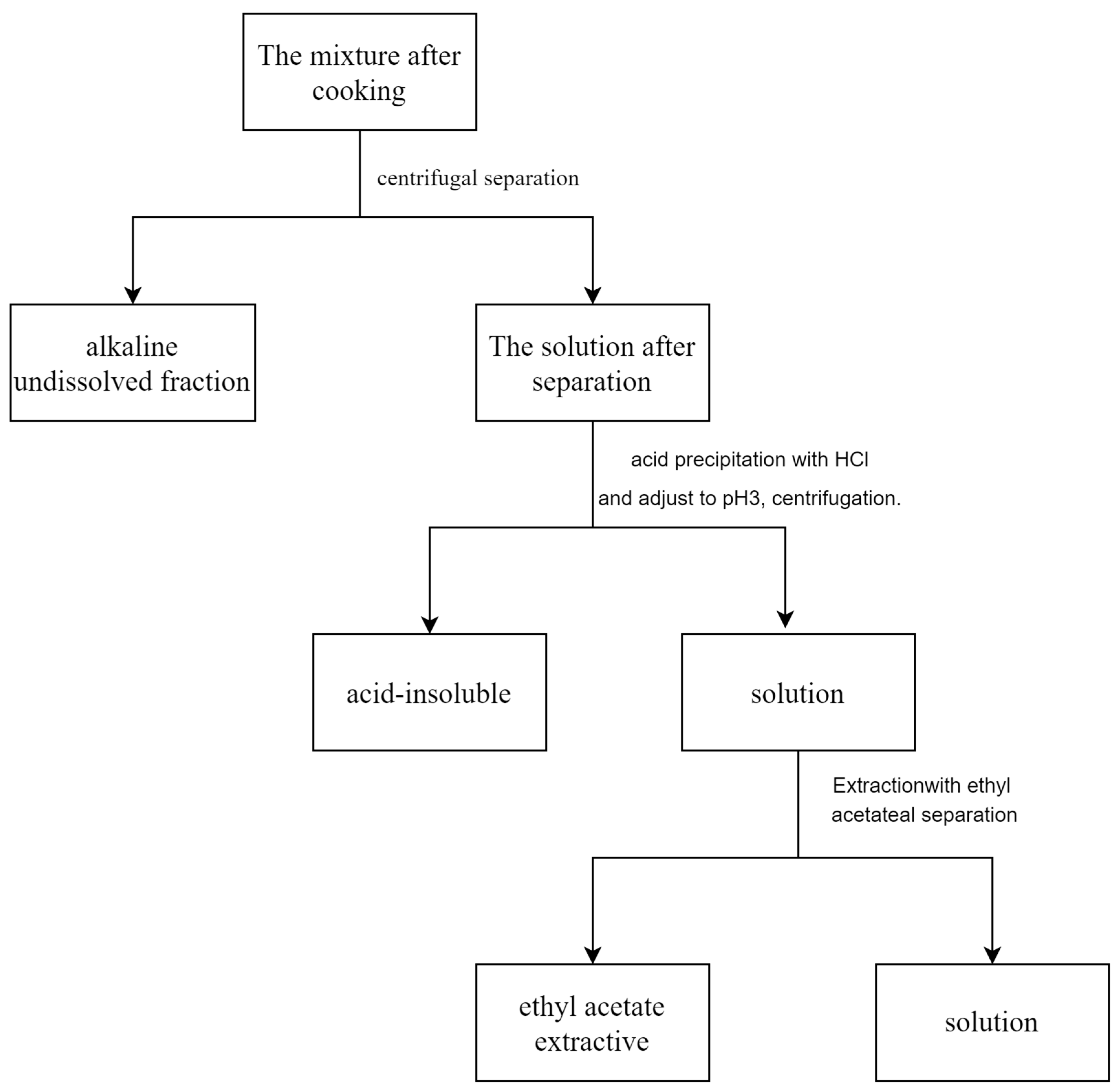
3. Experiment
3.1. Materials
3.2. Methods
3.2.1. Synthesis of lignin dimers-[α-13C]
3.2.2. Synthesis of 4-Acetyl Guaiacol-[α-13C]
3.2.3. Synthesis of 4-(α-Bromoacetyl)-Guaiacol-[α-13C]
3.2.4. Synthesis of 4-(α-(2-Methoxyphenoxy)-Acetyl)Guaiacol-[α-13C]
3.2.5. Synthesis of 4-(α-(2-Methoxyphenoxy)-β-Hydroxypropionyl)-Guaiacol-[α-13C]
3.2.6. Synthesis of Guaiacylglycerol-β-Guaiacyl Ether-[α-13C]
3.2.7. The Cooking Experiment of Lignin Dimer and Xylose
3.2.8. 13C-NMR and Two-Dimensional HMQC Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ekielski, A.; Mishra, P.K. Lignin for Bioeconomy: The Present and Future Role of Technical Lignin. Int. J. Mol. Sci. 2021, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Björkman, A. Studies on finely divided wood. Part 3. Extraction of lignin-carbohydrate complexes with neutral solvents. J. Sven. Papperstding 1957, 60, 243–251. [Google Scholar]
- Du, X.; Pérez-Boada, M.; Fernández, C.; Rencoret, J.; Del Río, J.C.; Jiménez-Barbero, J.; Martínez, A.T. Analysis of lignin-carbohydrate and lignin-lignin linkages after hydrolase treatment of xylan-lignin, glucomannan-lignin and glucan-lignin complexes from spruce wood. J. Planta 2014, 239, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Innocent, M.T.; Wang, Q.; Xiang, H.; Tang, J.; Zhu, M. Kraft lignin-based piezoresistive sensors: Effect of chemical structure on the micro-structure of ultrathin carbon fibers. Int. J. Biol. Macromol. 2020, 151, 730–739. [Google Scholar] [CrossRef]
- Xin, R.; Wang, C.; Zhang, Y.; Peng, R.; Li, R.; Wang, J.; Mao, Y.; Zhu, X.; Zhu, W.; Kim, M.; et al. Efficient Removal of Greenhouse Gases: Machine Learning-Assisted Exploration of Metal–Organic Framework Space. ACS Nano 2024, 18, 19403–19422. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, J.; Gordobil, O.; Robles, E.; González-Alriols, M.; Labidi, J. Lignin valorization from side-streams produced during agricultural waste pulping and total chlorine free bleaching. J. Clean. Prod. 2017, 142, 2609–2617. [Google Scholar] [CrossRef]
- Zhao, Y.; Shakeel, U.; Rehman, M.S.U.; Li, H.; Xu, X.; Xu, J. Lignin-carbohydrate complexes (LCCs) and its role in biorefinery. J. Clean. Prod. 2020, 253, 120076. [Google Scholar] [CrossRef]
- Shirke, A.N.; White, C.; Englaender, J.A.; Zwarycz, A.S.; Butterfoss, G.L.; Linhardt, R.J.; Gross, R.A. Stabilizing Leaf and Branch Compost Cutinase (LCC) with Glycosylation: Mechanism and Effect on PET Hydrolysis. Biochemistry 2018, 57, 1190–1200. [Google Scholar] [CrossRef]
- Aminzadeh, S.; Zhang, L.; Henriksson, G. A possible explanation for the structural inhomogeneity of lignin in LCC networks. Wood Sci. Technol. 2017, 51, 1365–1376. [Google Scholar] [CrossRef]
- You, T.T.; Zhang, L.M.; Zhou, S.K.; Xu, F. Structural elucidation of lignin–carbohydrate complex (LCC) preparations and lignin from Arundo donax Linn. J. Ind. Crops. Prod. 2015, 71, 65–74. [Google Scholar] [CrossRef]
- Zhao, B.C.; Xu, J.D.; Chen, B.Y.; Cao, X.F.; Yuan, T.Q.; Wang, S.F.; Sun, R.C. Selective precipitation and characterization of lignin–carbohydrate complexes (LCCs) from Eucalyptus. J. Planta 2018, 247, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Hasanbeigi, A.; Price, L. Assessment of emerging energy-efficiency technologies for the pulp and paper industry: A technical review. J. Clean. Prod. 2016, 122, 5–28. [Google Scholar] [CrossRef]
- Fan, J.Y.; Xie, Y.M.; Yang, H.T.; Wang, P. Study on the Bonds between Lignin and Cellulose by 13Carbon Isotope Tracer Method. J. Trans. China Pulp. Pap. 2006, 21, 4. [Google Scholar] [CrossRef]
- Cui, S.; Wei, X.; Chen, X.; Xie, Y. Investigation of chemical linkages between lignin and carbohydrates in cultured poplar cambium tissues via double isotope labeling. Int. J. Biol. Macromol. 2023, 231, 123250. [Google Scholar] [CrossRef]
- Hu, Z.J.; Xie, Y.M.; Ou, G.R.; Hong, W. A New Method of Synthesis of Guaiacylglycero-lβ-Guaiacyl Ether and Its Structural Analysis. J. Pap. Sci. Technol. 2002, 21, 4–7. [Google Scholar] [CrossRef]
- King, L.C.; Ostrum, G.K. Selective Bromination with copper (II) bromide1. J. Org. Chem. 1964, 29, 3459–3461. [Google Scholar] [CrossRef]
- Kolvari, E.; Koukabi, N.; Khoramabadi-Zad, A.; Shiri, A.; Zolfigol, M.A. Alternative Methodologies for Halogenation of Organic Compounds. Curr. Org. Synth. 2013, 10, 837–863. [Google Scholar] [CrossRef]
- Kosower, E.M.; Wu, G.S. Halogenation with copper (II). II. Unsaturated ketones. J. Org. Chem. 1963, 28, 633–638. [Google Scholar] [CrossRef]
- Qian, X.; Chen, Z.; Yang, X.; Zhao, W.; Liu, C.; Sun, T.; Zhou, D.; Yang, Q.; Wei, G.; Fan, M. Perovskite cesium lead bromide quantum dots: A new efficient photocatalyst for degrading antibiotic residues in organic system. J. Clean Prod. 2019, 249, 119335. [Google Scholar] [CrossRef]
- Chen, F.; Mo, K.; Zhang, Q.; Fei, S.; Zu, Y.; Yang, L. A novel approach for distillation of paeonol and simultaneous extraction of paeoniflorin by microwave irradiation using an ionic liquid solution as the reaction medium. Sep. Purif. Technol. 2017, 183, 73–82. [Google Scholar] [CrossRef]
- Vanucci, C.; De Violet, P.F.; Bouas-Laurent, H.; Castellan, A. Photodegradation of lignin: A photophysical and photochemical study of a non-phenolic α-carbonyl β-O-4 lignin model dimer, 3, 4-dimethoxy-α-(2′-methoxypenoxy) acetophenone. J. Photochem. Photobiol. A Chem. 1988, 41, 251–265. [Google Scholar] [CrossRef]
- Castellan, A.; Colombo, N.; Cucuphat, C.; de Violet, P.F. Photodegradation of Lignin: A Photochemical Study of a Phenolic α-Carbonyl β-O-4 Lignin Model Dimer 4-Hy droxy-3-Methoxy-α- (2′-Methoxyphenoxy)-Acetophenone. Holzforschung 1989, 43, 179–185. [Google Scholar] [CrossRef]
- Katahira, R.; Mittal, A.; McKinney, K.; Chen, X.; Tucker, M.P.; Johnson, D.K.; Beckham, G.T. Base-Catalyzed Depolymerization of Biorefinery Lignins. ACS Sustain. Chem. Eng. 2016, 4, 1474–1486. [Google Scholar] [CrossRef]
- Karhunen, P.; Rummakko, P.; Sipilä, J.; Brunow, G.; Kilpeläinen, I. Dibenzodioxocins; a novel type of linkage in softwood lignins. Tetrahedron Lett. 1995, 36, 169–170. [Google Scholar] [CrossRef]
- Hayashi, T.; Hosoya, T.; Miyafuji, H. Vanillin production pathways in alkaline nitrobenzene oxidation of guaiacyl glycer-ol-β-guaiacyl ether. J. Agric. Food Chem. 2023, 71, 10124–10132. [Google Scholar] [CrossRef]
- Niu, H.; Chen, X.; Zhao, Y.; Zhou, J.; Xie, Y. Exploration of the Linkages between Lignin and Carbohydrates in Kraft Pulp from Wheat Straw Using a 13C/2H Isotopic Tracer. Molecules 2023, 28, 7493. [Google Scholar] [CrossRef]
- Mkumbuzi, E.; Pillay, M.N.; van Zyl, W.E. Reaction mechanisms in microwave-assisted lignin depolymerisation in hydro-gen-donating solvents. J. Green Process. Synth. 2023, 12, 20230154. [Google Scholar] [CrossRef]
- Shi, Z.; Xu, G.; Deng, J.; Dong, M.; Murugadoss, V.; Liu, C.; Guo, Z. Structural characterization of lignin from D. sinicus by FTIR and NMR techniques. J. Green Chem. Lett. Rev. 2019, 12, 235–243. [Google Scholar] [CrossRef]
- Xue, Y.; Yu, C.; Kang, X. Quantitative and Structural Characterization of Native Lignin in Hardwood and Softwood Bark via Solid-State NMR Spectroscopy. J. Agric. Food Chem. 2024, 72, 18056–18066. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, Y.; Guo, T.; Zhao, H.; Jin, Y. Structural characterization of lignin and lignin-carbohydrate complex (LCC) from ginkgo shells (Ginkgo biloba L.) by comprehensive NMR spectroscopy. J. Polym. 2018, 10, 736. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Luo, N.; Wang, F. Visible-Light-Induced Oxidative Lignin C–C Bond Cleavage to Aldehydes Using Vanadium Catalysts. ACS Catal. 2019, 10, 632–643. [Google Scholar] [CrossRef]
- Beck, S.; Mushrif, S.H. Reaction pathways and energetics of the deconstruction of lignin carbohydrate complexes (LCCs) in lignocellulosic biomass. Sustain. Energy Fuels 2024, 8, 3113–3123. [Google Scholar] [CrossRef]
- Laskar, D.D.; Yang, B.; Wang, H.; Lee, J. Pathways for biomass-derived lignin to hydrocarbon fuels. J. Biofuels Bioprod. Biorefin. 2013, 7, 602–626. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Z.; Chen, J. Applications of lignin-derived catalysts for green synthesis. Green Energy Environ. 2019, 4, 210–244. [Google Scholar] [CrossRef]
- Nakatsubo, F.; Sato, K.; Higuchi, T. Synthesis of Guaiacylglycerol-β-guaiacyl Ether. Holzforschung 2009, 29, 165–168. [Google Scholar] [CrossRef]
- Nakano, J. (Ed.) Chemistry of Lignin; Uni Press: Tokyo, Japan, 1978; pp. 519–523. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Solvent | Time (h) | Cuprous Bromide Recovery Rate | Product Yield |
|---|---|---|---|
| Ethyl alcohol | 1 | 72.70% | 94.84% |
| 2 | 83.28% | 94.27% | |
| 3 | 84.28% | 93.01% | |
| Trichloromethane/ethyl acetate (1/1, v/v) | 1 | 92.04% | 81.08% |
| 2 | 92.44% | 81.37% | |
| 3 | 92.36% | 80.37% |
| Signal | δ (ppm) | Assignments |
|---|---|---|
| 1 | 7.59–7.61 | proton on C2 and C6 in guaiacyl |
| 2 | 6.85–7.57 | aromatic protons |
| 3 | 5.26 | methylene |
| 4 | 4.92 | bromomethyl |
| 5 | 3.82 | methoxy group |
| Signal | δ (ppm) | Assignments |
|---|---|---|
| 1 | 6.82–7.24 | aromatic protons |
| 2 | 4.94 | methylene in α position |
| 3 | 3.98–4.13 | proton in β position |
| 4 | 3.80 | methoxy group |
| 5 | 3.43 | proton in γ position |
| Signal | δ (ppm) | Assignments |
|---|---|---|
| 1 | 149.8–149.9 | C3 in guaiacyl |
| 2 | 148.2–148.5 | C3/C4 in guaiacyl |
| 3 | 145.6–147.1 | C3/C5 in guaiacyl |
| 4 | 133.1–133.4 | C1 in guaiacyl |
| 5 | 120.8–121.8 | C6 in |
| 6 | 119.1–119.6 | C5 in guaiacyl |
| 7 | 111.1–115.9 | C2 in guaiacyl |
| 8 | 83.8–84.6 | Cβ in β-O-4 |
| 9 | 70.3–71.7 | Cα in β-O-4 |
| 10 | 60.2 | Cγ in β-O-4 |
| 11 | 55.5–55.7 | methoxy group |
| Signal | δ (ppm) | Assignments | |
|---|---|---|---|
| A | B | ||
| 2 | 191.2 | 191.2 | C=O in α position |
| 4 | 147.8 | 147.8 | C4 in etherified guaiacyl |
| 6 | 133.0 | C1 in etherified guaiacyl | |
| 7 | 130.9 | 130.9 | C1 in etherified guaiacyl |
| 9 | 121.0 | 121.0 | C6 in etherified guaiacyl |
| 10 | 119.4 | 119.3 | C5 in etherified guaiacyl |
| 11 | 115.6 | 115.7 | C5 in etherified guaiacyl |
| 12 | 112.5 | 112.4 | C2 in etherifiedguaiacyl |
| 13 | 109.7 | C2 in guaiacyl | |
| 14 | 98.7 | C1 in xylose | |
| 15 | 81.5 | Cα (β-O-4) with ether bond to xylaose | |
| 16 | 79.3 | Cβ in guaiacyl | |
| 17 | 77.8 | C3/C4 in xylose | |
| 18 | 76.5 | C3 in xylose | |
| 19 | 75.6, 74.2 | C2 in xylose | |
| 20 | 72.8 | C2/C3 in xylose | |
| 21 | 71.6, 71.3 | 71.3, 70.9 | Cα in β-O-4 |
| 22 | 66.0 | C5 in xylose | |
| 23 | 55.8 | 55.9 | methoxyl group |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Xie, Y.; Zhao, B. Formation and Chemical Structure of Carbon-13 Tracer Lignin-Carbohydrate Complexes (LCCs) During Kraft Pulping. Molecules 2025, 30, 1077. https://doi.org/10.3390/molecules30051077
Wang Z, Xie Y, Zhao B. Formation and Chemical Structure of Carbon-13 Tracer Lignin-Carbohydrate Complexes (LCCs) During Kraft Pulping. Molecules. 2025; 30(5):1077. https://doi.org/10.3390/molecules30051077
Chicago/Turabian StyleWang, Zhi, Yimin Xie, and Boxuan Zhao. 2025. "Formation and Chemical Structure of Carbon-13 Tracer Lignin-Carbohydrate Complexes (LCCs) During Kraft Pulping" Molecules 30, no. 5: 1077. https://doi.org/10.3390/molecules30051077
APA StyleWang, Z., Xie, Y., & Zhao, B. (2025). Formation and Chemical Structure of Carbon-13 Tracer Lignin-Carbohydrate Complexes (LCCs) During Kraft Pulping. Molecules, 30(5), 1077. https://doi.org/10.3390/molecules30051077