
Thermodynamic Cards of Classic NADH Models and Their Related Photoexcited States Releasing Hydrides in Nine Elementary Steps and Their Applications


Abstract

1. Introduction
2. Results and Discussion
2.1. The Definitions, and Sources of Nine Potential Thermodynamic Driving Forces for XH and XH* Releasing Hydrides in Acetonitrile
2.2. Establishing Thermodynamic Cards of XH and XH* Releasing Hydrides in Acetonitrile
2.3. Thermodynamic Abilities of XH* and XH Releasing Electrons
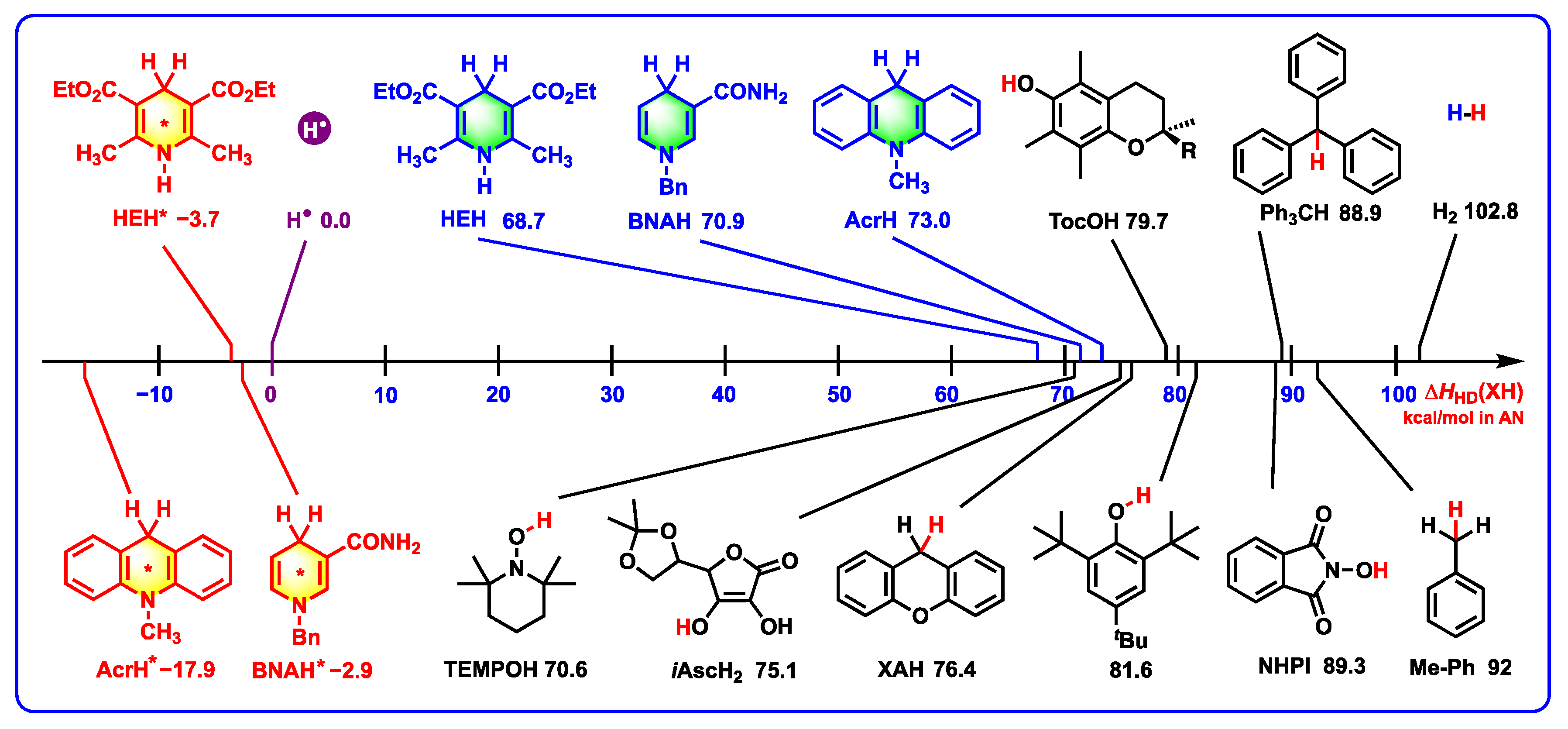
2.4. Thermodynamic Abilities of XH* and XH Releasing Hydrides
2.5. Thermodynamic Abilities of XH* and XH Releasing Hydrogen Atoms
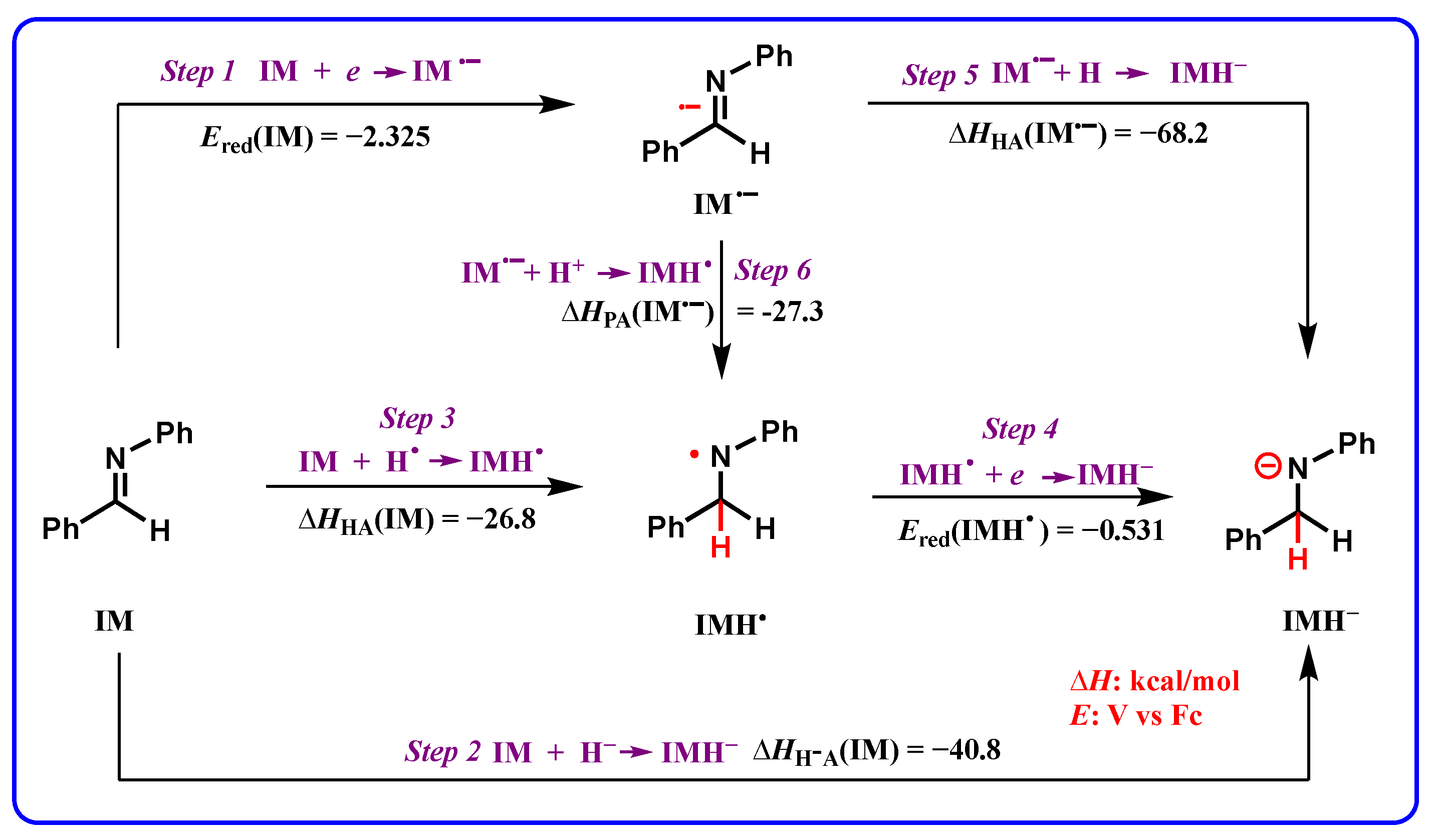
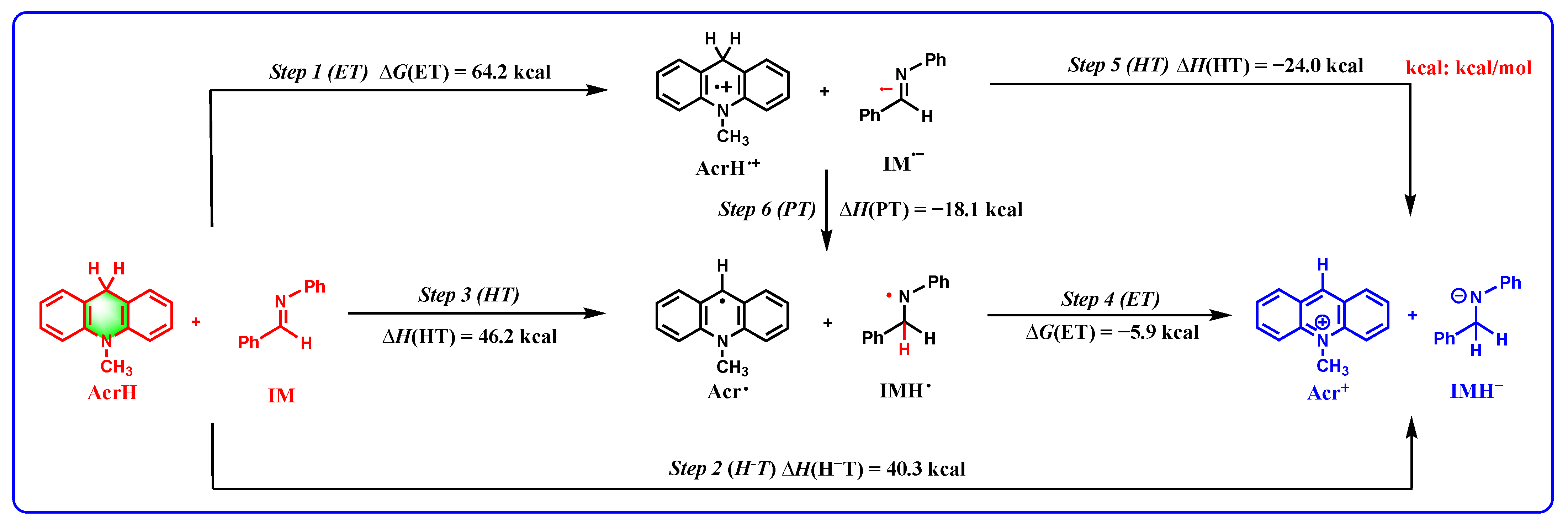
2.6. Application of Thermodynamic Cards of XH and XH* to Reduction Reactions
3. Experimental Section
3.1. Electrochemical Experiments
3.2. Isothermal Titration Experiments
3.3. The Eox(XH*) Values Determination
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [PubMed]
- Verkhovsky, M.I.; Bogachev, A.V.; Pivtsov, A.V.; Bertsova, Y.V.; Fedin, M.V.; Bloch, D.A.; Kulik, L.V. sodium-dependent movement of covalently bound FMN residue(s) in Na+-translocating NADH: Quinone oxidoreductase. Biochemistry 2012, 51, 5414–5421. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.-B.; Qian, B.-C.; Fu, Y.-H.; Zhu, X.-Q. Thermodynamics of the Elementary Steps of Organic Hydride Chemistry Determined in Acetonitrile and their Applications. Org. Chem. Front. 2022, 9, 6001–6062. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Tan, Y.; Cao, C.-T. Thermodynamic diagnosis of the properties and mechanism of dihydropyridine-type compounds as hydride source in acetonitrile with “Molecule ID Card”. J. Phys. Chem. B 2010, 114, 2058–2075. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Nam, W.; Fukuzumi, S. Redox Catalysis Via Photoinduced Electron Transfer. Chem. Sci. 2023, 14, 4205–4218. [Google Scholar] [CrossRef]
- Yedase, G.S.; Sreelakshmi Venugopal, S.; Arya, P.; Yatham, V.R. Catalyst-free Hantzsch Ester-mediated Organic Transformations Driven by Visible light. Asian J. Org. Chem. 2022, 11, e202200478. [Google Scholar] [CrossRef]
- Huang, W.; Cheng, X. Hantzsch Esters as Multifunctional Reagents in Visible-Light Pho- toredox Catalysis. Synlett 2017, 28, 148–158. [Google Scholar]
- Murray, P.R.D.; Cox, J.H.; Chiappini, N.D.; Roos, C.B.; McLoughlin, E.A.; Hejna, B.G.; Nguyen, S.T.; Ripberger, H.H.; Ganley, J.M.; Tsui, E.; et al. Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis. Chem. Rev. 2022, 122, 2017–2291. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef]
- Rueping, M.; Dufour, J.; Schoepke, F.R. Advances in Catalytic Metal-Free Reductions: From Bioinspired Concepts to Applications in the Organocatalytic Synthesis of Pharmaceuticals and Natural Products. Green Chem. 2011, 13, 1084–1105. [Google Scholar] [CrossRef]
- Huang, B.; Bu, X.-S.; Xu, J.; Dai, J.-J.; Feng, Y.-S.; Xu, H.-J. Metal-Free, Visible-Light-Mediated Direct C-H Trifluoromethylation of Hydrazones with NADH Coenzyme Model Catalyst. Asian J. Org. Chem. 2018, 7, 137–140. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, W.-M.; Dai, J.-J.; Xu, J.; Xu, H.-J. Visible-Light-Promoted C–H Arylation by Merging Palladium Catalysis with Organic Photoredox Catalysis. J. Org. Chem. 2017, 82, 3622–3630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-Z.; Zhai, X.-F.; Zhang, S.-L.; Xia, K.-J.; Ding, H.; Song, X.-R.; Tian, W.-F.; Liang, Y.-M.; Xiao, Q. Photo-Induced Nickel-Mediated Cross-Electrophile Coupling for Alkylated Allenes Via Electron Donor–Acceptor Complexes. Org. Chem. Front. 2023, 10, 298–303. [Google Scholar] [CrossRef]
- Kammer, L.M.; Badir, S.O.; Hu, R.-M.; Molander, G.A. Photoactive Electron Donor–Acceptor Complex Platform for Ni-Mediated C(sp3)–C(sp2) Bond Formation. Chem. Sci. 2021, 12, 5450–5457. [Google Scholar] [CrossRef]
- Xu, X.; Min, Q.-Q.; Lia, N.; Liu, F. Visible Light-Promoted Umpolung Coupling of Aryl Tri-/Difluoroethanones with 2-Alkenylpyridines. Chem. Commun. 2018, 54, 11017–11020. [Google Scholar] [CrossRef]
- Patel, S.S.; Kumara, D.; Tripathi, C.B. Brønsted Acid Catalyzed Radical Addition to Quinone Methides. Chem. Commun. 2021, 57, 5151–5154. [Google Scholar] [CrossRef]
- Yu, Z.; Kong, Y.; Li, B.; Su, S.; Rao, J.; Gao, Y.; Tu, T.; Chen, H.; Liao, K. HTE- and AI-Assisted Development of DHP-Catalyzed Decarboxylative Selenation. Chem. Commun. 2023, 59, 2935–2938. [Google Scholar] [CrossRef]
- Nagode, S.B.; Kant, R.; Rastogi, N. Hantzsch Ester-Mediated Benzannulation of Diazo Compounds under Visible Light Irradiation. Org. Lett. 2019, 21, 6249–6254. [Google Scholar] [CrossRef]
- Jung, J.; Kim, J.; Park, G.; You, Y.; Cho, E.J. Selective Debromination and a-Hydroxylation of a-Bromo Ketones Using Hantzsch Esters as Photoreductants. Adv. Synth. Catal. 2016, 358, 74–80. [Google Scholar] [CrossRef]
- Panferova, L.I.; Tsymbal, A.V.; Levin, V.V.; Struchkova, M.I.; Dilman, A.D. Reactions of gem-Difluorinated Phosphonium Salts Induced by Light. Org. Lett. 2016, 18, 996–999. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, S.; Zhang, D.; Song, B.; Jin, Y.; Hao, E.; Shi, L. Photochemical Nozaki−Hiyama−Kishi Coupling Enabled by Excited Hantzsch Ester. Org. Lett. 2022, 24, 3331–3336. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Grant, P.S.; Li, X.; Noble, A.; Aggarwal, V.K. Catalyst-Free Deaminative Functionalizations of Primary Amines via Photoinduced Single-Electron Transfer. Angew. Chem. Int. Ed. 2019, 58, 5697–5701. [Google Scholar] [CrossRef] [PubMed]
- Nagode, S.B.; Kant, R.; Rastogi, N. Hantzsch Ester-Mediated Synthesis of Phenanthridines under Visible Light Irradiation. Chem. Asian J. 2020, 15, 3513–3518. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yu, Z.; Tong, M.L.; Kohlhepp, S.V.; Yin, X.; Mendoza, A. Decarboxylative Alkyl Coupling Promoted by NADH and Blue Light. J. Am. Chem. Soc. 2020, 142, 20143–20151. [Google Scholar] [CrossRef]
- Johansen, C.M.; Boyd, E.A.; Peters, J.C. Catalytic Transfer Hydrogenation of N2 to NH3 via A Photoredox Catalysis Strategy. Sci. Adv. 2022, 8, eade3510. [Google Scholar] [CrossRef]
- Huang, W.; Chen, W.; Wang, G.; Li, J.; Cheng, X.; Li, G. Thiyl-Radical-Catalyzed Photoreductive Hydrodifluoroacetamidation of Alkenes with Hantzsch Ester as a Multifunctional Reagent. ACS Catal. 2016, 6, 7471–7474. [Google Scholar] [CrossRef]
- Yu, T.; Higashi, M.; Cembran, A.; Gao, J.; Truhlar, D.G. Concerted Hydrogen Atom and Electron Transfer Mechanism for Catalysis by Lysine-Specific Demethylase. J. Phys. Chem. B 2013, 117, 8422–8429. [Google Scholar] [CrossRef]
- Shen, G.-B.; Qian, B.-C.; Zhang, G.-S.; Luo, G.-Z.; Fu, Y.-H.; Zhu, X.-Q. Thermodynamics Regulated Organic Hydride/Acid Pairs as Novel Organic Hydrogen Reductants. Org. Chem. Front. 2022, 9, 6833–6848. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. Hydride, Hydrogen Atom, Proton, and Electron Transfer Driving Forces of Various Five-Membered Heterocyclic Organic Hydrides and their Reaction Intermediates in Acetonitrile. J. Am. Chem. Soc. 2008, 130, 2501–2516. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Zhang, M.; Liu, Q.-Y.; Wang, X.-X.; Zhang, J.-Y.; Cheng, J.-P. A Facile Experimental Method to Determine the Hydride Affinity of Polarized Olefins in Acetonitrile. Angew. Chem. Int. Ed. 2006, 45, 3954–3957. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Inada, O.; Suenobu, T. Mechanisms of Electron-Transfer Oxidation of NADH Analogues and Chemiluminescence. Detection of the Keto and Enol Radical Cations. J. Am. Chem. Soc. 2003, 125, 4808–4816. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Koumitsu, S.; Katsuhik Hironaka, K.; Tanaka, T. Energetic Comparison between Photoinduced Electron-Transfer Reactions from NADH Model Compounds to Organic and Inorganic Oxidants and Hydride-Transfer Reactions from NADH Model Compounds to p-Benzoquinone Derivatives. J. Am. Chem. Soc. 1987, 109, 305–316. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Hironaka, K.; Tanaka, T. Photoreduction of Alkyl Halides by an NADH Model Compound. An Electron-Transfer Chain Mechanism. J. Am. Chem. Soc. 1983, 105, 4722–4727. [Google Scholar] [CrossRef]
- Ji, P.; Zhang, Y.; Wei, Y.; Huang, H.; Hu, W.; Mariano, P.A.; Wang, W. Visible-Light-Mediated, Chemo- and Stereoselective Radical Process for the Synthesis of C-Glycoamino Acids. Org. Lett. 2019, 21, 3086–3092. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fukuzumi, S. 10-Methylacridine Derivatives Acting as Efficient and Stable Photocatalysts in Reductive Dehalogenation of Halogenated Compounds with Sodium Borohydride via Photoinduced Electron Transfer. J. Am. Chem. Soc. 1990, 112, 8864–8870. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Ishikawa, M.; Tanaka, T. Mechanisms of Photo-Oxidation of NADH Model Compounds by Oxygen. J. Chem. Soc. Perkin Trans. II 1989, 8, 1037–1045. [Google Scholar] [CrossRef]
- Strieth-Kalthoff, F.; James, M.J.; Teders, M.; Pitzer, L.; Glorius, F. Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev. 2018, 47, 7190–7202. [Google Scholar] [CrossRef]
- Zhu, D.-L.; Wu, Q.; Li, H.-Y.; Li, H.-X.; Lang, J.-P. Hantzsch Ester as a Visible-Light Photoredox Catalyst for Transition-Metal-Free Coupling of Arylhalides and Arylsulfinates. Chem. Eur. J. 2020, 26, 3484–3488. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion Constants for Redox Potentials Measured Versus Different Reference Electrodes in Acetonitrile Solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Arnett, E.M.; Amarnath, K.; Harvey, N.G.; Cheng, J.-P. Determination and Interrelation of Bond Heterolysis and Homolysis Energies in Solution. J. Am. Chem. Soc. 1990, 112, 344–355. [Google Scholar] [CrossRef]
- Wu, Y.; Kim, D.; Teets, T.S. Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations. Synlett 2022, 33, 1154–1179. [Google Scholar]
- Buzzetti, L.; Crisenza, G.E.M.; Melchiorre, P. Mechanistic Studies in Photocatalysis. Angew. Chem. Int. Ed. 2019, 58, 3730–3747. [Google Scholar] [CrossRef] [PubMed]
- Shon, J.-H.; Sittel, S.; Teets, T.S. Synthesis and Characterization of Strong Cyclometalated Iridium Photoreductants for Application in Photocatalytic Aryl Bromide Hydrodebromination. ACS Catal. 2019, 9, 8646–8658. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Chen, J.-R.; Xiao, W.-J. Visible Light-Driven Radical-Mediated C−C Bond Cleavage/Functionalization in Organic Synthesis. Chem. Rev. 2021, 121, 506–561. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef]
- Leitch, J.A.; Rossolini, T.; Rogova, T.; Maitland, J.A.P.; Dixon, D.J. α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. [Google Scholar] [CrossRef]
- Heiden, Z.M.; Lathem, A.P. Establishing the Hydride Donor Abilities of Main Group Hydrides. Organometallics 2015, 34, 1818–1827. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef]
- Agarwal, R.G.; Coste, S.C.; Groff, B.D.; Heuer, A.M.; Noh, H.; Parada, G.A.; Wise, C.F.; Nichols, E.M.; Warren, J.J.; Mayer, J.M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1–49. [Google Scholar] [CrossRef]
- Warburton, R.E.; Soudackov, A.V.; Hammes-Schiffer, S. Theoretical Modeling of Electrochemical Proton-Coupled Electron Transfer. Chem. Rev. 2022, 122, 10599–10650. [Google Scholar] [CrossRef]
- Yang, J.-D.; Xue, J.; Cheng, J.-P. Understanding the Role of Thermodynamics in Catalytic Imine Reductions. Chem. Soc. Rev. 2019, 48, 2913–2926. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-Q.; Liu, Q.-Y.; Chen, Q.; Mei, L.-R. Hydride, Hydrogen, Proton, and Electron Affinities of Imines and Their Reaction Intermediates in Acetonitrile and Construction of Thermodynamic Characteristic Graphs (TCGs) of Imines as a “Molecule ID Card”. J. Org. Chem. 2010, 75, 789–808. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step X | Process | Parameters | Unit | BNAH | HEH | AcrH |
|---|---|---|---|---|---|---|
| Step 1 | XH → XH•+ + e | Eox(XH) | V vs. Fc | 0.219 | 0.479 | 0.460 |
| Step 2 | XH → X+ + H− | ΔHH−R(XH) | kcal/mol | 64.2 | 69.3 | 81.1 |
| Step 3 | XH → X• + H• | ΔHHR(XH) | kcal/mol | 70.9 | 68.7 | 73.0 |
| Step 4 | X• → X+ + e | Eox(X•) | V vs. Fc | −1.419 | −1.112 | −0.789 |
| Step 5 | XH•+ → X+ + H• | ΔHHR(XH•+) | kcal/mol | 32.9 | 32.0 | 44.2 |
| Step 6 | XH•+ → X• + H+ | ΔHPR(XH•+) | kcal/mol | 12.4 | 4.5 | 9.2 |
| Step 7 | XH* → XH•+ + e | Eox(XH*) | V vs. Fc | −2.98 | −2.66 | −3.48 |
| Step 8 | XH* → X+ + H− XH* → X+ + ECH | ΔHH−R(XH*) | kcal/mol | −9.6 | −3.1 | −9.8 |
| Step 9 | XH* → X• + H• XH* → X• + PCE | ΔHHR(XH*) | kcal/mol | −2.9 | −3.7 | −17.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, B.-C.; Zhu, X.-Q.; Shen, G.-B. Thermodynamic Cards of Classic NADH Models and Their Related Photoexcited States Releasing Hydrides in Nine Elementary Steps and Their Applications. Molecules 2025, 30, 1053. https://doi.org/10.3390/molecules30051053
Qian B-C, Zhu X-Q, Shen G-B. Thermodynamic Cards of Classic NADH Models and Their Related Photoexcited States Releasing Hydrides in Nine Elementary Steps and Their Applications. Molecules. 2025; 30(5):1053. https://doi.org/10.3390/molecules30051053
Chicago/Turabian StyleQian, Bao-Chen, Xiao-Qing Zhu, and Guang-Bin Shen. 2025. "Thermodynamic Cards of Classic NADH Models and Their Related Photoexcited States Releasing Hydrides in Nine Elementary Steps and Their Applications" Molecules 30, no. 5: 1053. https://doi.org/10.3390/molecules30051053
APA StyleQian, B.-C., Zhu, X.-Q., & Shen, G.-B. (2025). Thermodynamic Cards of Classic NADH Models and Their Related Photoexcited States Releasing Hydrides in Nine Elementary Steps and Their Applications. Molecules, 30(5), 1053. https://doi.org/10.3390/molecules30051053