
Finely-Tuned Bis(imino)pyridylcobalt Complexes Enhance Ethylene Polymerization: The Role of Bulky and Halogen Substituents

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
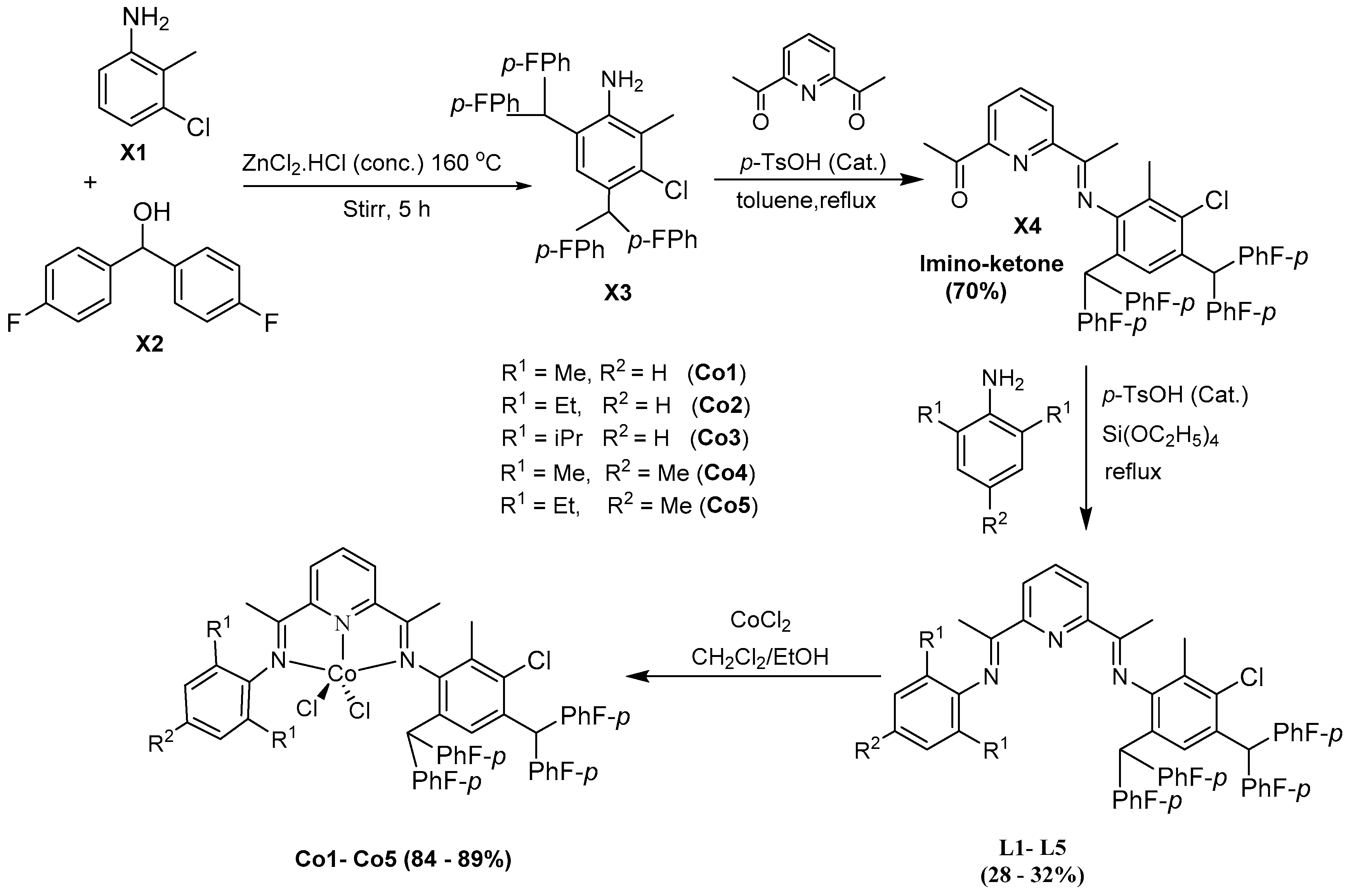
2.1. Synthesis and Characterizations of Ligands and Cobalt Complexes
2.2. Ethylene Polymerization
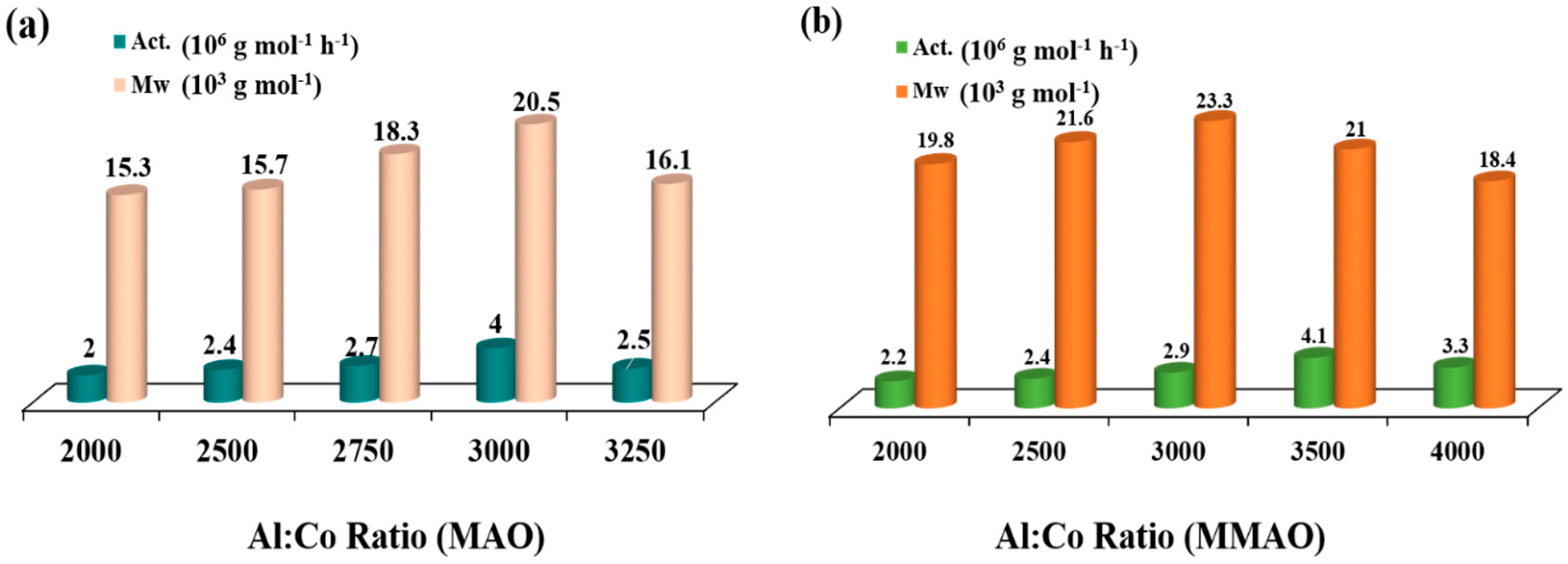
2.2.1. Optimization of Polymerization Conditions Using Co4/MAO

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | T (°C) | t (min) | Al/Co | PE (g) | Activity b | Mw c | Mw/M nc | Tm (°C) d |
|---|---|---|---|---|---|---|---|---|
| 1 | 30 | 30 | 2000 | 2.0 | 2.0 | 15.3 | 2.3 | 131.6 |
| 2 | 30 | 30 | 2500 | 2.4 | 2.4 | 15.7 | 1.9 | 130.7 |
| 3 | 30 | 30 | 2750 | 2.7 | 2.7 | 18.3 | 1.7 | 130.9 |
| 4 | 30 | 30 | 3000 | 4.0 | 4.0 | 20.8 | 2.2 | 131.6 |
| 5 | 30 | 30 | 3250 | 2.5 | 2.5 | 16.1 | 2.4 | 131.6 |
| 6 | 40 | 30 | 3000 | 4.1 | 4.1 | 20.5 | 2.6 | 131.8 |
| 7 | 50 | 30 | 3000 | 4.5 | 4.5 | 16.3 | 2.0 | 130.7 |
| 8 | 60 | 30 | 3000 | 4.9 | 4.9 | 14.6 | 2.2 | 130.4 |
| 9 | 70 | 30 | 3000 | 8.9 | 8.9 | 10.9 | 2.0 | 131.8 |
| 10 | 80 | 30 | 3000 | 7.2 | 7.2 | 7.5 | 2.0 | 129.1 |
| 11 | 90 | 30 | 3000 | 5.0 | 5.0 | 7.1 | 2.2 | 128.1 |
| 12 | 100 | 30 | 3000 | 1.6 | 1.6 | 6.4 | 2.9 | 128.3 |
| 13 | 70 | 5 | 3000 | 3.0 | 18.0 | 8.5 | 2.1 | 128.0 |
| 14 | 70 | 15 | 3000 | 6.9 | 13.7 | 10.0 | 2.4 | 129.1 |
| 15 | 70 | 45 | 3000 | 12.9 | 8.6 | 11.4 | 2.1 | 129.0 |
| 16 | 70 | 60 | 3000 | 13.3 | 6.6 | 12.7 | 2.7 | 128.8 |
| 17 e | 70 | 30 | 3000 | 4.3 | 4.3 | 8.2 | 2.5 | 129.0 |
| 18 f | 70 | 30 | 3000 | 0.9 | 0.9 | 2.2 | 2.3 | 128.0 |
| Entry | T (°C) | t (min) | Al/Co | PE (g) | Activity b | Mw c | Mw/Mn c | Tm (°C) d |
|---|---|---|---|---|---|---|---|---|
| 1 | 30 | 30 | 2000 | 2.2 | 2.2 | 19.8 | 1.9 | 130.8 |
| 2 | 30 | 30 | 2500 | 2.4 | 2.4 | 21.6 | 2.0 | 130.7 |
| 3 | 30 | 30 | 3000 | 2.9 | 2.9 | 23.3 | 1.9 | 132.0 |
| 4 | 30 | 30 | 3500 | 4.1 | 4.1 | 21.0 | 2.0 | 131.5 |
| 5 | 30 | 30 | 4000 | 3.3 | 3.3 | 18.4 | 2.1 | 132.0 |
| 6 | 40 | 30 | 3500 | 4.5 | 4.5 | 20.0 | 2.1 | 131.6 |
| 7 | 50 | 30 | 3500 | 4.6 | 4.6 | 15.9 | 2.2 | 130.7 |
| 8 | 60 | 30 | 3500 | 4.8 | 4.8 | 15.0 | 2.2 | 130.5 |
| 9 | 70 | 30 | 3500 | 3.2 | 3.2 | 10.3 | 2.3 | 129.4 |
| 10 | 80 | 30 | 3500 | 0.9 | 0.9 | 9.0 | 2.6 | 128.9 |
| 11 | 90 | 30 | 3500 | 0.8 | 0.8 | 8.1 | 2.4 | 128.7 |
| 12 | 60 | 5 | 3500 | 2.1 | 13.0 | 11.8 | 1.1 | 130.0 |
| 13 | 60 | 15 | 3500 | 2.3 | 4.7 | 13.0 | 1.7 | 130.9 |
| 14 | 60 | 45 | 3500 | 5.3 | 3.5 | 19.3 | 2.3 | 131.9 |
| 15 | 60 | 60 | 3500 | 6.1 | 3.1 | 22.1 | 2.9 | 131.8 |
| 16 e | 60 | 60 | 3500 | 2.0 | 2.0 | 19.1 | 2.0 | 128.6 |
| 17 f | 60 | 60 | 3500 | 0.5 | 0.5 | 6.5 | 1.9 | 128.0 |
| Entry | Precat. | Co-Cat. | Activity b | Mw c | Mw/Mn c | Tm (°C) d |
|---|---|---|---|---|---|---|
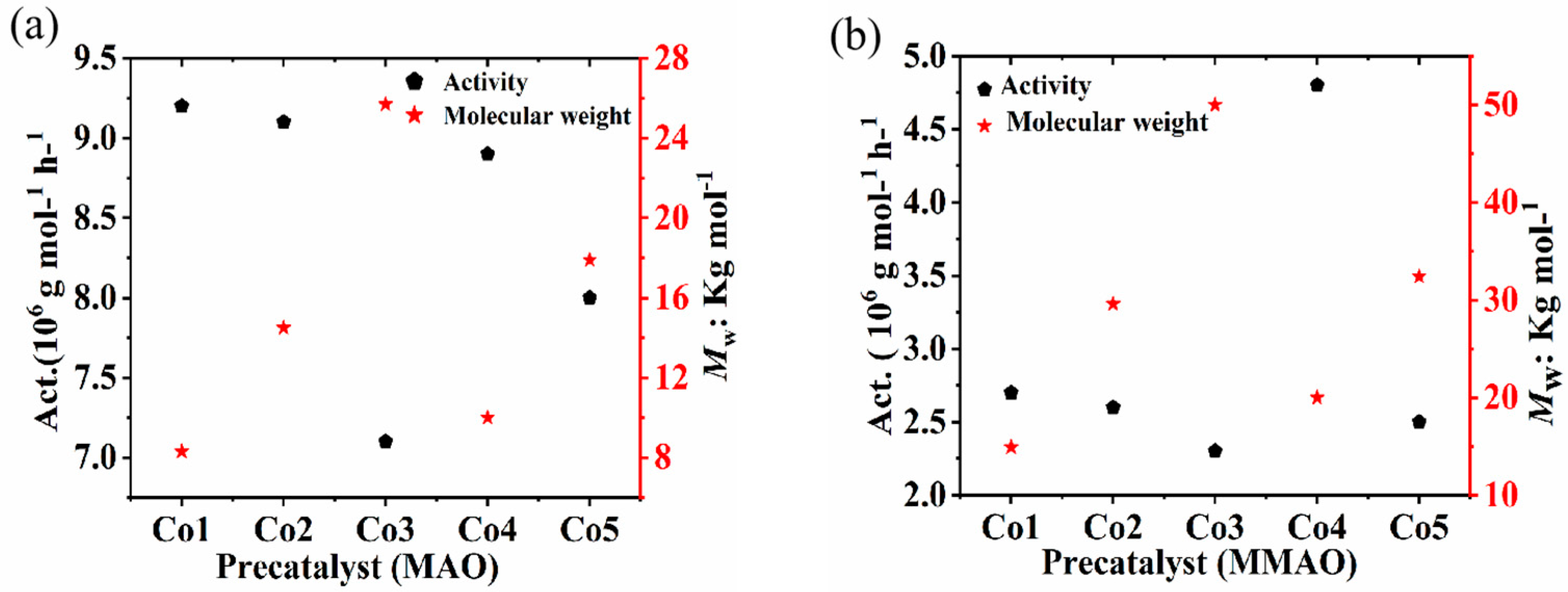
| 1 | Co1 | MAO | 9.2 | 8.3 | 2.6 | 129.5 |
| 2 | Co2 | MAO | 9.1 | 14.5 | 2.4 | 130.7 |
| 3 | Co3 | MAO | 7.1 | 25.7 | 2.3 | 131.8 |
| 4 | Co4 | MAO | 8.9 | 10.9 | 2.0 | 131.8 |
| 5 | Co5 | MAO | 8.0 | 17.9 | 2.1 | 131.3 |
| 6 | Co1 | MMAO | 2.7 | 14.9 | 2.2 | 130.7 |
| 7 | Co2 | MMAO | 2.6 | 29.6 | 2.1 | 131.9 |
| 8 | Co3 | MMAO | 2.3 | 50.0 | 1.9 | 132.0 |
| 9 | Co4 | MMAO | 4.8 | 15.0 | 2.2 | 131.7 |
| 10 | Co5 | MMAO | 2.5 | 32.4 | 1.8 | 132.3 |
2.2.2. Optimization of Polymerization Conditions Using Co4/MMAO
2.2.3. Evaluation of Co1 to Co5 with Either Methyl Aluminoxane (MAO) or Modified-Methyl Aluminoxane (MMAO) Under Optimized Conditions
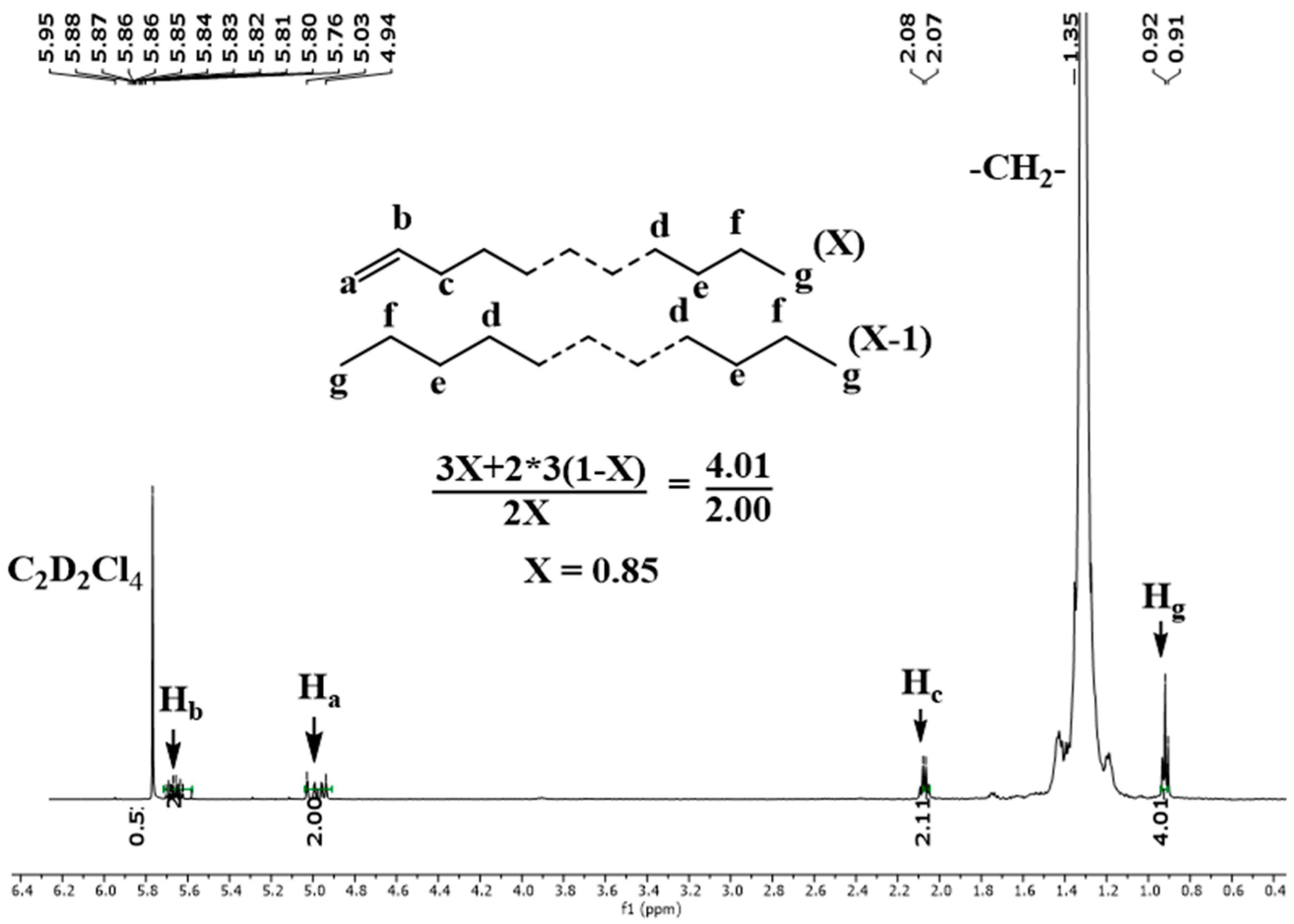
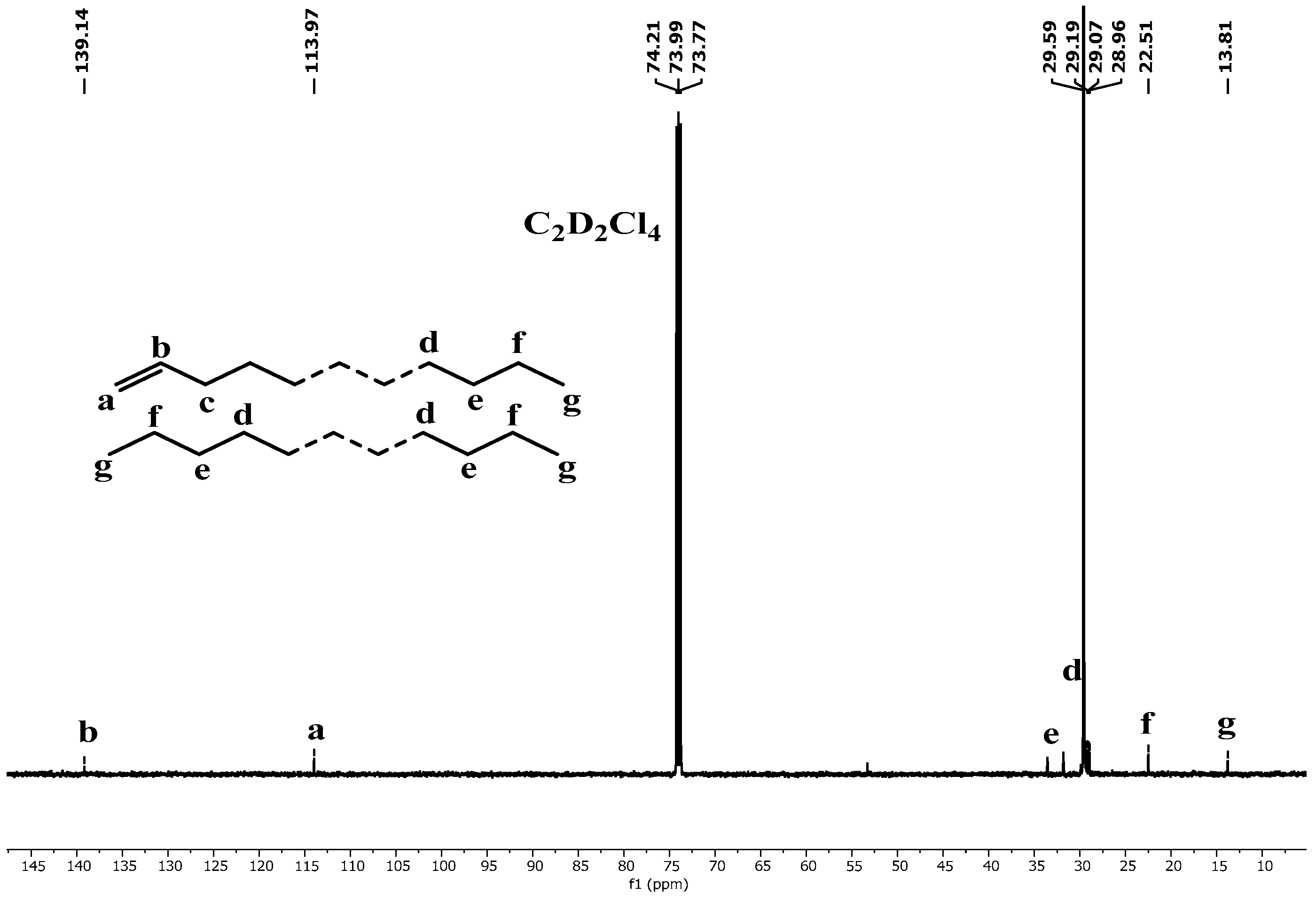
2.3. Microstructural Attributes of the Produced Polyethylenes
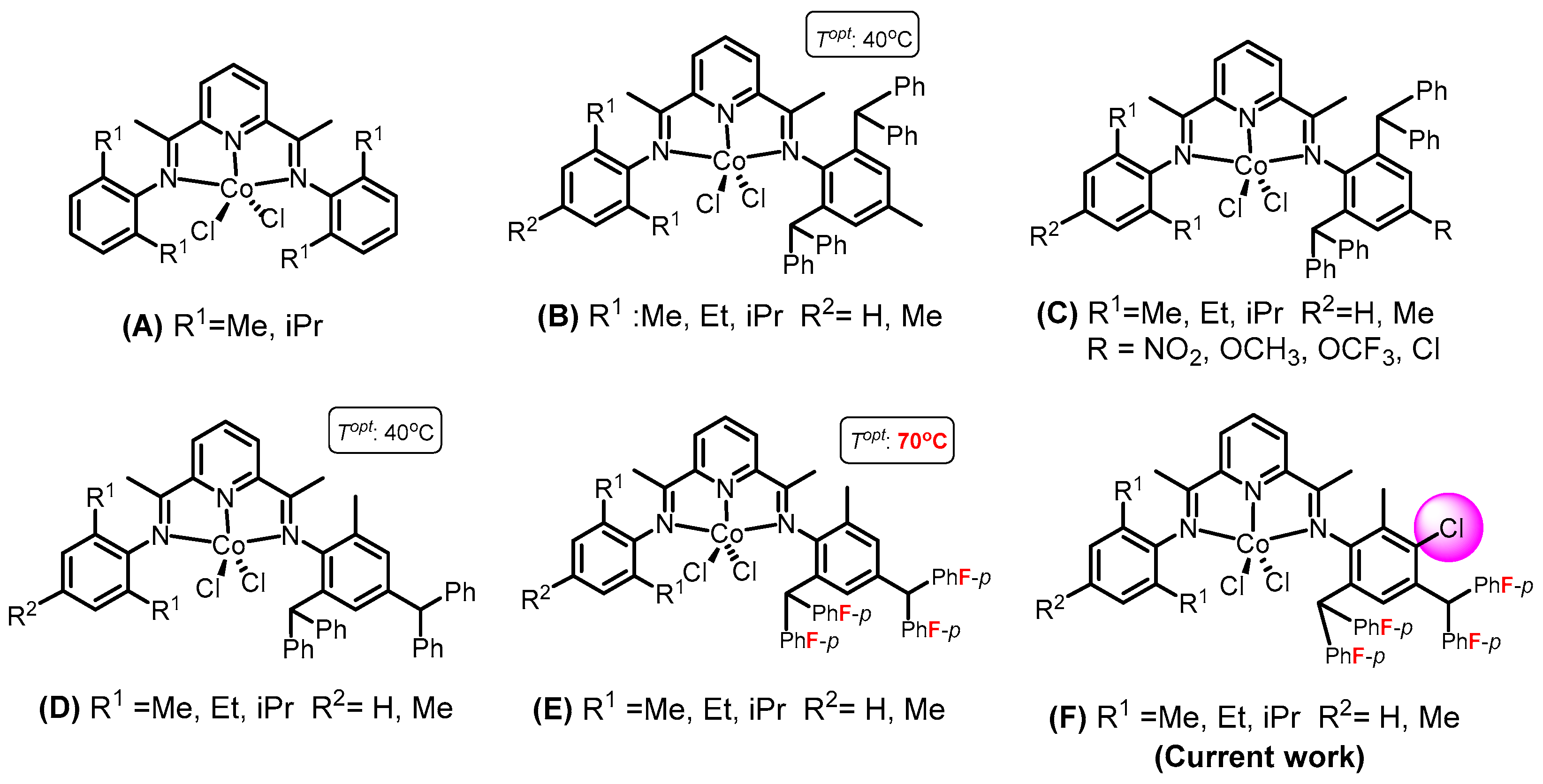
2.4. Comparative Analysis of the Present Catalyst System with Previously Documented Examples
3. Experimental Section
3.1. Synthesis of 4,6-Bis(bis(4-fluorophenyl)methyl)-3-chloro-2-methylaniline (X3)
3.2. Synthesis of 2-Acetyl-6-[1-(3-chloro-4,6-bis((di(4-fluorophenyl)methyl)-2-methylphenylimino)-ethyl]pyridine (X4)
3.3. Synthesis of 2-[1-(3-Chloro-4,6-bis((di(4-fluorophenyl)methyl)-2-methylphenylimino)-ethyl]-6-(arylimino)ethyl] (L1 to L5)
- L1 (Ar = 2,6-Me2C6H3)
- L2 (Ar = 2,6-Et2C6H3)
- L3 (Ar = 2,6-iPr2C6H3)
- L4 (Ar = 2,4,6-Me3C6H2)
- L5 (Ar = 2,6-Et2-4-MeC6H2)
3.4. Synthesis of 2-[1-(3-Chloro-4,6-bis((di(4-fluorophenyl)methyl)-2-methylphenylimino)-ethyl]-6-[1-(arylimino)ethyl]pyridylcobalt(II) Chlorides (Co1 to Co5)
- Co1 (Ar = 2,6-Me2C6H3)
- Co2 (Ar = 2,6-Et2C6H3)
- Co3 (Ar = 2,6-iPr2C6H3)
- Co4 (Ar = 2,4,6-Me3C6H2)
- Co5 (Ar = 2,6-Et2-4-MeC6H2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joseph, T.; Azat, S.; Ahmadi, Z.; Moini Jazani, O.; Esmaeili, A.; Kianfar, E.; Haponiuk, J.; Thomas, S. Polyethylene Terephthalate (PET) Recycling: A Review. Case Stud. Chem. Environ. Eng. 2024, 9, 100673. [Google Scholar] [CrossRef]
- Hutanu, D. Recent Applications of Polyethylene Glycols (PEGs) and PEG Derivatives. Modern Chem. Appl. 2014, 2, 1000132. [Google Scholar] [CrossRef]
- Vasile, C. (Ed.) Handbook of Polyolefins, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Göpperl, L.; Pernusch, D.; Schwarz, J.; Paulik, C. Impact of Polymerization Process Parameters on Improved Comonomer Incorporation Behavior in Ziegler-Natta Catalysis. Macromol. React. Eng. 2021, 16, 202100042. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Gibson, V.C.; Hoarau, O.D.; Spitzmesser, S.K.; White, A.J.P.; Williams, D.J. Iron and Cobalt Ethylene Polymerization Catalysts: Variations on the Central Donor. Inorg. Chem. 2003, 42, 3454–3465. [Google Scholar] [CrossRef]
- Goller, A.; Obenauf, J.; Kretschmer, W.; Kempe, R. The Highly Controlled and Efficient Polymerization of Ethylene. Angew. Chem. 2022, 135, e202216464. [Google Scholar] [CrossRef]
- Wang, Z.; Solan, G.A.; Zhang, W.; Sun, W.-H. Carbocyclic-fused NNN-pincer ligands as ring-strain adjustable supports for iron and cobalt catalysts in ethylene oligo-/polymerization. Coord. Chem. Rev. 2018, 363, 92–108. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, S.; Liu, Y.; Kang, X.; Liu, S.; Li, Z.; Braunstein, P. Controlling Polyethylene Molecular Weights and Distributions Using Chromium Complexes Supported by SNN-Tridentate Ligands. Macromolecules 2022, 55, 2433–2443. [Google Scholar] [CrossRef]
- Wang, Z.; Mahmood, Q.; Zhang, W.; Sun, W. Chapter Two—Recent Progress on the Tridentate Iron Complex Catalysts for Ethylene Oligo-/Polymerization. Adv. Organomet. Chem. 2023, 79, 41–86. [Google Scholar]
- Hu, S.; Zhao, Q.; Luo, M.; Xin, S.; Mao, G. Research Progress of Iron Catalysts with N,N,N-Tridentate Ligands for Ethylene Polymerization. ChemistrySelect 2024, 9, e202303611. [Google Scholar] [CrossRef]
- Ma, Z.; Sun, W.-H.; Li, Z.; Shao, C.; Hu, Y.; Li, X. Ethylene polymerization by iron complexes with symmetrical and unsymmetrical ligands. Polym. Int. 2002, 51, 994–997. [Google Scholar] [CrossRef]
- Yu, J.; Huang, W.; Wang, L.; Redshaw, C.; Sun, W.-H. 2-[1-(2,6-Dibenzhydryl-4-methylphenylimino)ethyl]-6-[1-(arylimino)ethyl]pyridylcobalt(ii) dichlorides: Synthesis, characterization and ethylene polymerization behavior. Dalton Trans. 2011, 40, 10209–10214. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Baugh, S.P.D.; Hoarau, O.; Gibson, V.C.; Wass, D.F.; White, A.J.P.; Williams, D.J. The Role of Bulky Substituents in the Polymerization of Ethylene Using Late Transition Metal Catalysts: A Comparative Study of Nickel and Iron Catalyst Systems. Inorg. Chim. Acta 2003, 345, 279–291. [Google Scholar] [CrossRef]
- Appukuttan, V.K.; Liu, Y.; Son, B.C.; Ha, C.-S.; Suh, H.; Kim, I. Iron and Cobalt Complexes of 2,3,7,8-Tetrahydroacridine-4,5(1H,6H)-diimine Sterically Modulated by Substituted Aryl Rings for the Selective Oligomerization to Polymerization of Ethylene. Organometallics 2011, 30, 2285–2294. [Google Scholar] [CrossRef]
- Mitchell, N.E.; Anderson, W.C., Jr.; Long, B.K. Mitigating Chain-Transfer and Enhancing the Thermal Stability of Co-Based Olefin Polymerization Catalysts through Sterically Demanding Ligands. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3990–3995. [Google Scholar] [CrossRef]
- Tomov, A.K.; Gibson, V.C.; Britovsek, G.J.P.; Long, R.J.; Meurs, M.V.; Jones, D.J.; Tellmann, K.P.; Chirinos, J.J. Distinguishing Chain Growth Mechanisms in Metal-catalyzed Olefin Oligomerization and Polymerization Systems: C2H4/C2D4 Cooligomerization/Polymerization Experiments Using Chromium, Iron, and Cobalt Catalysts. Organometallics 2009, 28, 7033–7040. [Google Scholar] [CrossRef]
- Semikolenova, N.V.; Sun, W.-H.; Soshnikov, I.E.; Matsko, M.A.; Kolesova, O.V.; Zakharov, V.A.; Bryliakov, K.P. Origin of “Multisitelike” Ethylene Polymerization Behavior of the Single-Site Nonsymmetrical Bis(imino)pyridine Iron(II) Complex in the Presence of Modified Methylaluminoxane. ACS Catal. 2017, 7, 2868–2877. [Google Scholar] [CrossRef]
- Zada, M.; Guo, L.; Ma, Y.; Zhang, W.; Flisak, Z.; Sun, Y.; Sun, W.-H. Activity and Thermal Stability of Cobalt(II)-Based Olefin Polymerization Catalysts Adorned with Sterically Hindered Dibenzocycloheptyl Groups. Molecules 2019, 24, 2007. [Google Scholar] [CrossRef] [PubMed]
- Small, B.L.; Brookhart, M.; Bennett, A.M.A. Highly Active Iron and Cobalt Catalysts for the Polymerization of Ethylene. J. Am. Chem. Soc. 1998, 120, 4049–4050. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Gibson, V.C.; Kimberley, B.S.; Maddox, P.J.; McTavish, S.J.; Solan, G.A.; White, A.J.P.; Williams, D.J. Novel olefin polymerization catalysts based on iron and cobalt. Chem. Commun. 1998, 7, 849–850. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Bruce, M.; Gibson, V.C.; Kimberley, B.S.; Maddox, P.J.; Mastroianni, S.; McTavish, S.J.; Redshaw, C.; Solan, G.A.; Strömberg, S.; et al. Iron and Cobalt Ethylene Polymerization Catalysts Bearing 2,6-Bis(Imino)Pyridyl Ligands: Synthesis, Structures, and Polymerization Studies. J. Am. Chem. Soc. 1999, 121, 8728–8740. [Google Scholar] [CrossRef]
- Yan, Y.; Yuan, S.-F.; Liu, M.; Solan, G.A.; Ma, Y.-P.; Liang, T.-L.; Sun, W.-H. Investigating the Effects of Para-methoxy Substitution in Sterically Enhanced Unsymmetrical Bis(arylimino)pyridine- cobalt Ethylene Polymerization Catalysts. Chin. J. Polym. Sci. 2022, 40, 266–279. [Google Scholar] [CrossRef]
- Mahmood, Q.; Ma, Y.; Hao, X.; Sun, W.-H. Substantially Enhancing the Catalytic Performance of Bis(imino)pyridylcobaltous Chloride Pre-Catalysts Adorned with Benzhydryl and Nitro Groups for Ethylene Polymerization. Appl. Organomet. Chem. 2019, 33, e4857. [Google Scholar] [CrossRef]
- Liu, M.; Jiang, S.; Ma, Y.; Solan, G.A.; Sun, Y.; Sun, W.-H. CF3O-Functionalized Bis(arylimino)pyridine–Cobalt Ethylene Polymerization Catalysts: Harnessing Solvent Effects on Performance and Polymer Properties. Organometallics 2022, 41, 3237–3248. [Google Scholar] [CrossRef]
- Lai, J.; Zhao, W.; Yang, W.; Redshaw, C.; Liang, T.; Liu, Y.; Sun, W.-H. 2-[1-(2,4-Dibenzhydryl-6-methylphenylimino)ethyl]-6-[1-(arylimino)ethyl]pyridylcobalt(II) Dichlorides: Synthesis, Characterization, and Ethylene Polymerization Behavior. Polym. Chem. 2012, 3, 787–793. [Google Scholar] [CrossRef]
- Helldörfer, M.; Milius, W.; Alt, H.G. The Influence of Halogen Substituents at the Ligand Framework of (α-Diimine)Nickel(II) Catalyst Precursors on Their Behavior in Ethylene Oligomerization and Polymerization. J. Mol. Catal. A Chem. 2003, 197, 1–13. [Google Scholar] [CrossRef]
- Bariashir, C.; Zhang, R.; Vignesh, A.; Ma, Y.; Liang, T.; Sun, W. Enhancing Ethylene Polymerization of NNN-Cobalt(II) Precatalysts Adorned with a Fluoro-substituent. ACS Omega 2021, 6, 4448–4460. [Google Scholar] [CrossRef]
- Tahir, F.; Ma, Y.; Mahmood, Q.; Ren, G.; Khalid, A.; Wang, Y.; Zou, S.; Liang, T.; Sun, W.-H. Achieving Linear α-Macro-olefins in Ethylene Polymerization through Precisely Tuned Bis(imino)pyridylcobalt Precatalysts with Steric and Electronic Parameters. Precision Chem. 2024, 2, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ma, Y.; Liu, T.; Zhang, Q.; Solan, G.A.; Liang, T.; Sun, W.-H. Exploring Fluoride Effects in Sterically Enhanced Cobalt Ethylene Polymerisation Catalysts; A Combined Experimental and DFT Study. RSC Adv. 2023, 13, 14–24. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Du, S.; Guo, C.Y.; Hao, X.; Sun, W.-H. 2-(1-(2,4-Bis((di(4- fluorophenyl)methyl)-6-methylphenylimino)ethyl)-6-(1-(arylimino)ethyl)pyridylmetal (Iron or Cobalt) Complexes: Synthesis, Characterization, and Ethylene Polymerization Behavior. Macromol. Chem. Phys. 2014, 215, 1797–1809. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Z.; Han, M.; Xiang, J.; Solan, G.A.; Ma, Y.; Liang, T.; Sun, W.-H. Fluorinated Cobalt Catalysts and Their Use in Forming Narrowly Dispersed Polyethylene Waxes of High Linearity and Incorporating Vinyl Functionality. Catal. Sci. Technol. 2021, 11, 656–670. [Google Scholar] [CrossRef]
- Beaufort, L.; Benvenuti, F.; Noels, A.F. Iron(II)–Ethylene Polymerization Catalysts Bearing 2,6-Bis(imino)pyrazine Ligands: Part I. Synthesis and Characterization. J. Mol. Catal. A Chem. 2006, 260, 210–214. [Google Scholar] [CrossRef]
- Zhang, Q.; Zuo, Z.; Ma, Y.; Liang, T.; Yang, X.; Sun, W.-H. Fluorinated 2,6-Bis(arylimino)pyridyl Iron Complexes Targeting Bimodal Dispersive Polyethylenes: Probing Chain Termination Pathways via a Combined Experimental and DFT Study. Dalton Trans. 2022, 51, 8290–8302. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Zhang, Q.; Zou, S.; Ma, Y.; Solan, G.A.; Zhang, W.; Sun, W.-H. Exploring Long Range para-Phenyl Effects in Unsymmetrically Fused bis(imino)pyridine-Cobalt Ethylene Polymerization Catalysts. Catalysts 2023, 13, 1387. [Google Scholar] [CrossRef]
- Matsko, M.; Semikolenova, N.; Zakharov, V.; Soshnikov, I.; Shundrina, I.; Sun, W. Formation of Branched Polyethylenes by Ethylene Homopolymerization Using LNiBr2 Homo- and Heterogeneous Precatalysts: Interpretation of the Polymer Structures in Comparison with Commercial LLDPE. J. Appl. Polym. Sci. 2021, 138, 50436. [Google Scholar] [CrossRef]
- Chiefari, J.; Rizzardo, E. Control of Free-Radical Polymerization by Chain Transfer Methods. Handb. Radic. Polym. 2002, 629, 690. [Google Scholar]
- Blencowe, A.; Tan, J.F.; Goh, T.K.; Qiao, G.G. Core Cross-Linked Star Polymers via Controlled Radical Polymerisation. Polymer 2009, 50, 5–32. [Google Scholar] [CrossRef]
- Valencia, L.; Enríquez-Medrano, F.; Textle, H.; Soriano, F.; Lopez Gonzalez, H.R.; Thomas, C.; Gámez, F.; Olivares Romero, J.L.; Gómez, R.E.D. The Influence of Co-catalyst in the Polymerization of 1,3-butadiene Catalyzed by Neodymium Chloride Tripentanolate. J. Mex. Chem. Soc. 2016, 60, 141–147. [Google Scholar] [CrossRef]
- Jiang, T.; Ning, Y.; Hu, W.; Wang, L.; Huang, Z.; Liu, X. Ethylene Polymerization by Novel Highly Active Iron/Acetyl(imino)pyridyl Complex. Chin. Sci. Bull. 2006, 51, 2197–2200. [Google Scholar] [CrossRef]
- Sun, W.-H.; Hao, P.; Zhang, S.; Shi, Q.; Zuo, W.; Tang, X.; Lu, X. Iron(II) and Cobalt(II) 2-(Benzimidazolyl)-6-(1-(arylimino)ethyl)pyridyl Complexes as Catalysts for Ethylene Oligomerization and Polymerization. Organometallics 2007, 26, 2720–2734. [Google Scholar] [CrossRef]
- Zhao, W.; Yu, J.; Song, S.; Yang, W.; Liu, H.; Hao, X.; Redshaw, C.; Sun, W.-H. Controlling the ethylene polymerization parameters in iron pre-catalysts of the type 2-[1-(2,4-dibenzhydryl-6-methylphenylimino)ethyl]-6-[1-(arylimino)ethyl] pyridyliron dichloride. Polymer 2012, 53, 130–137. [Google Scholar] [CrossRef]
- Han, M.; Zuo, Z.; Ma, Y.; Solan, G.A.; Hu, X.; Liang, T.; Sun, W.-H. Bis(imino)-6,7-dihydro-5H-quinoline-cobalt complexes as highly active catalysts for the formation of vinyl-terminated PE waxes; steps towards inhibiting deactivation pathways through targeted ligand design. RSC Adv. 2021, 11, 39869–39878. [Google Scholar] [CrossRef]
- Liu, T.; Ma, Y.; Solan, G.A.; Sun, Y.; Sun, W.-H. Unimodal Polyethylenes of High Linearity and Narrow Dispersity by Using Ortho-4,4′-Dichlorobenzhydryl-Modified Bis(imino)pyridyl-Iron Catalysts. New J. Chem. 2023, 47, 5786–5795. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]

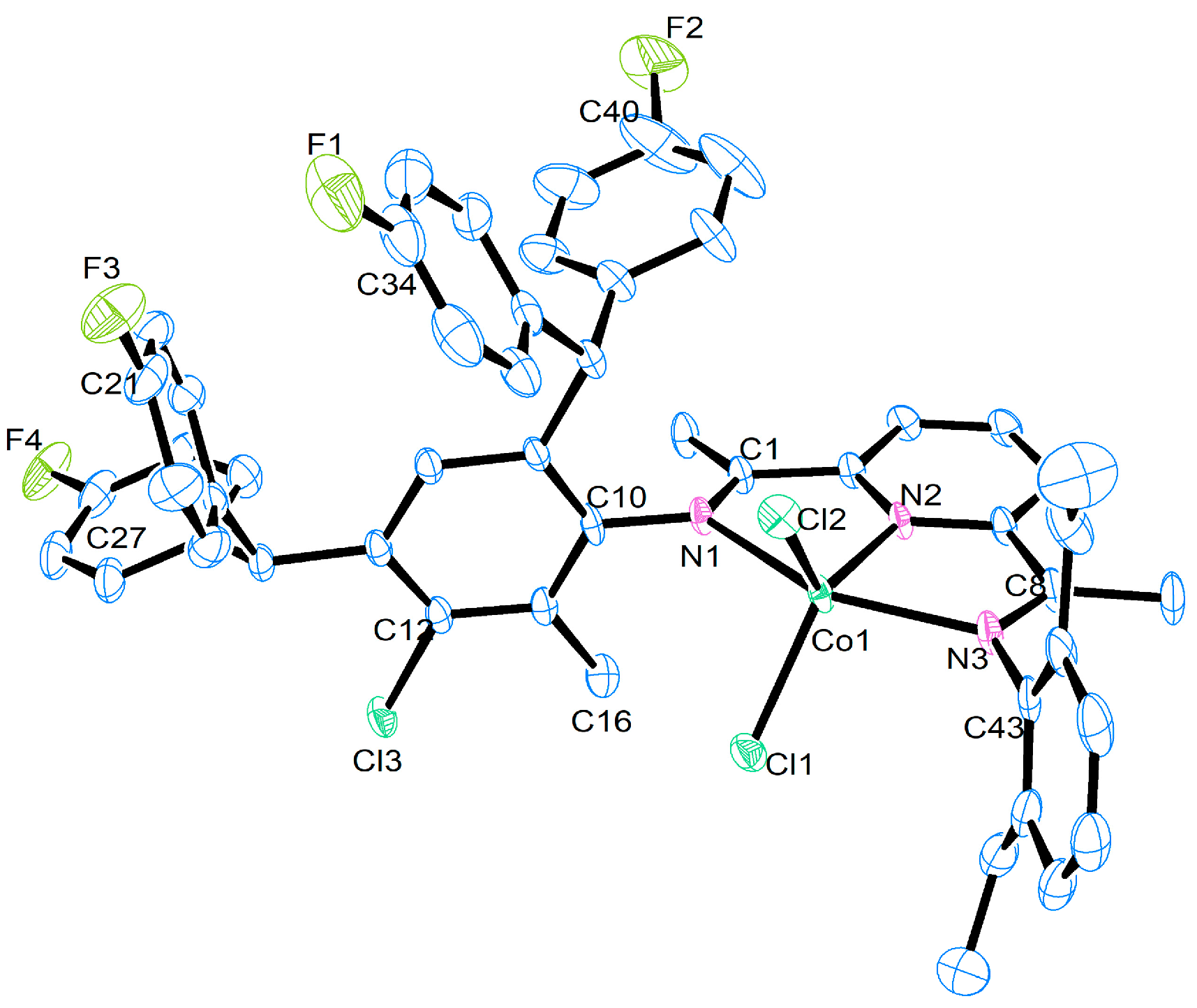
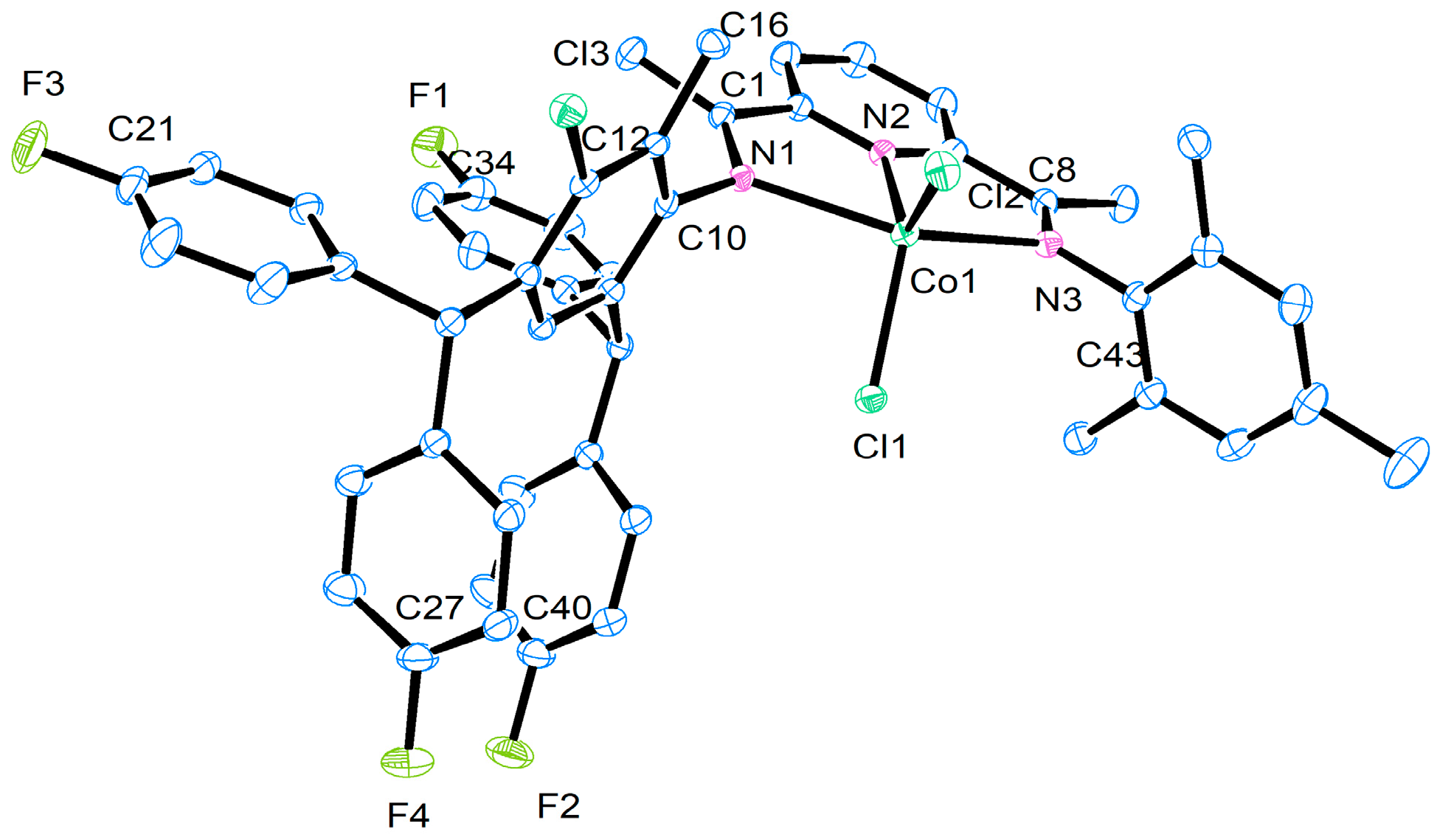
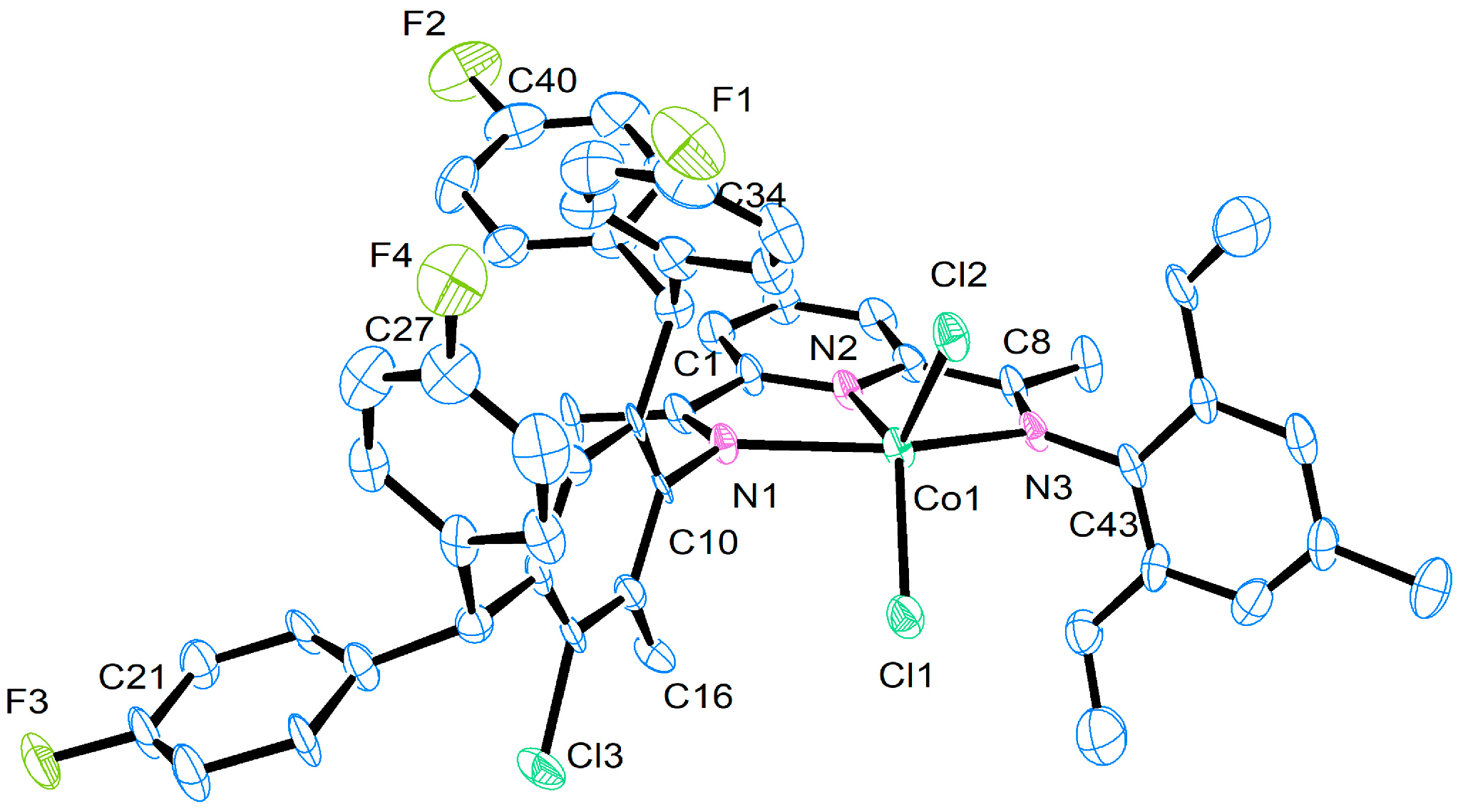


| Selected Bond Lengths (Å) | ||||
|---|---|---|---|---|
| Co1 | Co2 | Co4 | Co5 | |
| Co(1)-N(1) | 2.2918 (15) | 2.271 (15) | 2.3090 (19) | 2.294 (8) |
| Co(1)-N(2) | 2.0321 (15) | 2.033 (5) | 2.0362 (19) | 2.021 (8) |
| Co(1)-N(3) | 2.2398 (15) | 2.228 (5) | 2.204 (2) | 2.233 (9) |
| Co(1)-Cl(1) | 2.2470 (5) | 2.2542 (18) | 2.2567 (7) | 2.242 (3) |
| Co(1)-Cl(2)) | 2.2689 (5) | 2.2538 (18) | 2.2979 (7) | 2.259 (3) |
| N(1)-C(10) | 1.432 (2) | 1.440 (7) | 1.435 (3) | 1.453 (11) |
| N(3)-C(43) | 1.435 (2) | 1.447 (8) | 1.445 (3) | 1.410 (14) |
| F(1)-C(34) | 1.361 (2) | 1.354 (10) | 1.357 (3) | 1.405 (16) |
| Selected Bond Angles (°) | ||||
| N(1)-Co(1)-N(2) | 74.57 (6) | 74.40 (18) | 73.65 (7) | 75.5 (3) |
| N(1)-Co(1)-N(3) | 149.96 (6) | 150.44 (18) | 150.53 (7) | 101.8 (2) |
| N(2)-Co(1)-N(3) | 75.46 (6) | 76.05 (19) | 76.90 (8) | 75.1 (3) |
| N(1)-Co(1)-Cl(1) | 97.67 (4) | 92.86 (14) | 96.86 (5) | 92.2 (2) |
| N(2)-Co(1)-Cl(1) | 122.45 (4) | 123.07 (16) | 122.57 (6) | 126.4 (3) |
| N(3)-Co(1)-Cl(1) | 99.91 (4) | 102.52 (16) | 97.92 (5) | 101.8 (2) |
| N(2)-Co(1)-Cl(2) | 125.16 (4) | 123.49 (16) | 126.23 (5) | 118.2 (3) |
| N(3)-Co(1)-Cl(2) | 96.50 (4) | 96.96 (2) | 105.59 (6) | 96.2 (2) |
| Cl(1)-Co(1)-Cl(2) | 112.39 (19) | 113.26 (7) | 110.44 (3) | 115.32 (12) |
| N(1)-Co(1)-Cl(2) | 99.05 (4) | 99.93 (14) | 92.82 (5) | 101.3 (2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogbe, E.; Ma, Y.; Wang, Y.; Gao, J.; Sun, Y.; Sun, W.-H. Finely-Tuned Bis(imino)pyridylcobalt Complexes Enhance Ethylene Polymerization: The Role of Bulky and Halogen Substituents. Molecules 2025, 30, 859. https://doi.org/10.3390/molecules30040859
Ogbe E, Ma Y, Wang Y, Gao J, Sun Y, Sun W-H. Finely-Tuned Bis(imino)pyridylcobalt Complexes Enhance Ethylene Polymerization: The Role of Bulky and Halogen Substituents. Molecules. 2025; 30(4):859. https://doi.org/10.3390/molecules30040859
Chicago/Turabian StyleOgbe, Elizabeth, Yanping Ma, Yizhou Wang, Jiahao Gao, Yang Sun, and Wen-Hua Sun. 2025. "Finely-Tuned Bis(imino)pyridylcobalt Complexes Enhance Ethylene Polymerization: The Role of Bulky and Halogen Substituents" Molecules 30, no. 4: 859. https://doi.org/10.3390/molecules30040859
APA StyleOgbe, E., Ma, Y., Wang, Y., Gao, J., Sun, Y., & Sun, W.-H. (2025). Finely-Tuned Bis(imino)pyridylcobalt Complexes Enhance Ethylene Polymerization: The Role of Bulky and Halogen Substituents. Molecules, 30(4), 859. https://doi.org/10.3390/molecules30040859