
In Vitro and In Silico Studies of Maculosin as a Melanogenesis and Tyrosinase Inhibitor



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Biological Evaluations
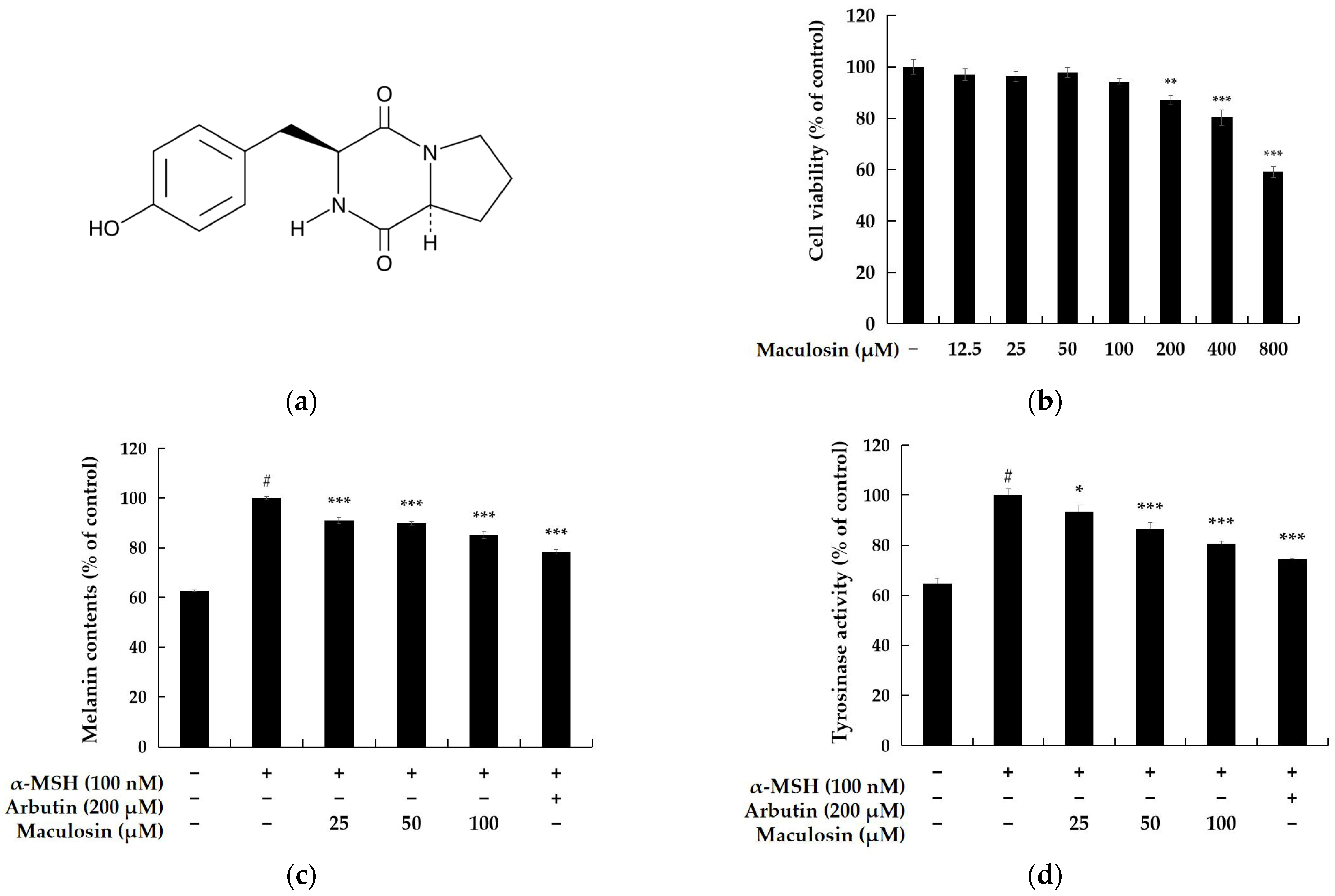
2.1.1. Cell Viability
2.1.2. Melanin Contents and Tyrosinase Inhibiton Activity of Maculosin
2.2. Molecular Properties and Drug Likeness
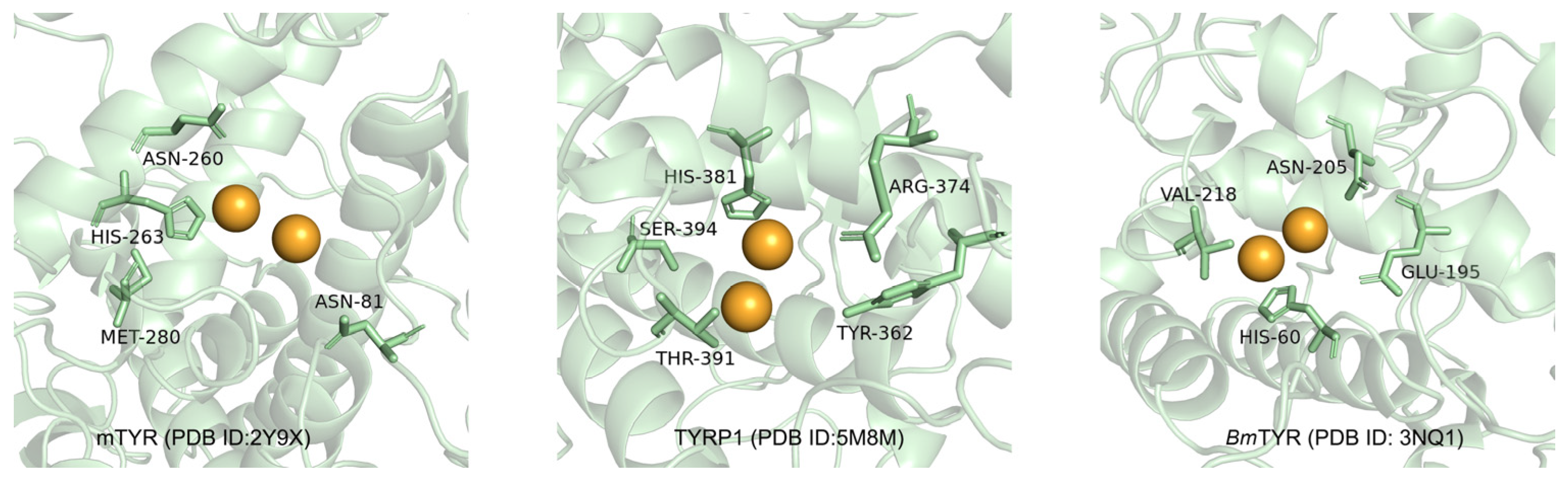
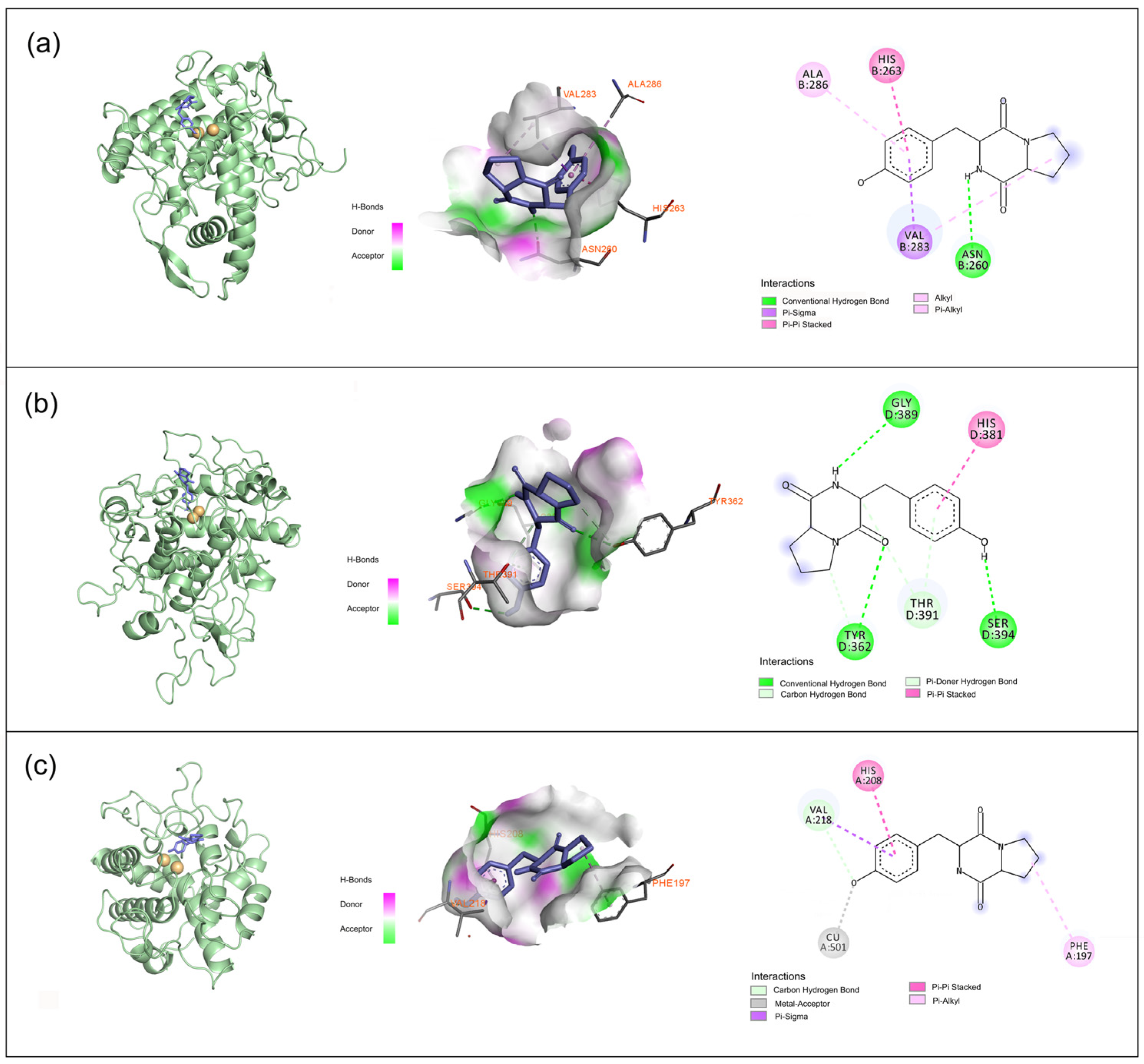
2.3. Molecular Docking Simulation
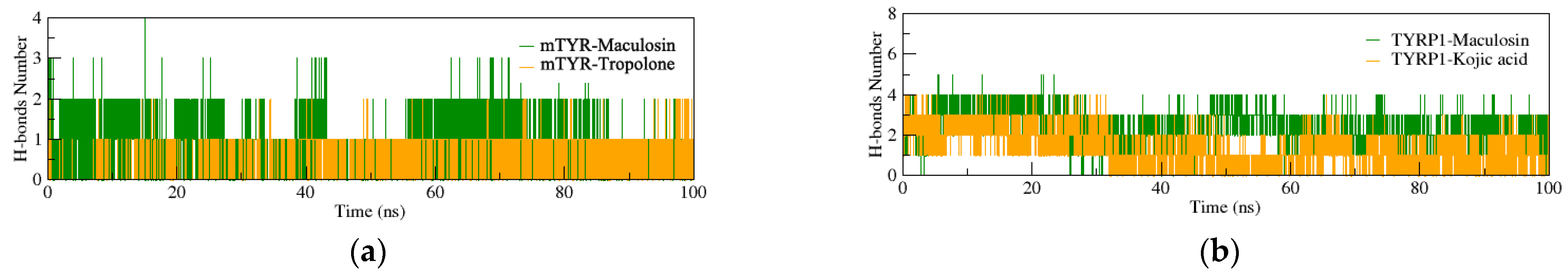
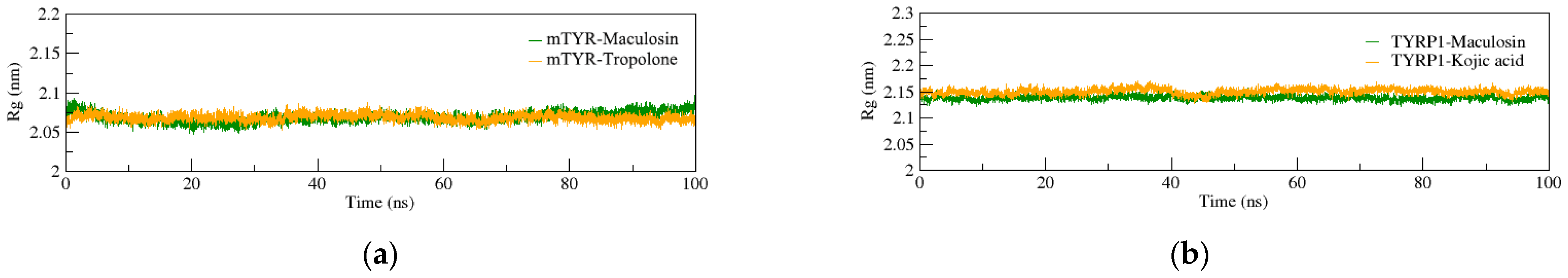
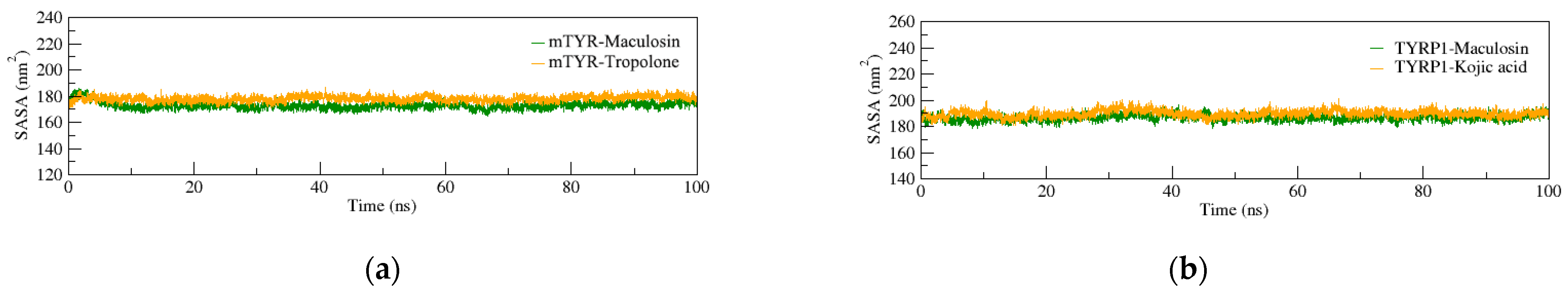
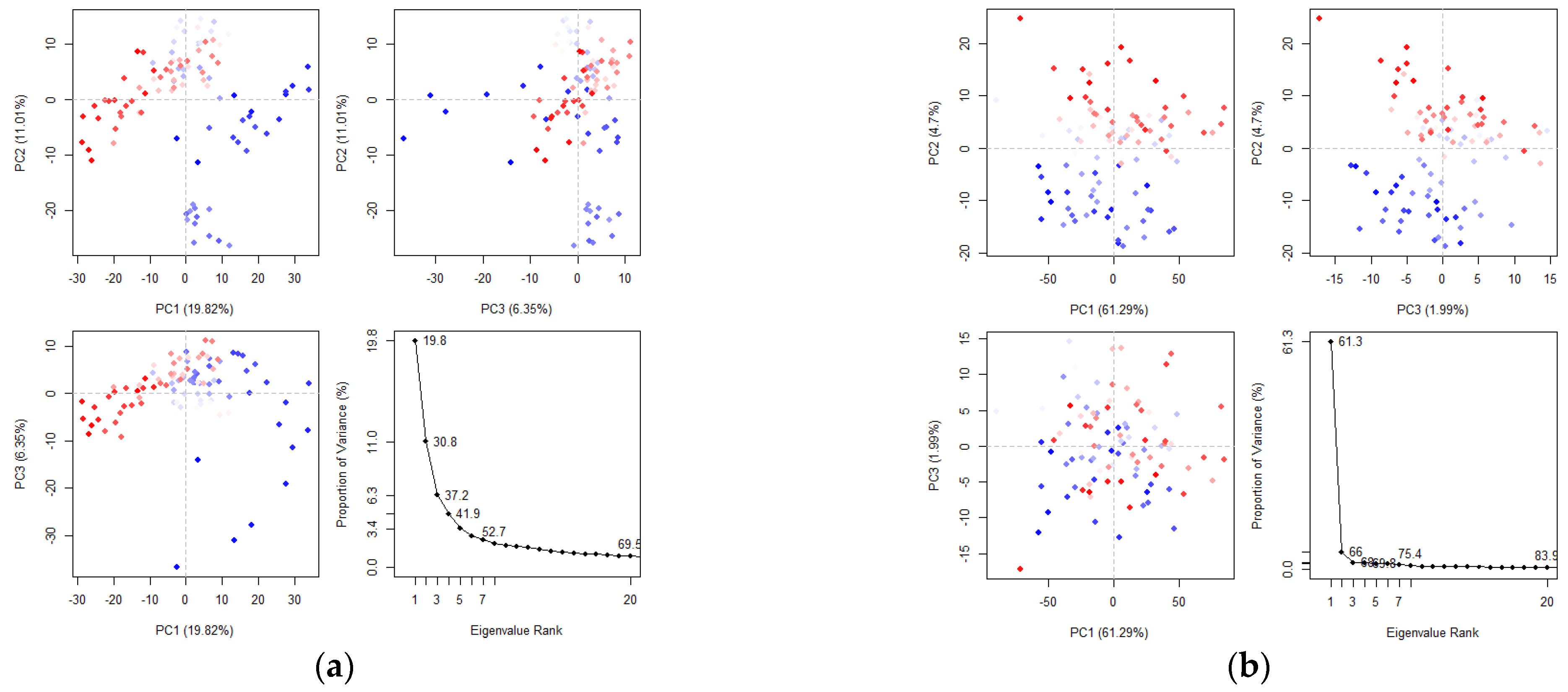
2.4. Molecular Dynamics (MD) Simulation
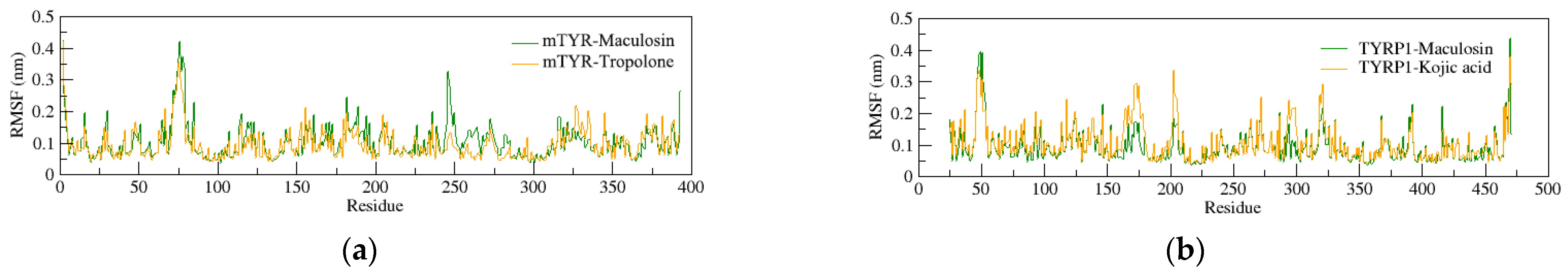
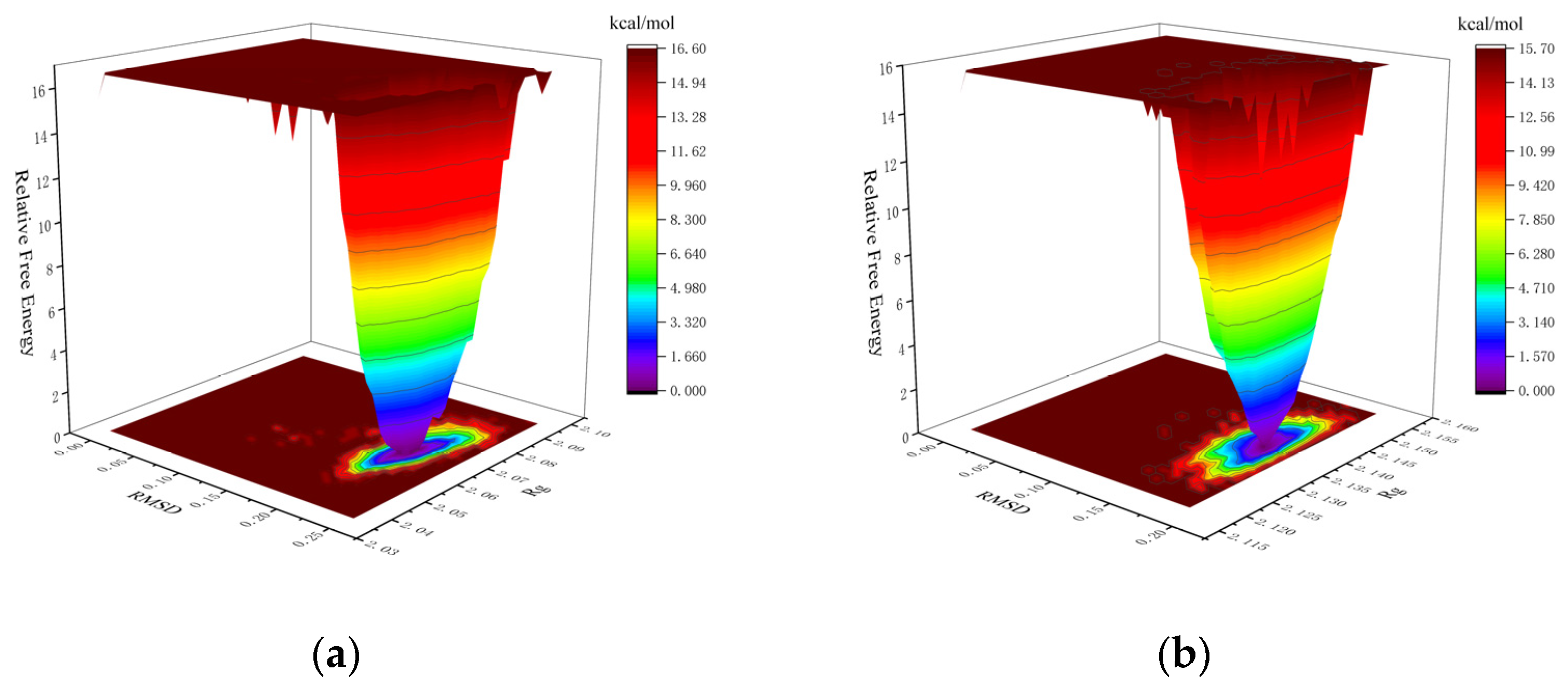
2.4.1. Structural Stability of the Complexes
2.4.2. Dynamic Structural Conformational Analysis of the Complexes
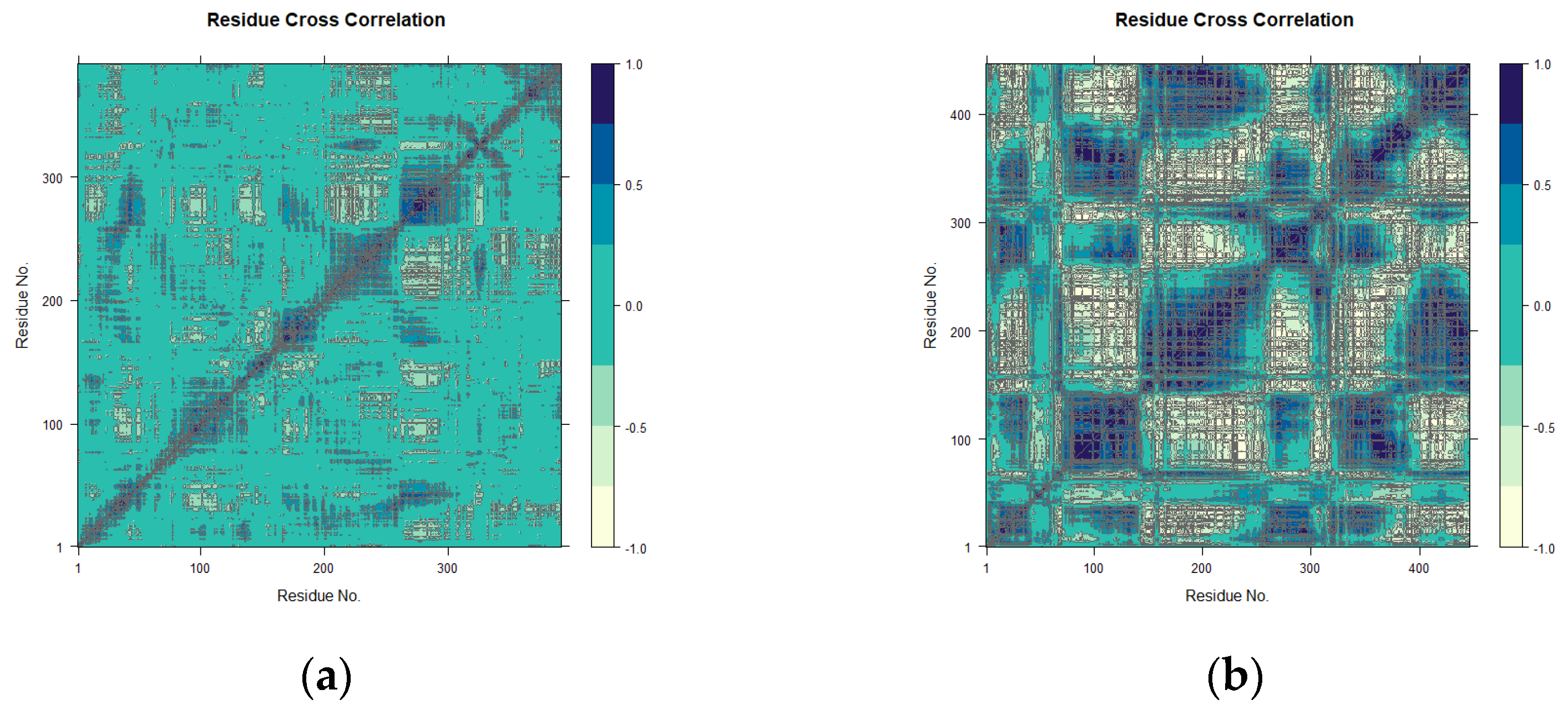
2.4.3. Principal Component and Dynamical Residues Cross-Correlation Binding Analysis
2.4.4. Binding Free Energy Evaluation of the Complexes
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Cell Culture and Materials
3.3. Melanin Contents of Maculosin
3.4. Tyrosinase Inhibition Activity of Maculosin
3.5. Statistical Analysis
3.6. Computational Methodology
3.6.1. Molecular Properties and Drug Likeness
3.6.2. Molecular Docking Simulation
3.6.3. Molecular Dynamics (MD) Simulations
3.6.4. PCA and DCCM Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, H.W.; Kohli, I.; Ruvolo, E. Impact of visible light on skin health: The role of antioxidants and free radical quenchers in skin protection. J. Am. Acad. Dermatol. 2022, 86, S27–S37. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, W.; Gu, Z. Recent advances and progress on melanin: From source to application. Int. J. Mol. Sci. 2023, 24, 4360. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, H.; Soundhararajan, R.; Srinivasan, H. Interaction of various types of bisphenols with enzymes involved in melanin synthesis. Toxicol. Environ. Health Sci. 2021, 14, 19–24. [Google Scholar] [CrossRef]
- Hushcha, Y.; Blo, I.; Oton-Gonzalez, L. microRNAs in the Regulation of Melanogenesis. Int. J. Mol. Sci. 2021, 22, 6104. [Google Scholar] [CrossRef]
- Kapoor, R.; Dhatwalia, S.K.; Kumar, R. Emerging role of dermal compartment in skin pigmentation: Comprehensive review. J. Eur. Acad. Dermatol. 2020, 34, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Fisher, D.E.; Rizos, H. Biology of melanocytes and primary melanoma. In Cutaneous Melanoma; Taylor & Francis: Abingdon, UK, 2020; pp. 3–40. [Google Scholar] [CrossRef]
- Lee, A.Y. Skin pigmentation abnormalities and their possible relationship with skin aging. Int. J. Mol. Sci. 2021, 22, 3727. [Google Scholar] [CrossRef]
- Wang, Y.; Viennet, C.; Robin, S. Precise role of dermal fibroblasts on melanocyte pigmentation. J. Dermatol. Sci. 2017, 88, 159–166. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan, K.M.T. A comprehensive review on tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Qian, W.; Liu, W.; Zhu, D. Natural skin whitening compounds for the treatment of melanogenesis. Exp. Ther. Med. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Huang, T.; Duan, Y.; Zou, Y. NRPS protein MarQ catalyzes flexible adenylation and specific S-methylation. ACS Chem. Biol. 2018, 13, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 2016, 33, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Gondry, M.; Jacques, I.B.; Thai, R. A comprehensive overview of the cyclodipeptide synthase family enriched with the characterization of 32 new enzymes. Front. Microbiol. 2018, 9, 46. [Google Scholar] [CrossRef]
- Borgman, P.; Lopez, R.D.; Lane, A.L. The expanding spectrum of diketopiperazine natural product biosynthetic pathways containing cyclodipeptide synthases. Org. Biomol. Chem. 2019, 17, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.A.; Niwa, K.; Perlatti, B. Genome mining for unknown–unknown natural products. Nat. Chem. Biol. 2023, 19, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Belin, P. The nonribosomal synthesis of diketopiperazines in tRNA-dependent cyclodipeptide synthase pathways. Nat. Prod. Rep. 2012, 29, 961–979. [Google Scholar] [CrossRef] [PubMed]
- Harken, L.; Li, S.M. Modifications of diketopiperazines assembled by cyclodipeptide synthases with cytochrome P450 enzymes. Appl. Microbiol. Biotechnol. 2021, 105, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Andreas, M.P.; Giessen, T.W. Cyclodipeptide oxidase is an enzyme filament. Nat. Commun. 2024, 15, 3574. [Google Scholar] [CrossRef]
- Lautru, S.; Gondry, M.; Genet, R. The albonoursin gene cluster of S. noursei: Biosynthesis of diketopiperazine metabolites independent of nonribosomal peptide synthetases. Chem. Biol. 2002, 9, 1355–1364. [Google Scholar] [CrossRef]
- Belin, P.; Le, D.M.H.; Fielding, A. Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7426–7431. [Google Scholar] [CrossRef] [PubMed]
- Cryle, M.J.; Bell, S.G.; Schlichting, I. Structural and biochemical characterization of the cytochrome P450 CypX (CYP134A1) from Bacillus subtilis: A cyclo-L-leucyl-L-leucyl dipeptide oxidase. Biochemistry 2010, 49, 7282–7296. [Google Scholar] [CrossRef]
- Vior, N.M.; Lacret, R.; Chandra, G. Discovery and biosynthesis of the antibiotic bicyclomycin in distantly related bacterial classes. Appl. Environ. Microbiol. 2018, 84, e02828-17. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; von Tesmar, A.M.; Marahiel, M.A. Insights into the generation of structural diversity in a tRNA-dependent pathway for highly modified bioactive cyclic dipeptides. Chem. Biol. 2013, 20, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Grant, T.M.; Rennison, D.; Cervin, G. Towards eco-friendly marine antifouling biocides–Nature inspired tetrasubstituted 2, 5-diketopiperazines. Sci. Total Environ. 2022, 812, 152487. [Google Scholar] [CrossRef] [PubMed]
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M. Cyclic dipeptides: The biological and structural landscape with special focus on the anti-cancer proline-based scaffold. Biomolecules 2021, 11, 1515. [Google Scholar] [CrossRef] [PubMed]
- Winyakul, C.; Phutdhawong, W.; Tamdee, P. 2, 5-diketopiperazine derivatives as potential anti-influenza (H5N2) agents: Synthesis, biological evaluation, and molecular docking study. Molecules 2022, 27, 4200. [Google Scholar] [CrossRef]
- Jia, J.; Yao, J.; Kong, J. 2, 5-Diketopiperazines: A review of source, synthesis, bioactivity, structure, and MS fragmentation. Curr. Med. Chem. 2023, 30, 1060–1085. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lu, R.; Chen, N. Research Progress on Activity and Biosynthesis of Diketopiperazines. Mini-Rev. Org. Chem. 2024, 21, 891–906. [Google Scholar] [CrossRef]
- Adamczeski, M.; Reed, A.R.; Crews, P. New and known diketopiperazines from the Caribbean sponge, Calyx cf. podatypa. J. Nat. Prod. 1995, 58, 201–208. [Google Scholar] [CrossRef] [PubMed]
- de Moura, G.G.D.; de Barros, A.V.; Machado, F. Endophytic bacteria from strawberry plants control gray mold in fruits via production of antifungal compounds against Botrytis cinerea L. Microbiol. Res. 2021, 251, 126793. [Google Scholar] [CrossRef]
- Xu, Y.; Liang, X.; Hyun, C.G. Isolation, Characterization, Genome Annotation, and Evaluation of Tyrosinase Inhibitory Activity in Secondary Metabolites of Paenibacillus sp. JNUCC32: A Comprehensive Analysis through Molecular Docking and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2024, 25, 2213. [Google Scholar] [CrossRef] [PubMed]
- Paudel, B.; Maharjan, R.; Rajbhandari, P. Maculosin, a non-toxic antioxidant compound isolated from Streptomyces sp. KTM18. Pharm. Biol. 2021, 59, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ku, S.K.; Park, S. Inhibitory effect of three diketopiperazines from marine-derived bacteria on HMGB1-induced septic responses in vitro and in vivo. Am. J. Chin. Med. 2016, 44, 1145–1166. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Puopolo, G.; Perazzolli, M. Cyclo (L-PRO-L-TYR), The fungicide isolated from Lysobacter capsici AZ78: A structure–activity relationship study. Chem. Heterocycl. Compd. 2014, 50, 290–295. [Google Scholar] [CrossRef]
- Sekino, Y.; Yamamoto, I.; Watanabe, M. Cyclo (L-Pro-L-Tyr) isolated from the human skin commensal Corynebacterium tuberculostearicum inhibits tyrosinase. Int. J. Mol. Sci. 2024, 25, 7365. [Google Scholar] [CrossRef] [PubMed]
- Goenka, S.; Simon, R.S. Inhibitory Effects of the Bioactive Thermorubin Isolated from the Fungus Thermoactinomyces antibioticus on Melanogenesis. Cosmetics 2020, 7, 61. [Google Scholar] [CrossRef]
- Pintus, F.; Matos, M.J.; Vilar, S. New insights into highly potent tyrosinase inhibitors based on 3-heteroarylcoumarins: Anti-melanogenesis and antioxidant activities, and computational molecular modeling studies. Bioorgan. Med. Chem. 2017, 25, 1687–1695. [Google Scholar] [CrossRef]
- Nazir, Y.; Rafique, H.; Roshan, S. Molecular docking, synthesis, and tyrosinase inhibition activity of acetophenone amide: Potential inhibitor of melanogenesis. BioMed Res. Int. 2022, 2022, 1040693. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M. Structure of human tyrosinase related protein 1 reveals a binuclear zinc active site important for melanogenesis. Angew. Chem. Int. Ed. 2017, 56, 9812–9815. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Faisal, S.; Ahmad, S. Structure-based virtual screening of furan-1, 3, 4-oxadiazole tethered N-phenylacetamide derivatives as novel class of hTYR and hTYRP1 inhibitors. Pharmaceuticals 2023, 16, 344. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liang, X.; Hyun, C.G. Discovery of Indole–Thiourea Derivatives as Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Kinetic Studies, and In Silico Analysis. Int. J. Mol. Sci. 2024, 25, 9636. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Kirschner, K.N. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11· 23S subdomain. J. Mol. Model. 2013, 19, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Post, M.; Wolf, S.; Stock, G. Principal component analysis of nonequilibrium molecular dynamics simulations. J. Chem. Phys. 2019, 150, 204110. [Google Scholar] [CrossRef] [PubMed]
- Meeuwsen, S.M.; Hodac, A.N.; Adams, L.M. Investigation of intrinsic dynamics of enzymes involved in metabolic pathways using coarse-grained normal mode analysis. Cogent Biol. 2017, 3, 1291877. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Liang, X.; Kim, H.-M.; Hyun, C.-G. In Vitro and In Silico Studies of Maculosin as a Melanogenesis and Tyrosinase Inhibitor. Molecules 2025, 30, 860. https://doi.org/10.3390/molecules30040860
Xu Y, Liang X, Kim H-M, Hyun C-G. In Vitro and In Silico Studies of Maculosin as a Melanogenesis and Tyrosinase Inhibitor. Molecules. 2025; 30(4):860. https://doi.org/10.3390/molecules30040860
Chicago/Turabian StyleXu, Yang, Xuhui Liang, Hyeon-Mi Kim, and Chang-Gu Hyun. 2025. "In Vitro and In Silico Studies of Maculosin as a Melanogenesis and Tyrosinase Inhibitor" Molecules 30, no. 4: 860. https://doi.org/10.3390/molecules30040860
APA StyleXu, Y., Liang, X., Kim, H.-M., & Hyun, C.-G. (2025). In Vitro and In Silico Studies of Maculosin as a Melanogenesis and Tyrosinase Inhibitor. Molecules, 30(4), 860. https://doi.org/10.3390/molecules30040860