
Clicked H-Shaped Arylopeptoids


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
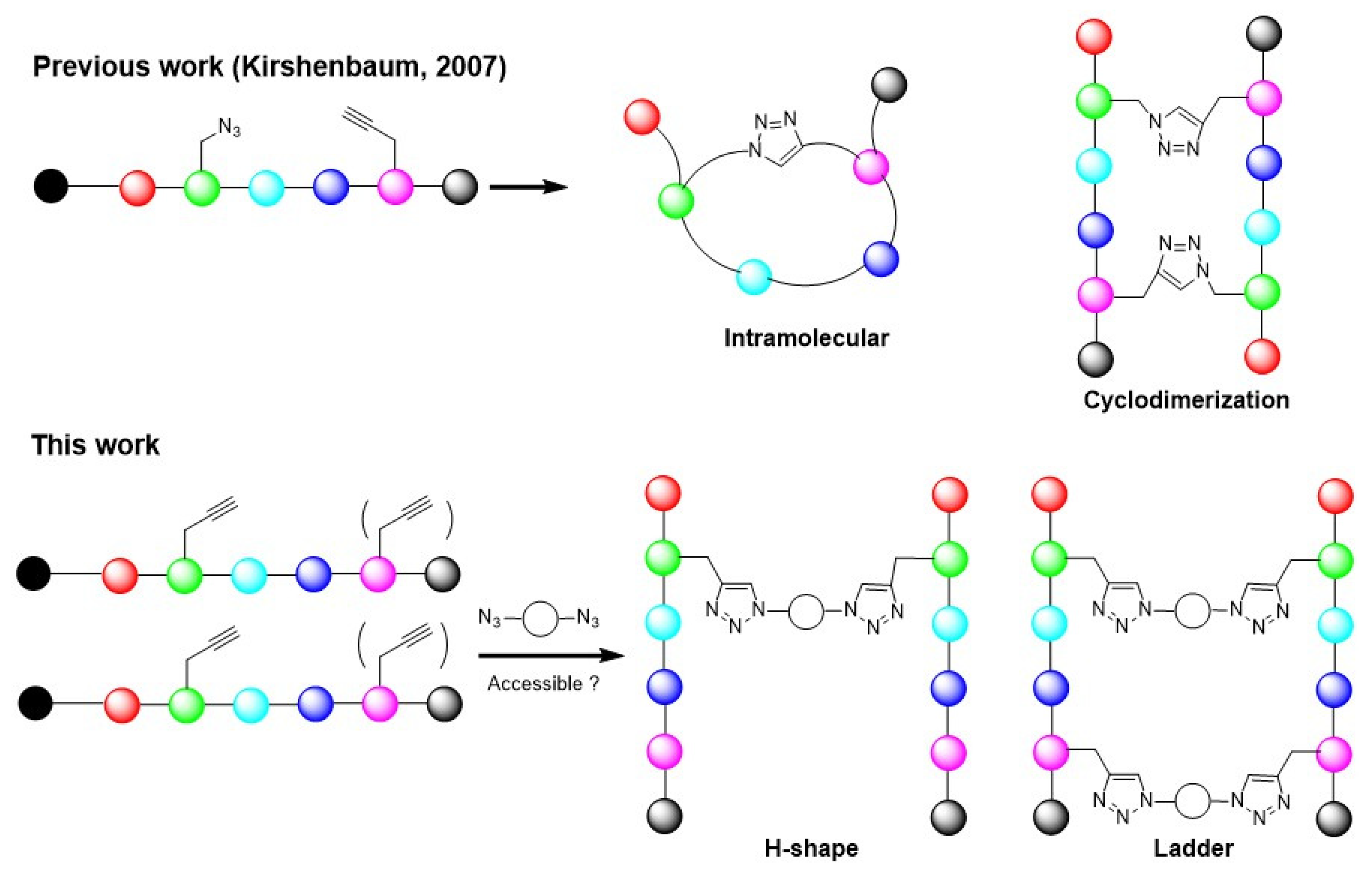
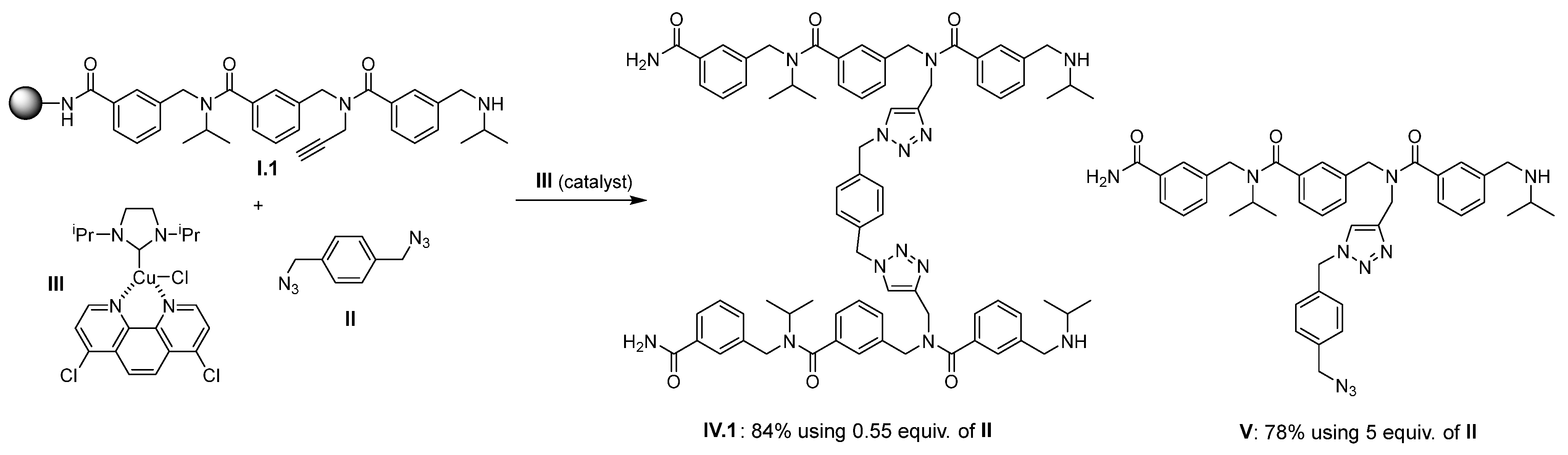
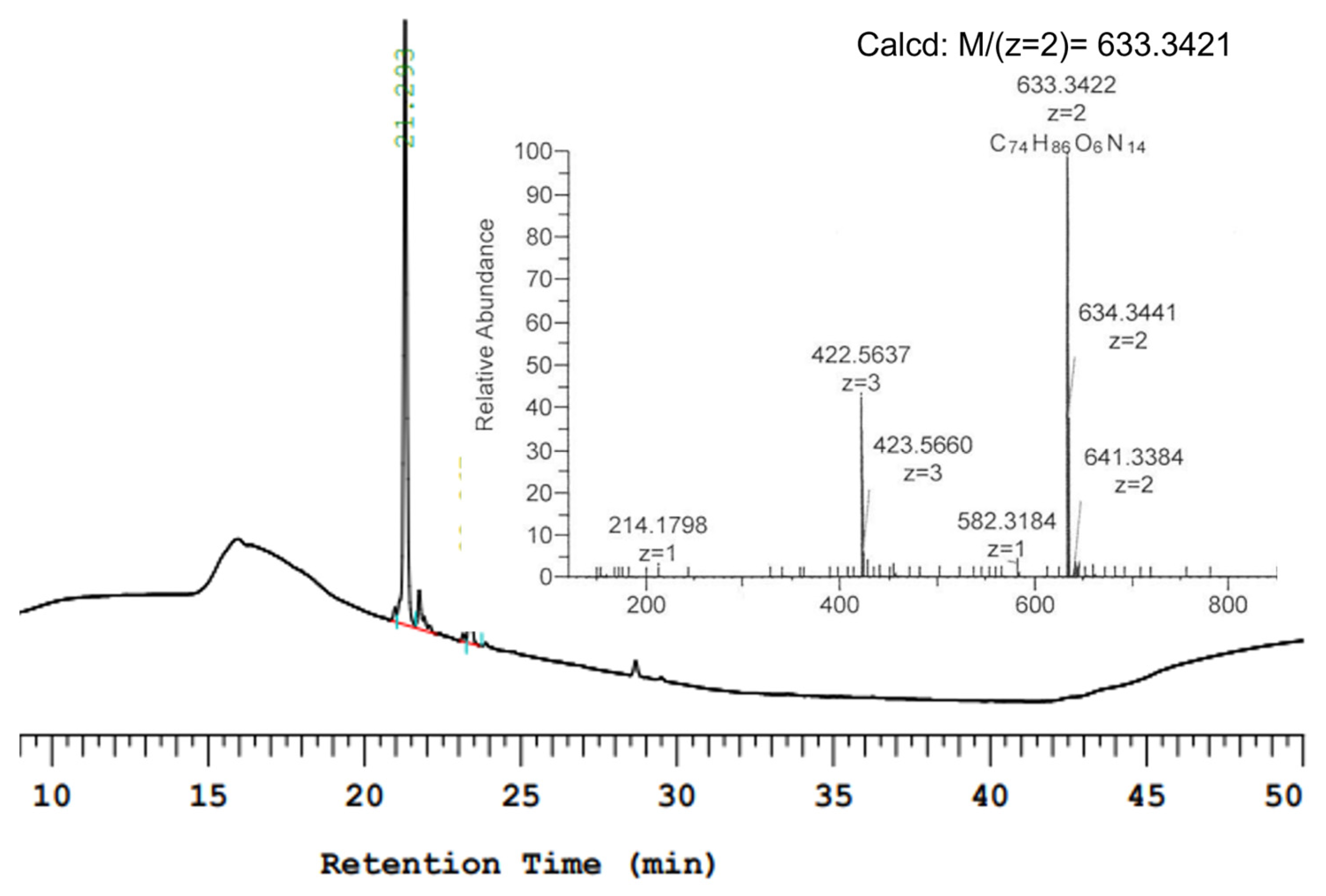
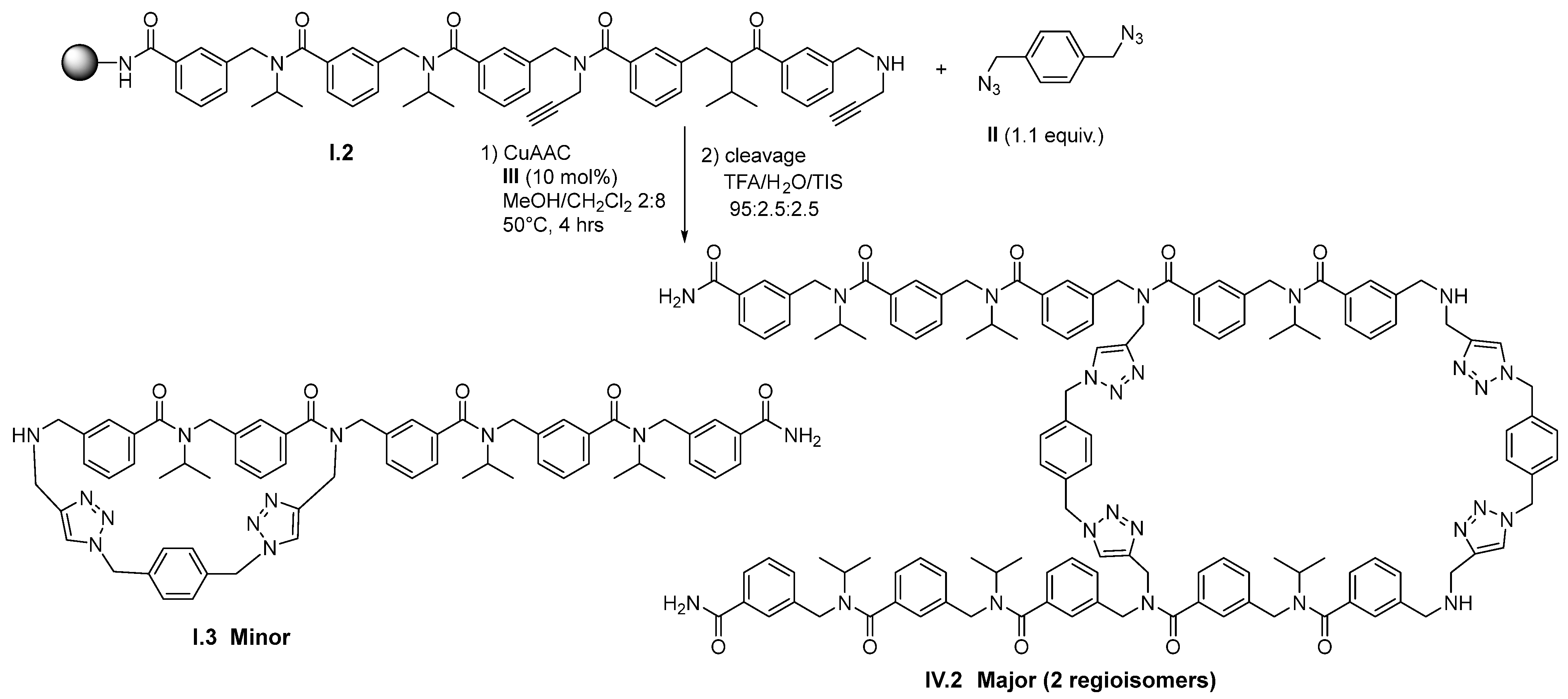
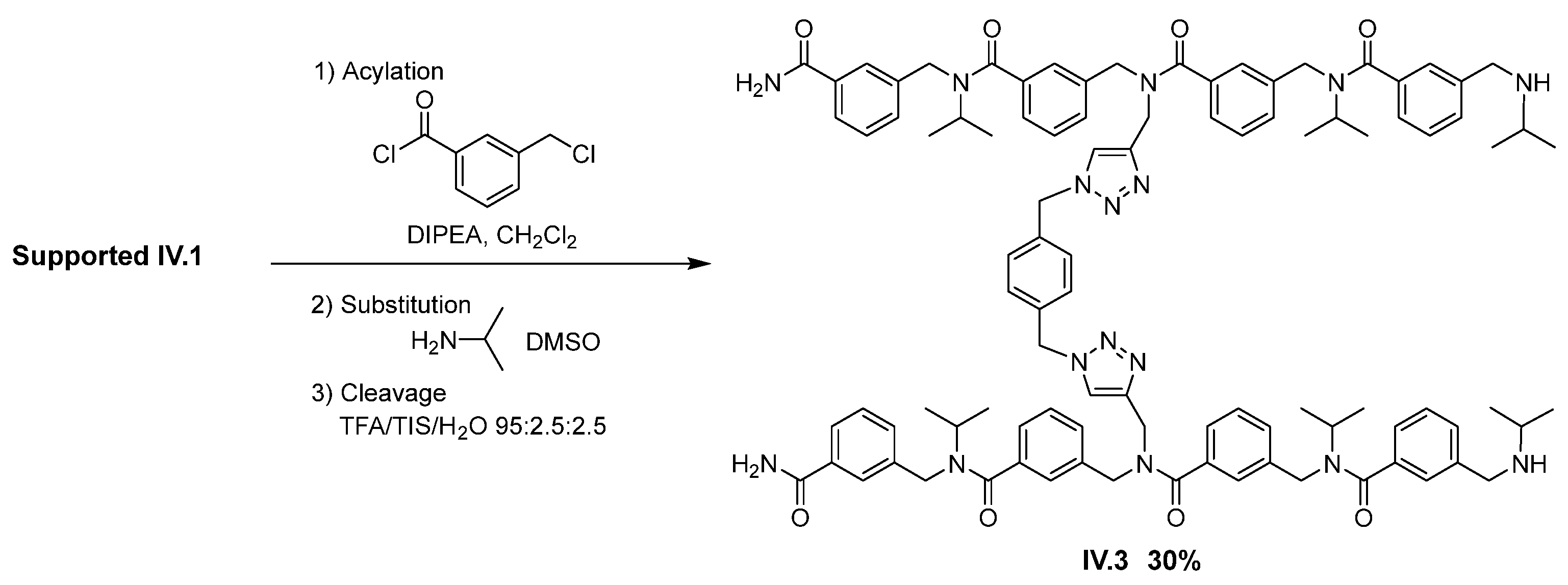
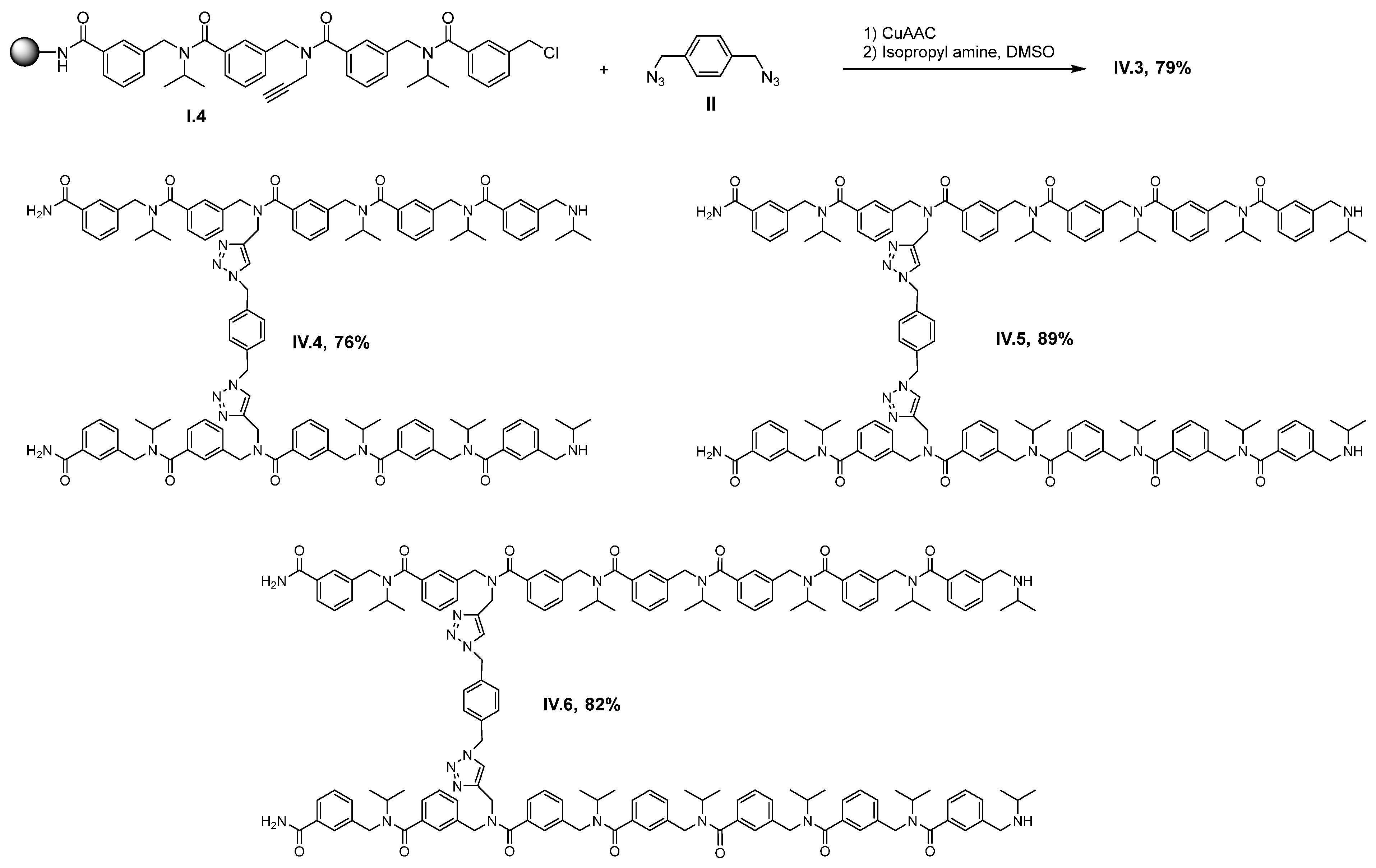
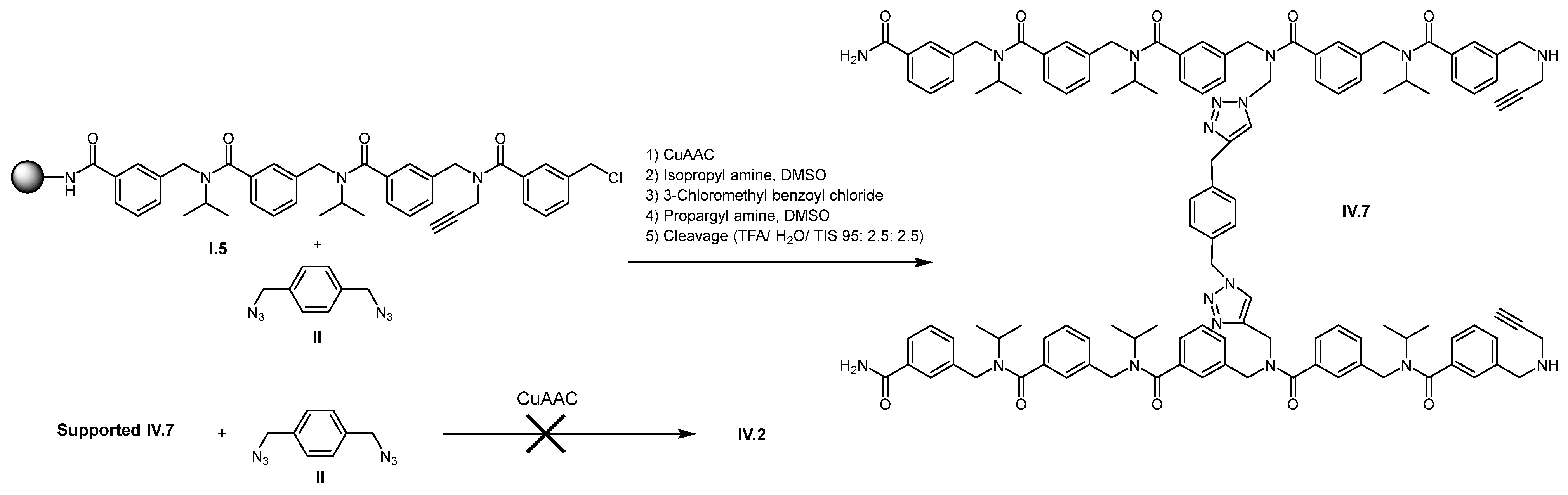
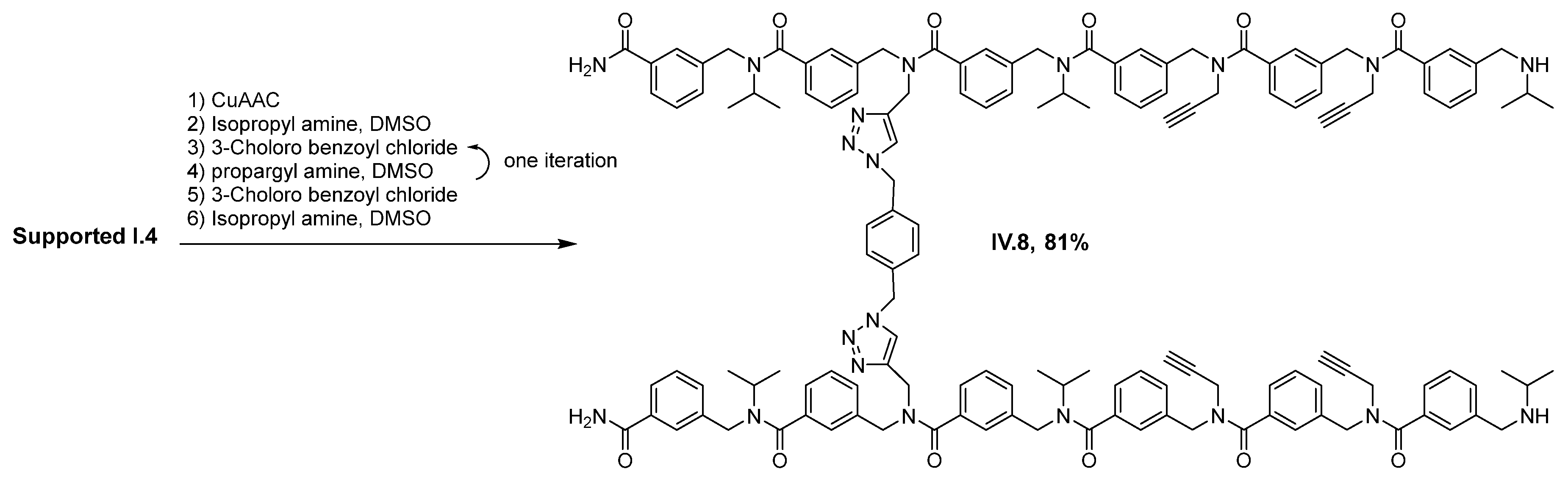
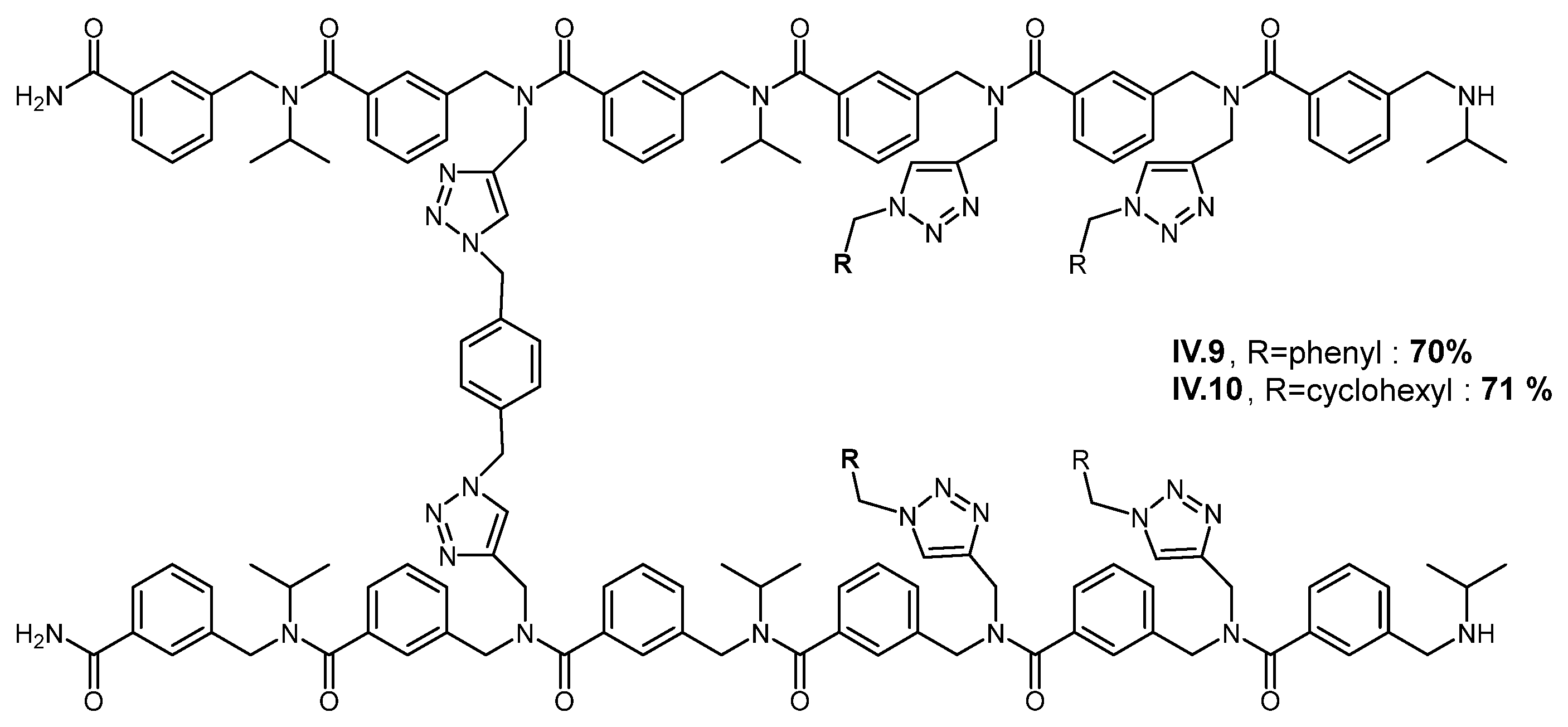
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Protocols for On-Resin Arylopeptoid Oligomers Synthesis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huc, I. Aromatic Oligoamide Foldamers. Eur. J. Org. Chem. 2004, 2004, 17–29. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Zhao, X.; Hou, J.-L.; Li, Z.T. Aromatic amide foldamers: Structures, properties, and functions. Chem. Rev. 2012, 112, 5271–5316. [Google Scholar] [CrossRef] [PubMed]
- Saraogi, I.; Hamilton, D.A. Recent advances in the development of aryl-based foldamers. Chem. Soc. Rev. 2009, 38, 1726–1774. [Google Scholar] [CrossRef] [PubMed]
- Huc, I.; Cuccia, L. Foldamers: Structures, Properties and Applications; Hecht, S., Huc, I., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp. 3–34. [Google Scholar]
- Rinaldi, S. The Diverse World of Foldamers: Endless Possibilities of Self-Assembly. Molecules 2020, 25, 3276. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.-K.; Whitesides, G.M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Bednarek, C.; Wehl, I.; Jung, N.; Schepers, U.; Bräse, S. The Staudinger Ligation. Chem. Rev. 2020, 120, 4301–4354. [Google Scholar] [CrossRef]
- Lossouarn, A.; Renard, P.-Y.; Sabo, C. Tailored Bioorthogonal and Bioconjugate Chemistry: A Source of Inspiration for Developing Kinetic Target-Guided Synthesis Strategies. Bioconjugate Chem. 2021, 32, 63–72. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargöet, M.; Monbaliu, J.-C.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef]
- Kolb, M.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1–3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Holub, J.M.; Kirshenbaum, K. Tricks with clicks: Modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3 + 2] cycloaddition. Chem. Soc. Rev. 2010, 39, 1325–1337. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yan, L.; Xiao, H.; Li, H.; Jing, X. Application of microwave-assisted click chemistry in the preparation of functionalized copolymers for drug conjugation. J. Appl. Polym. Sci. 2012, 127, 3365–3373. [Google Scholar] [CrossRef]
- Barge, A.; Tagliapietra, S.; Binello, A.; Cravotto, G. Click chemistry under microwave or ultrasound irradiation. Curr. Org. Chem. 2011, 15, 189–203. [Google Scholar] [CrossRef]
- Jang, H.; Fafarman, A.; Holub, J.; Kirshenbaum, K. Click to Fit: Versatile Polyvalent Display on a Peptidomimetic Scaffold. Org. Lett. 2005, 7, 1951–1954. [Google Scholar] [CrossRef]
- Li, S.; Cai, H.; He, J.; Chen, H.; Lam, S.; Cai, T.; Zhu, Z.; Bark, S.J.; Cai, C. Extent of the Oxidative Side Reactions to Peptides and Proteins During the CuAAC Reaction. Bioconjugate Chem. 2016, 27, 2315–2322. [Google Scholar] [CrossRef]
- Kennedy, D.C.; McKay, C.S.; Legault, M.C.B.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef]
- Van Hest, J.C.M.; van Delft, F.L. Protein modification by strain-promoted alkyne-azide cycloaddition. ChemBioChem 2011, 12, 1309–1312. [Google Scholar] [CrossRef]
- Hong, V.; Pressolski, S.I.; Ma, C.; Finn, M.G. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. [Google Scholar] [CrossRef]
- Uttamapinant, C.; Tangpeerachaikul, A.; Grecian, S.; Clarke, S.; Singh, U.; Slade, P.; Gee, K.R.; Ting, A.Y. Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem. Int. Ed. 2012, 51, 5852–5856. [Google Scholar] [CrossRef]
- Bevilaqua, V.; King, M.; Chaumontet, M.; Nothisen, M.; Gallibet, S.; Buisson, D.; Puente, C.; Wagner, A.; Tran, F. Copper-Chelating Azides for Efficient Click Conjugation Reactions in Complex Media. Angew. Chem. Int. Ed. 2014, 53, 5872–5876. [Google Scholar] [CrossRef] [PubMed]
- Jewett, J.C.; Sletten, E.M.; Bertozzi, C. Rapid Cu-Free Click Chemistry with Readily Synthesized Biarylazacyclooctynones. J. Am. Chem. Soc. 2010, 132, 3688–3690. [Google Scholar] [CrossRef] [PubMed]
- Teyssot, M.-L.; Chevry, A.; Traïkia, M.; El-Ghozzi, M.; Avignant, D.; Gautier, A. Improved Copper(I)–NHC Catalytic Efficiency on Huisgen Reaction by Addition of Aromatic Nitrogen Donors. Chem. Eur. J. 2009, 15, 6322–6326. [Google Scholar] [CrossRef] [PubMed]
- Teyssot, M.-L.; Nauton, L.; Canet, J.-L.; Cisnetti, F.; Chevry, A.; Gautier, A. Aromatic Nitrogen Donors for Efficient Copper(I)–NHC CuAAC under Reductant-Free Conditions. Eur. J. Org. Chem. 2010, 2010, 3507–3515. [Google Scholar] [CrossRef]
- Akhdar, A.; Gautier, A.; Hjelmgaard, T.; Faure, S. N-Alkylated Aromatic Poly- and Oligoamides. ChemPlusChem 2021, 86, 298–312. [Google Scholar] [CrossRef]
- Akhdar, A.; Faure, S.; Gautier, A. Arylopeptoid oligomers functionalised by combinatorial or sequential on-resin click chemistry using a copper(I)–N-heterocyclic carbene catalyst. Org. Biomol. Chem. 2022, 20, 2402–2406. [Google Scholar] [CrossRef]
- Punna, S.; Kuzelka, J.; Wang, Q.; Finn, M.G. Head-to-tail peptide cyclodimerization by copper-catalyzed azide-alkyne cycloaddition. Angew. Chem. Int. Ed. Engl. 2005, 44, 2215–2220. [Google Scholar] [CrossRef]
- Holub, J.M.; Jang, H.; Kirshenbaum, K. Fit to Be Tied: Conformation-Directed Macrocyclization of Peptoid Foldamers. Org. Lett. 2007, 9, 3275–3278. [Google Scholar] [CrossRef]
- Gaulier, C.; Hospital, A.; Legeret, B.; Delmas, A.F.; Aucagne, V.; Cisnetti, F.; Gautier, A. A water-soluble CuI–NHC for CuAAC ligation of unprotected peptides under open air conditions. Chem. Commun. 2012, 48, 4005–4007. [Google Scholar] [CrossRef]
- Bruyat, P.; Gautier, A.; Jean, L.; Renard, P.-Y. Use of an Air-Stable Cu(I)-NHC Catalyst for the Synthesis of Peptidotriazoles. J. Org. Chem. 2018, 83, 13515–13522. [Google Scholar] [CrossRef]
- Akhdar, A.; Nauton, L.; Jouffret, L.; Faure, S.; Gautier, A. Cage-like structures based on constrained cyclic arylopeptoids. Chem. Commun. 2023, 59, 8087–8090. [Google Scholar] [CrossRef] [PubMed]
- Hjelmgaard, T.; Faure, S.; De Santis, E.; Staerk, D.; Alexander, B.D.; Edwards, A.A.; Taillefumier, C.; Nielsen, J. Improved solid-phase synthesis and study of arylopeptoids with conformation-directing side chains. Tetrahedron 2012, 68, 4444–4454. [Google Scholar] [CrossRef]
- Shyam, R.; Charbonnel, N.; Job, A.; Blavignac, C.; Forestier, C.; Taillefumier, C.; Faure, S. 1,2,3-Triazolium-Based Cationic Amphipathic Peptoid Oligomers Mimicking Antimicrobial Helical Peptides. Chem. Med. Chem. 2018, 13, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamas, Z.E.A.; Akhdar, A.; Charnay-Pouget, F.; Faure, S.; Gautier, A. Clicked H-Shaped Arylopeptoids. Molecules 2025, 30, 724. https://doi.org/10.3390/molecules30030724
Chamas ZEA, Akhdar A, Charnay-Pouget F, Faure S, Gautier A. Clicked H-Shaped Arylopeptoids. Molecules. 2025; 30(3):724. https://doi.org/10.3390/molecules30030724
Chicago/Turabian StyleChamas, Zein El Abidine, Ayman Akhdar, Florence Charnay-Pouget, Sophie Faure, and Arnaud Gautier. 2025. "Clicked H-Shaped Arylopeptoids" Molecules 30, no. 3: 724. https://doi.org/10.3390/molecules30030724
APA StyleChamas, Z. E. A., Akhdar, A., Charnay-Pouget, F., Faure, S., & Gautier, A. (2025). Clicked H-Shaped Arylopeptoids. Molecules, 30(3), 724. https://doi.org/10.3390/molecules30030724