
Copper(I)-Photocatalyzed Addition of Trichloromethanesulfenyl Chloride to Olefinic Compounds

 , ,
, ,  , ,
, ,  and
and 
Abstract
1. Introduction
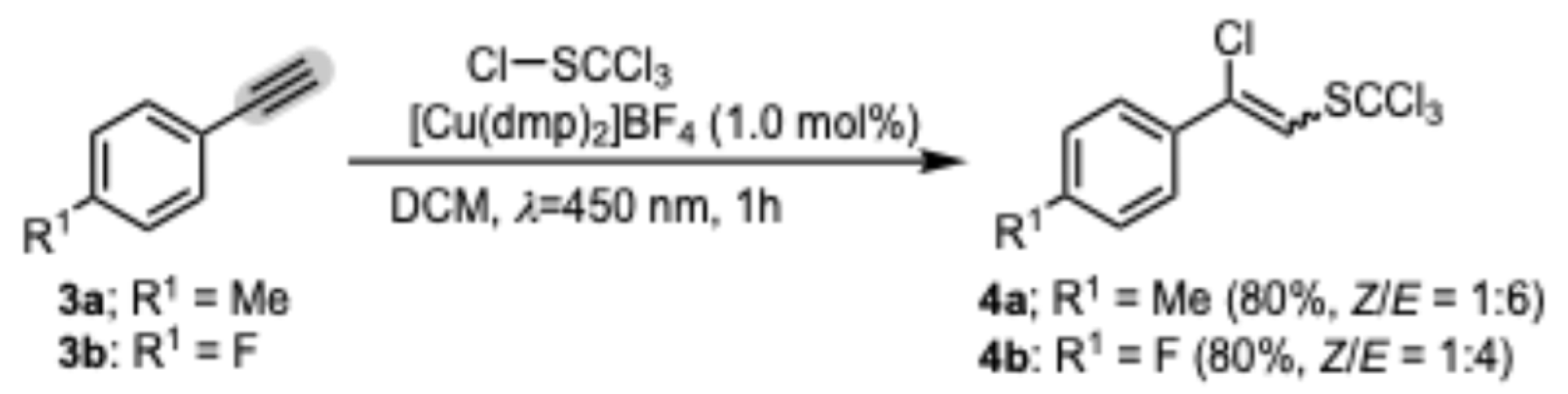
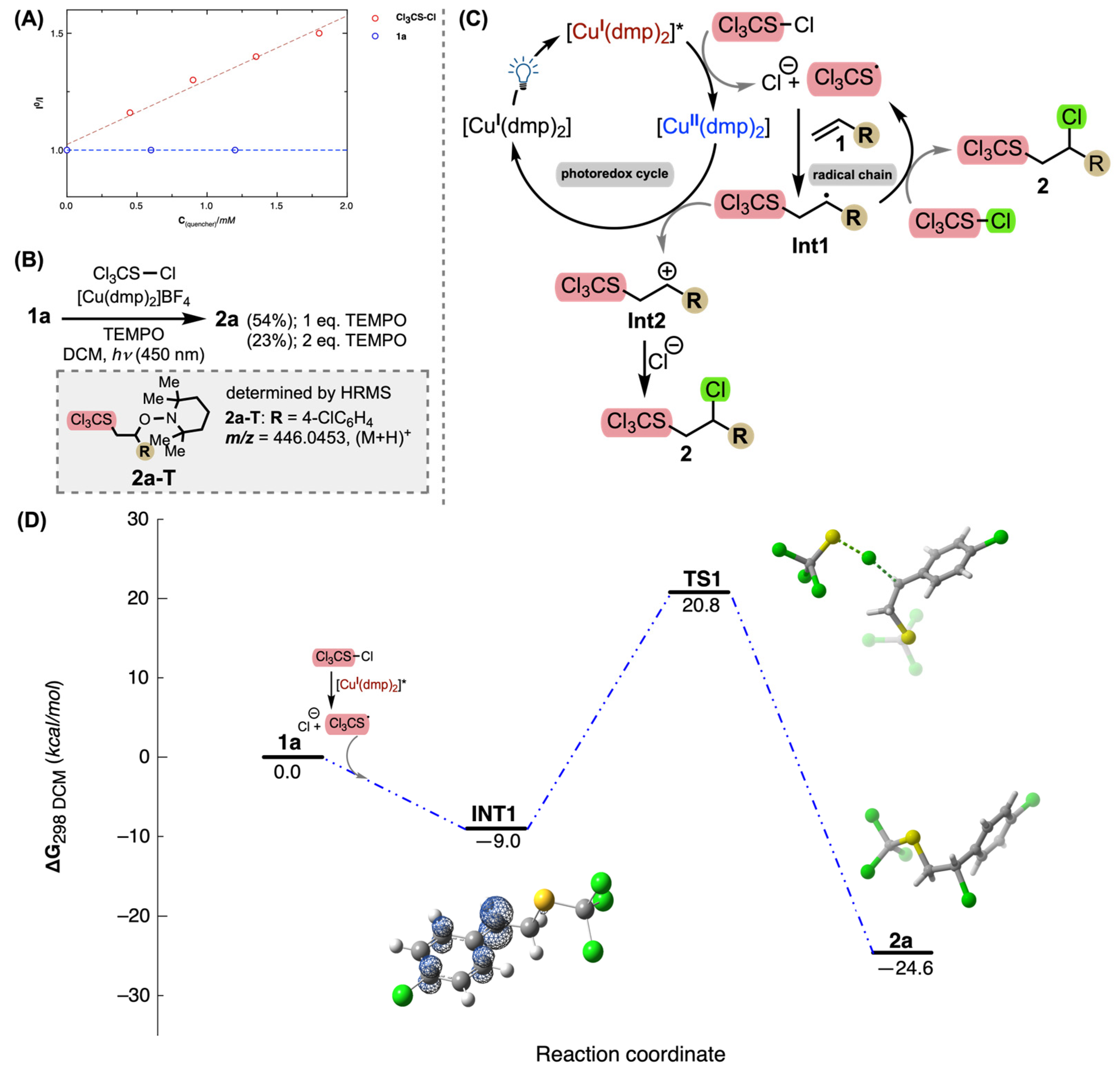
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barattucci, A.; Aversa, M.; Mancuso, A.; Sa lerno, T.; Bonaccorsi, P. Transient Sulfenic Acids in the Synthesis of Biologically Relevant Products. Molecules 2018, 23, 1030. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X. (Ed.) Topics in Current Chemistry Collections. In Sulfur Chemistry; Springer Nature Switzerland AG: Cham, Switzerland, 2019; pp. 1–475. [Google Scholar] [CrossRef]
- Mori, H.; Nishihara, Y. Low-bandgap semiconducting polymers based on sulfur-containing phenacene-type molecules for transistor and solar cell applications. Polym. J. 2018, 50, 615–625. [Google Scholar] [CrossRef]
- Diez, S.; Hoefling, A.; Theato, P.; Pauer, W. Mechanical and Electrical Properties of Sulfur-Containing Polymeric Materials Prepared via Inverse Vulcanization. Polymers 2017, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Gil, C. Heterocycles Containing Nitrogen and Sulfur as Potent Biologically Active Scaffolds. In Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Bräse, S., Ed.; The Royal Society of Chemistry: London, UK, 2016; pp. 231–261. [Google Scholar] [CrossRef]
- Benson, S.W. Thermochemistry and kinetics of sulfur-containing molecules and radicals. Chem. Rev. 1978, 78, 23–35. [Google Scholar] [CrossRef]
- Chu, J.C.K.; Rovis, T. Amide-directed photoredox-catalysed C–C bond formation at unactivated sp3 C–H bonds. Nature 2016, 539, 272–275. [Google Scholar] [CrossRef]
- Jeffrey, J.L.; Terrett, J.A.; MacMillan, D.W.C. O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1636. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.J.; Nguyen, L.Q.; Beard, G.H.; Knowles, R.R. Catalytic Olefin Hydroamination with Aminium Radical Cations: A Photoredox Method for Direct C–N Bond Formation. J. Am. Chem. Soc. 2014, 136, 12217–12220. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Ahneman, D.T.; Chu, L.; Terrett, J.A.; Doyle, A.G.; MacMillan, D.W.C. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angew. Chem. Int. Ed. 2011, 50, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Beaudelot, J.; Oger, S.; Peruško, S.; Phan, T.-A.; Teunens, T.; Moucheron, C.; Evano, G. Photoactive Copper Complexes: Properties and Applications. Chem. Rev. 2022, 122, 16365–16609. [Google Scholar] [CrossRef] [PubMed]
- Bagal, D.B.; Kochkovskyi, G.; Knorn, M.; Rawner, T.; Bhanage, B.M.; Reiser, O. Trifluoromethylchlorosulfonylation of Alkenes: Evidence for an Inner-Sphere Mechanism by a Copper Phenanthroline Photoredox Catalyst. Angew. Chem. Int. Ed. 2015, 54, 6999–7002. [Google Scholar] [CrossRef] [PubMed]
- Pagire, S.K.; Paria, S.; Reiser, O. Synthesis of β-Hydroxysulfones from Sulfonyl Chlorides and Alkenes Utilizing Visible Light Photocatalytic Sequences. Org. Lett. 2016, 18, 2106–2109. [Google Scholar] [CrossRef] [PubMed]
- Chakrasali, P.; Kim, K.; Jung, Y.-S.; Kim, H.; Han, S.B. Visible-Light-Mediated Photoredox-Catalyzed Regio- and Stereoselective Chlorosulfonylation of Alkynes. Org. Lett. 2018, 20, 7509–7513. [Google Scholar] [CrossRef]
- Petek, N.; Brodnik, H.; Reiser, O.; Štefane, B. Copper- and Photoredox-Catalyzed Cascade to Trifluoromethylated Divinyl Sulfones. J. Org. Chem. 2023, 88, 6538–6547. [Google Scholar] [CrossRef]
- Alkan-Zambada, M.; Hu, X. Cu-Catalyzed Photoredox Chlorosulfonylation of Alkenes and Alkynes. J. Org. Chem. 2019, 84, 4525–4533. [Google Scholar] [CrossRef]
- Hossain, A.; Engl, S.; Lutsker, E.; Reiser, O. Visible-Light-Mediated Regioselective Chlorosulfonylation of Alkenes and Alkynes: Introducing the Cu(II) Complex [Cu(dap)Cl2] to Photochemical ATRA Reactions. ACS Catal. 2019, 9, 1103–1109. [Google Scholar] [CrossRef]
- Engl, S.; Reiser, O. Making Copper Photocatalysis Even More Robust and Economic: Photoredox Catalysis with [CuII(dmp)Cl]Cl. Eur. J. Org. Chem. 2020, 1523–1533. [Google Scholar] [CrossRef]
- Sookezian, A.; Molander, G.A. Photoinduced Vicinal 1,2-Difunctionalization of Olefins for the Synthesis of Alkyl Sulfonamides. Org. Lett. 2023, 25, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Hell, M.; Meyer, C.F.; Laudadio, G.; Misale, A.; Willis, M.C.; Noël, T.; Trabanco, A.A.; Gouverneur, V. Silyl Radical Mediated Activation of Sulfamoyl Chlorides Enables Direct Access to Aliphatic Sulfonamides from Alkenes. J. Am. Chem. Soc. 2020, 142, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Mao, M.; Zhu, Y.; Wang, Y. Photoredox-Catalyzed Generation of Sulfamyl Radicals: Sulfonamidation of Enol Silyl Ether with Chlorosulfonamide. J. Org. Chem. 2019, 84, 13897–13907. [Google Scholar] [CrossRef] [PubMed]
- Wanga, X.; Zhanga, J.; Chena, Q.; Zhoub, W.; Wu, J. Generation of sulfonylureas under photoredox catalysis and their biological evaluations. Chin. Chem. Lett. 2022, 33, 4860–4864. [Google Scholar] [CrossRef]
- Petek, N.; Brodnik, H.; Grošelj, U.; Svete, J.; Štefane, B. Access to β-Lactam Sulfonamides from Sulfamoyl Chlorides via Photoredox Catalyzed C−S Bond Formation. Adv. Synth. Cat. 2024, 366, 3310–3315. [Google Scholar] [CrossRef]
- Kühle, E. One Hundred Years of Sulfenic Acid Chemistry II a. Oxidation, Reduction, and Addition Reactions of Sulfenyl Halides. Synthesis 1971, 1971, 563–586. [Google Scholar] [CrossRef]
- Block, E.; Schwan, A.L. Addition of X–Y Reagents to Alkenes and Alkynes. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1991; Volume 4, pp. 329–362. [Google Scholar]
- Sheppard, W.A.; Harries, J.F. Dichlorofluoromethanesulfenyl Chloride. J. Am. Chem. Soc. 1960, 82, 5106–5107. [Google Scholar] [CrossRef]
- He, J.; Chen, C.; Fu, G.C.; Peters, J.C. Visible-Light-Induced, Copper-Catalized Tree-Component Coupling of Alkyl Halides, Olefines, and Trifluoromethylthiolate to Generate Trifluoromethyl Thioethers. ACS Catal. 2018, 8, 11741–11748. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Malpani, Y.R.; Ha, N.; Jung, Y.S.; Han, S.B. Vicinall Difunctionalization of Alkenes: Chlorotrifluoromethylation with CF3SO2Cl by Photoredox Catalysis. Org. Lett. 2014, 16, 1310–1313. [Google Scholar] [CrossRef]
- Tang, X.-J.; Dolbier, W.R., Jr. Efficient Cu-catalyzed Atom Transfer Radical Addition Reactions of Fluoroalkylsulfonyl Chlorides with Electron-deficient Alkenes Induced by Visible Light. Angew. Chem. Int. Ed. 2015, 54, 4246–4249. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.N.; Olding, A.; Ho, C.C.; Bissember, A.C.; Coote, M.L. Investigating Competing Inner- and Outer-Sphere Electron-Transfer Pathways in Copper Photoredox-Catalyzed Atom-Transfer Radical Addition: Closing the Cycle. Angew. Chem. Int. Ed. 2024, 64, e202415792. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}

| Entry | Deviation from Standard Conditions | Yield (%) 2 |
|---|---|---|
| 1 | none 1 | 99 (98) 3 |
| 2 | [Cu(dmp)(Xsantphos)]BF4 | 97 |
| 3 | [Cu(dap)2]BF4 | 96 |
| 4 | [Ru(bpy)3](PF6)2 | 38 |
| 5 | [Acr+-Mes]ClO4– | 10 |
| 6 | [Cu(dmp)2]BF4, no light | n.d. |
| 7 | [Cu(dmp)2]BF4, no light, 60 °C | <5 |
| 8 | 520 nm | n.d. |
| 9 | [Cu(dmp)2]BF4, 0.5 mol% | 99 |
| 10 | [Cu(dmp)2]BF4, 0.1 mol% | 87 |
| 11 | 1 min | 9 |
| 12 | 0.5 h | 82 |
| 13 | 16 h | 99 |
| 14 | MeCN instead of DCM | 32 |
| 15 | toluene instead of DCM | 45 |
| 16 | THF instead of DCM | 48 |
| 17 | 1,4-dioxane instead of DCM | 50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petek, N.; Zorko, T.; Škrinjar, M.; Grošelj, U.; Svete, J.; Kočar, D.; Štefane, B. Copper(I)-Photocatalyzed Addition of Trichloromethanesulfenyl Chloride to Olefinic Compounds. Molecules 2025, 30, 661. https://doi.org/10.3390/molecules30030661
Petek N, Zorko T, Škrinjar M, Grošelj U, Svete J, Kočar D, Štefane B. Copper(I)-Photocatalyzed Addition of Trichloromethanesulfenyl Chloride to Olefinic Compounds. Molecules. 2025; 30(3):661. https://doi.org/10.3390/molecules30030661
Chicago/Turabian StylePetek, Nejc, Tilen Zorko, Martin Škrinjar, Uroš Grošelj, Jurij Svete, Drago Kočar, and Bogdan Štefane. 2025. "Copper(I)-Photocatalyzed Addition of Trichloromethanesulfenyl Chloride to Olefinic Compounds" Molecules 30, no. 3: 661. https://doi.org/10.3390/molecules30030661
APA StylePetek, N., Zorko, T., Škrinjar, M., Grošelj, U., Svete, J., Kočar, D., & Štefane, B. (2025). Copper(I)-Photocatalyzed Addition of Trichloromethanesulfenyl Chloride to Olefinic Compounds. Molecules, 30(3), 661. https://doi.org/10.3390/molecules30030661