4.1. Epoxidation
A recent development in stereoselective epoxidation reactions came from an unexpected area of sulfoxonium ylide chemistry. The quickly developing area of interrupted Johnson–Corey–Chaykovsky reactions has seen recent successes in mostly stereoselective cyclopropanation reactions [
40,
41,
42]. However, success has also been observed in stereoselective epoxidation reactions [
43]. In 2024, Ramasastry and colleagues developed an interrupted Johnson–Corey–Chaykovsky reaction methodology involving the unexpected formation of 2,3-epoxyhexahydrofluoren-9-ones
23 from tethered bis-enones
22 (
Scheme 16) [
43]. In the majority of reactions of dimethylsulfoxonium methylide and tethered bis-enone, the expected cyclopropanation of either
α,
β-unsaturated ketone group is not observed. The reaction proceeds through the expected 1,4-addition of ylide
2 to the methylene of
22, as opposed to the competing methine. Another, and unexpected, conjugate addition occurs to the methine resulting in an arene-fused cyclopentanone
A. The ketone group reforms through a 1,5-H
+-transfer, resulting in another sulfoxonium ylide
B. A hexahydrofluorenone
C is formed through a nucleophilic addition reaction whereby the
α-carbanion adjacent to the sulfoxonium group acts as a nucleophile to attack the carbonyl group of the ketone. Epoxidation occurs through the quick cleavage of DMSO
7, resulting in 2,3-epoxyhexahydrofluoren-9-one
23.
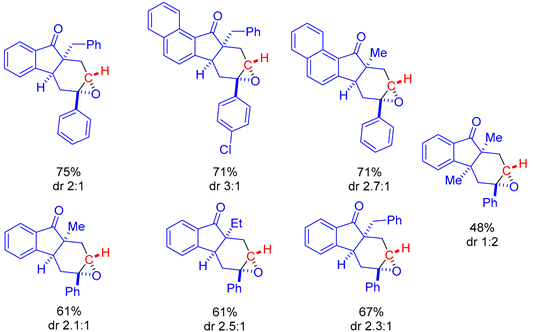
The initial investigation started with optimization of the reaction. They probed the reaction of 22 and dimethylsulfoxonium methylide 2 using different bases and solvent systems at room temperature for 5–10 min. The reaction proceeded best in terms of yield and diastereoselectivity (dr) in DMF using 1.2 equivalents of a base. Bases included NaH (84%, dr 2:1), potassium t-butoxide (63%, dr 1.7:1), caesium carbonate (76%, dr 1.7:1) and lithium bis(trimethylsilyl)amide (65%, dr 3.5:1). The reaction proceeded moderately in CH3CN using NaH (66%, dr 1.3:1). Poor yield and diastereoselectivity was observed when NaH was tested in THF (19%, dr 1:1) and 1,4-dioxane (22%, dr 1:1) and no reaction was observed when Et3N and 1,1,3,3-tetramethylguanidine were explored in DMF. Despite lithium bis(trimethylsilyl)amide enabling the best diastereoselectivity, the combination of NaH in DMF at room temperature was found to be the most effective overall.
The scope of the reaction was examined, resulting in 23 examples with reaction yields of up to 84% and diastereoselectivity of up to 3:1 (
Scheme 17,
Table 1). The R
1 substituent responded well when substituted with alkyl groups and sterically large alkyl groups bonded to phenyl rings. The arene backbone was investigated, the R
2 substituent was found to be tolerant of naphthalene, activated aryls and deactivated aryls bearing fluorine. The R
4 substituent performed well where aliphatic ketones and aryl ketones were probed. Both activated and deactivated aryl ketones were tolerated. In most cases, the R
3 substituent remained a hydrogen atom, however, a methyl group was tolerated in one example, resulting in two contiguous chiral quaternary carbon centres being formed.
4.2. Aziridination
An aziridine is a three-membered nitrogen-containing heterocycle of considerable interest in terms of organic synthesis due to its potential role in performing as a precursor to complex bioactive molecules. Considering ring strain exists within the atoms of the heterocycle, this can provide a way to interesting synthetic pathways which could potentially formulate structurally important pharmaceuticals. The chemistry pertaining to the field of aziridine synthesis has followed many avenues since the first synthesis by Gabriel in 1888 [
44,
45,
46,
47,
48,
49,
50,
51]. Aziridines are susceptible to ring opening reactions due to the ring strain and the polarized nature of the carbon–nitrogen bond. Aziridines have the capability of partaking in stereoselective ring-opening reactions and cycloadditions, similar to their epoxide analogues, resulting in structurally interesting heterocycles [
46,
47,
48,
49,
50,
51]. Due to the fact that stereoselective aziridination methodologies are sparse and lacking in efficacy, aziridines are less commonly used in synthetic applications compared to their epoxide counterparts [
52,
53,
54]. The utility of sulfonium ylides and sulfoxonium ylides in facilitating asymmetric aziridinations has been described in several reviews in previous years [
8,
55,
56,
57,
58,
59,
60,
61,
62,
63]. In analogous fashion to epoxidation, the reaction proceeds through a zwitterionic intermediate formed through nucleophilic addition of the ylidic carbanion to an electrophilic imine site. An intramolecular cyclization occurs, constructing the aziridine.
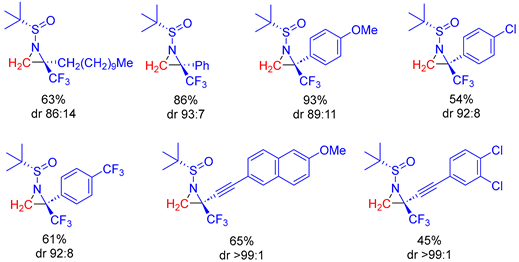
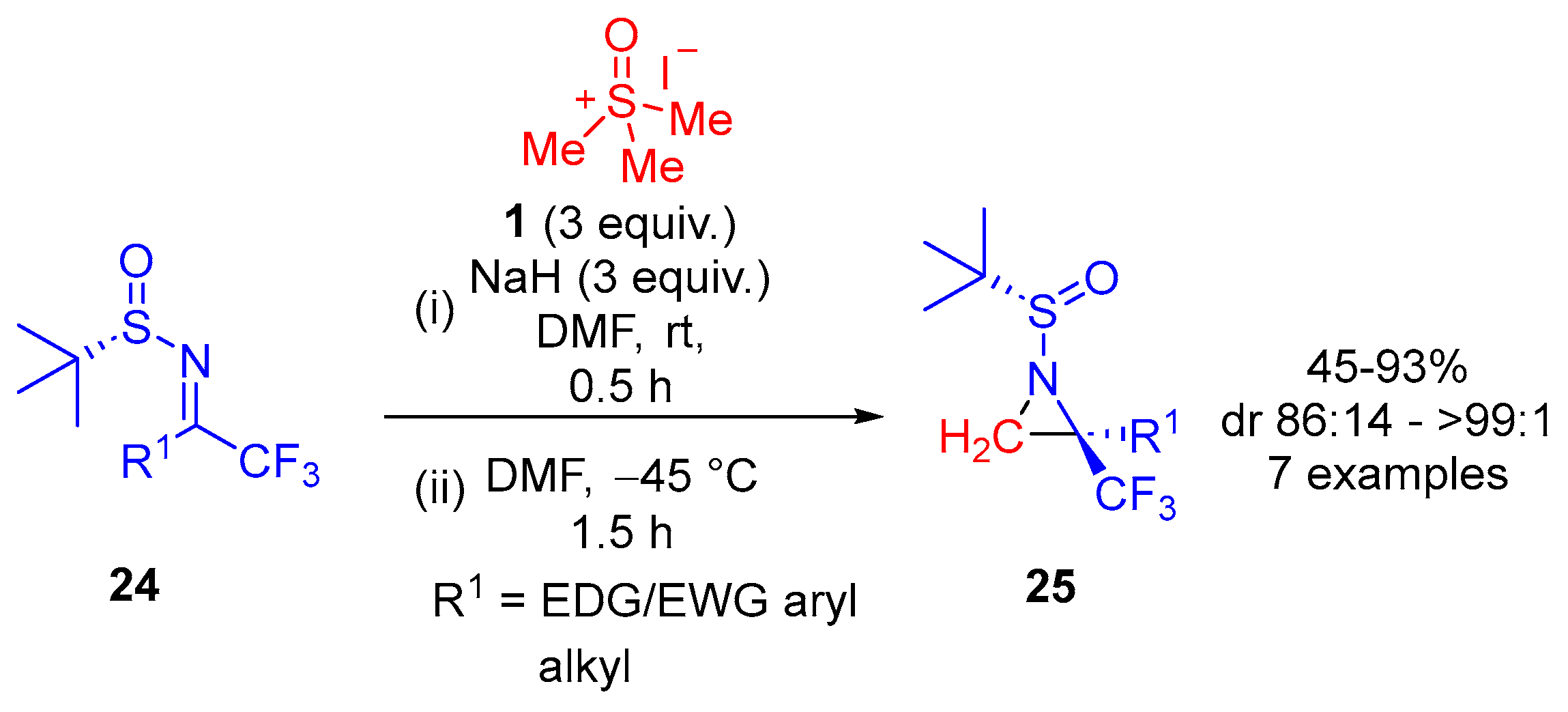
In 2013, Huang and colleagues developed a convenient and practical method for the asymmetric synthesis of trifluoromethylated aziridines by employing reactions of sulfoxonium ylide
2 with (
S)-
N-tert-butanesulfinyl ketimines
24 (
Scheme 18) [
64]. The reaction of ylide generated from trimethylsulfoxonium iodide
1 in the presence of base with chiral tert-butanesulfinyl ketimines
24 furnished trifluoromethylated aziridines
25 in moderate to excellent yields and with very good diastereoselectivities (
Table 2). Aromatic as well as aliphatic ketimines underwent the reaction smoothly to afford the corresponding aziridines in good yields (61–93%). In general, electron-withdrawing groups were beneficial to the construction of trifluoromethylated aziridines with good diastereoselectivities but lower yields (45%, 54% and 61%), whereas ketimines bearing an aryl group with an electron-donating group gave the highest yield but with lower diastereoselectivity. The highest diastereoselectivity of de >98% was reached in the cases of the formation of acetylenic trifluoromethylated aziridines, which were afforded in moderate to good yields.
In 2015, Marsini and colleagues, building on work from Huang, developed a highly diastereoselective aza-Johnson–Corey–Chaykovsky reaction of chiral
N-tert-butanesulfinyl ketimino esters
26 with dimethylsulfoxonium methylide, to form
α-quaternary aziridine-2-carboxylates
27 (
Scheme 19) [
65]. The reaction displayed moderate to excellent yields, exhibiting a broad scope with respect to substrates, and was tolerant of alkyl, alkenyl, aryl and heteroaryl groups substituted at the R
1 position. The reaction mechanistic pathway is expected to proceed through the conventional route whereby a methylene group undergoes transfer to the imine, forming an aziridine, in <10 min reaction time at 0 °C. They observed, through mechanistic studies, that the reaction proceeds to only form a single diastereomer (
S,
S). In terms of which substrates performed most predominantly, the results suggested bulky or aryl-based substituents produced the best yields and diastereoselectivities [
65] (
Table 3), with smaller substituents showing poorer yields and diastereoselectivities. This methodology provided an avenue to synthesize unnatural heterocyclic amino esters and
α-quaternary conjugated peptides through follow-up reactions.
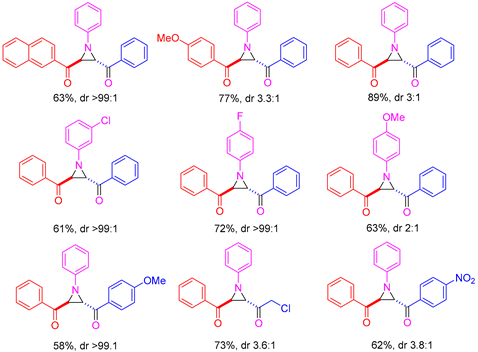
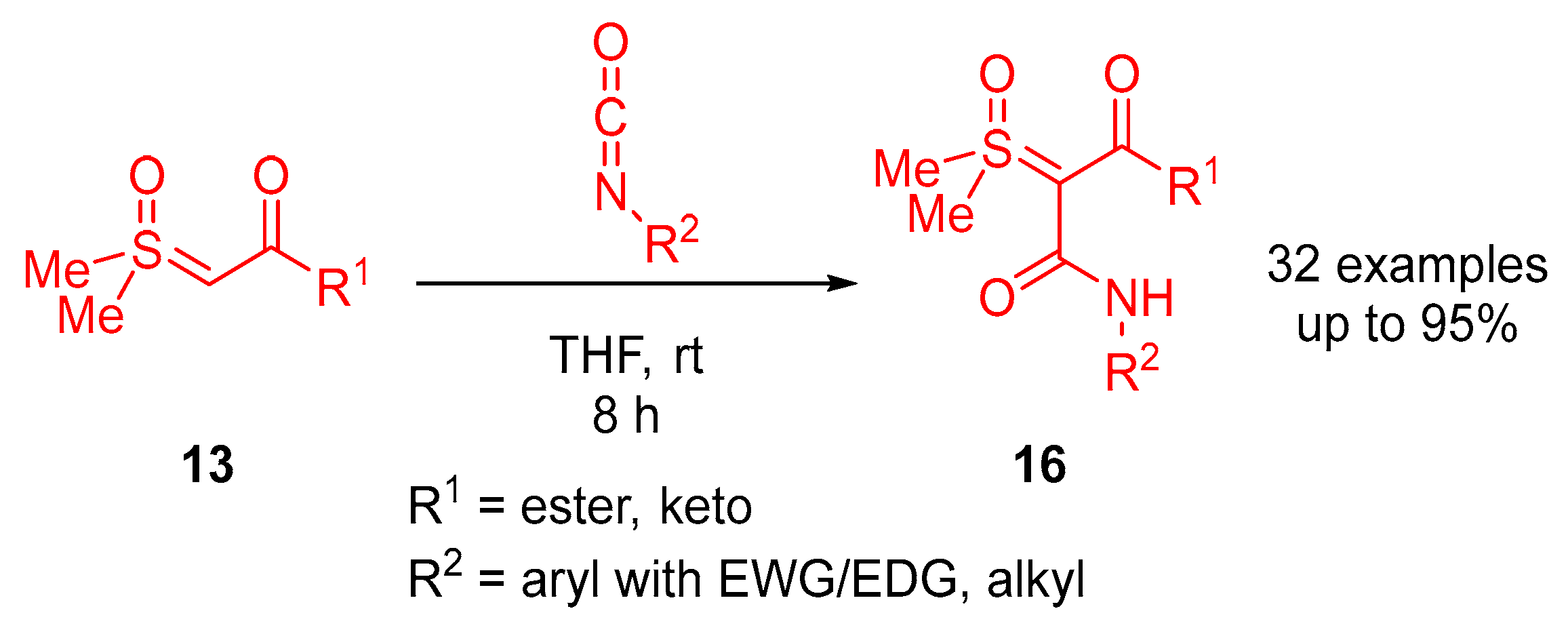
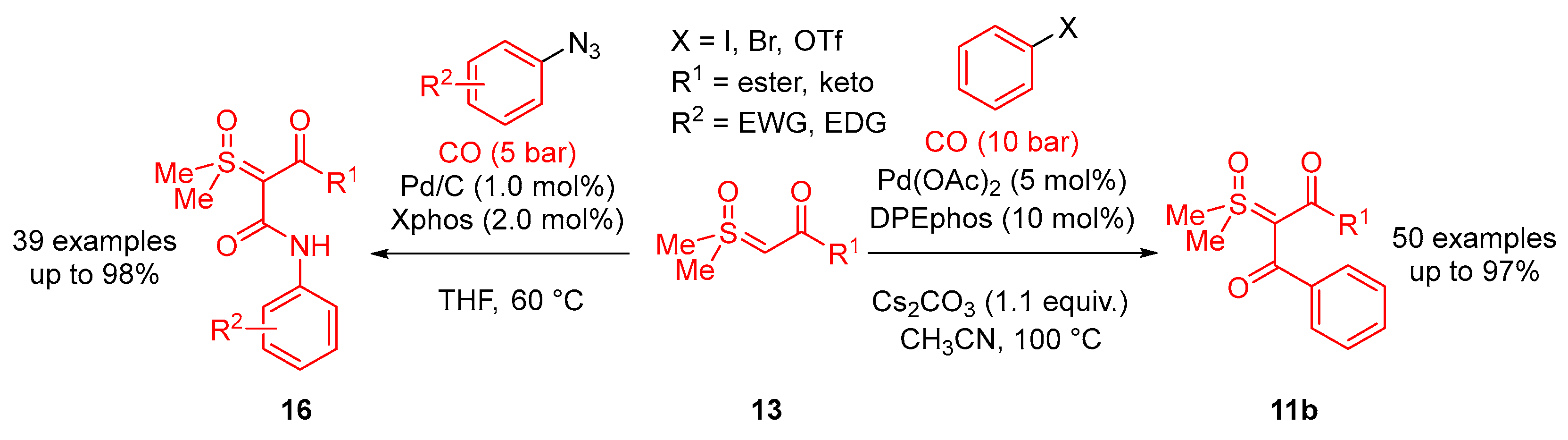
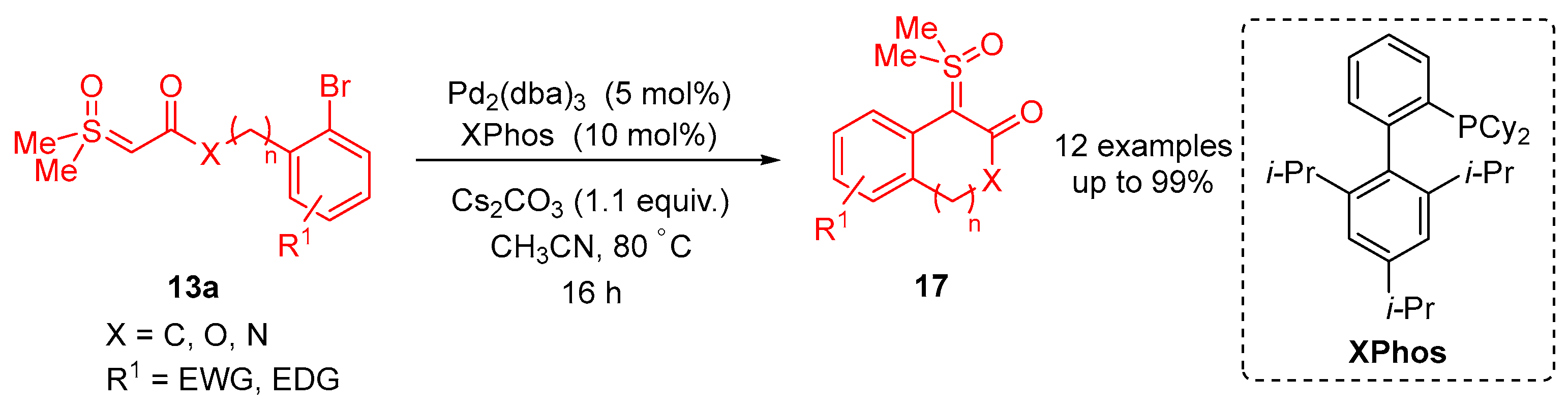
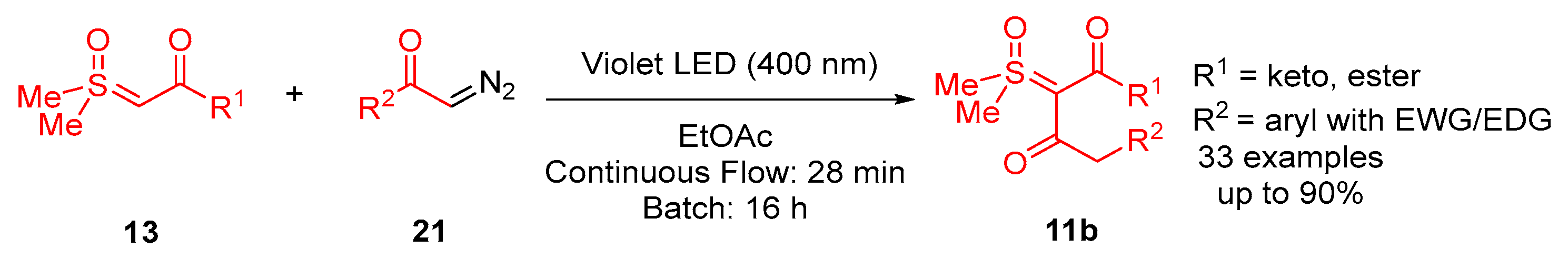
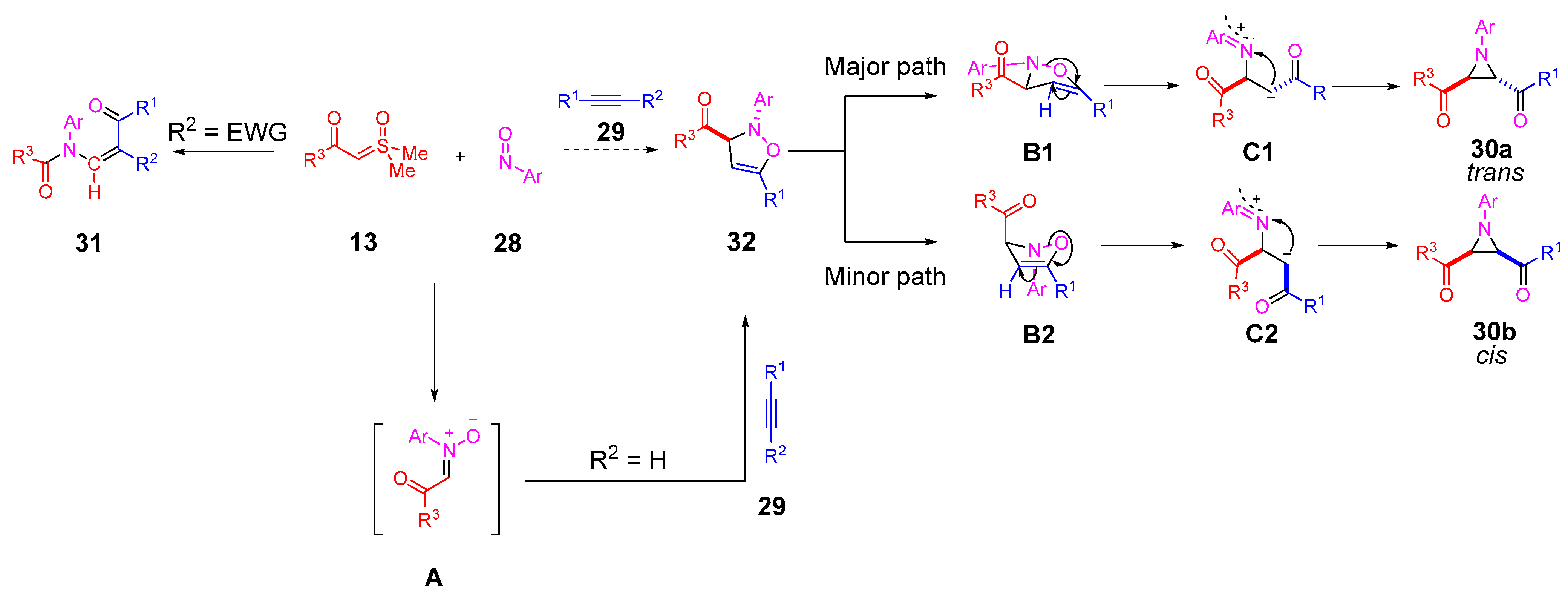
In 2023, Li and colleagues reported a catalyst-free one-pot synthesis of chiral diketo aziridines
30 and alkenes
31 [
66]. A three-component reaction of
α-ketosulfoxonium ylides
13, nitrosoarenes
28 and terminal alkynes
29 was observed to facilitate access to diketo aziridines
30 with moderate to good
trans-diastereoselectivities and with moderate to good yields (41–89%) (
Scheme 20). They initially investigated the effect of solvent variation in the reaction, and it was clear that CH
3CN resulted in the best yields (up to 89%) and diastereoselectivity (dr up to >99:1). In contrast, toluene, CH
2Cl
2 and THF resulted in yields of 31%, 42% and 50%, respectively, and as a stereorandom mixture of
cis- and
trans-diastereomers. Following this, the R
3 substituent scope of the
α-ketosulfoxonium ylide was investigated, with the reaction demonstrating tolerance for alkyl, heteroaryl and aryl (with EDG/EWG) groups (
Table 4). Activated and deactivated aryl-substituted nitroso substrates were tolerated. However, when 4-nitrosopyridine was investigated under the optimal reaction conditions, no aziridine was formed.
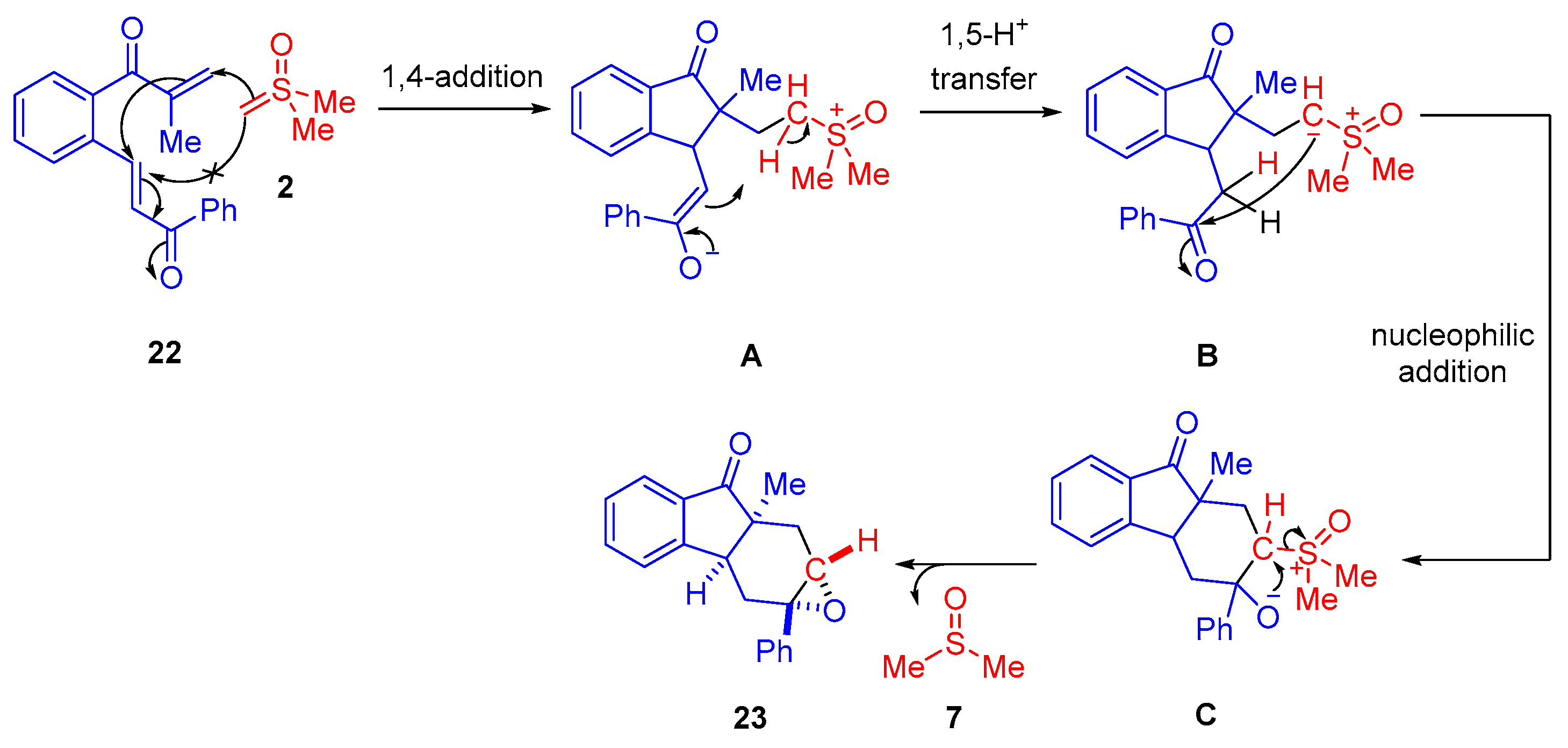
The proposed mechanism proceeds via an initial nitrone formation step involving reaction of the
α-ketosulfoxonium ylide
13 and the nitrosoarene
28 (
Scheme 21). The nitrone intermediate
A reacts with a terminal alkyne
29 in a (3 + 2)-cycloaddition step to form an isoxazoline intermediate
32. At this point, the reaction pathway can take three possible routes. In the case of terminal alkynes
29 (R
3 = H) in the reaction, the formation of diketo aziridines has been reported. The isoxazoline intermediate
32 can exist in two different conformations (
B1 and
B2), and the selectivity for either a
trans- or
cis-diastereomer is believed to be due to N-O bond Baldwin rearrangement of the preferred isoxazoline conformation
B1 [
66,
67]. The preferred
trans-diastereomer
30a forms as a result of the two keto carbonyl groups avoiding steric interactions in intermediate
C1, whereas the formation of the lesser observed
cis-diastereomer
30b is due to the slight tolerance of steric repulsion of these groups. In the case of an internal alkyne with an electron-withdrawing group (R
2) on
29, the formation of
α,
β-substituted ketones
31 was observed. The rationale for this transformation over aziridine formation is due to the stabilizing effect of the electron-withdrawing group during the Baldwin rearrangement step.
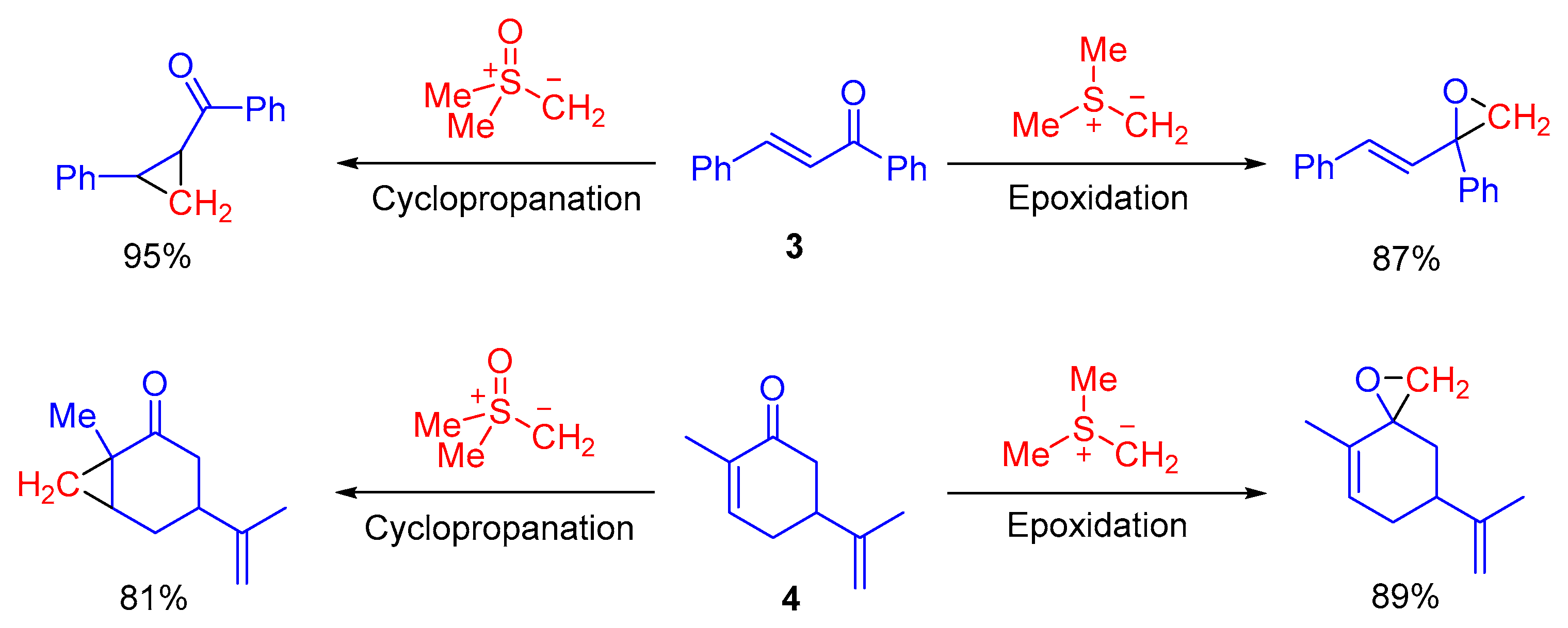
4.3. Cyclopropanation
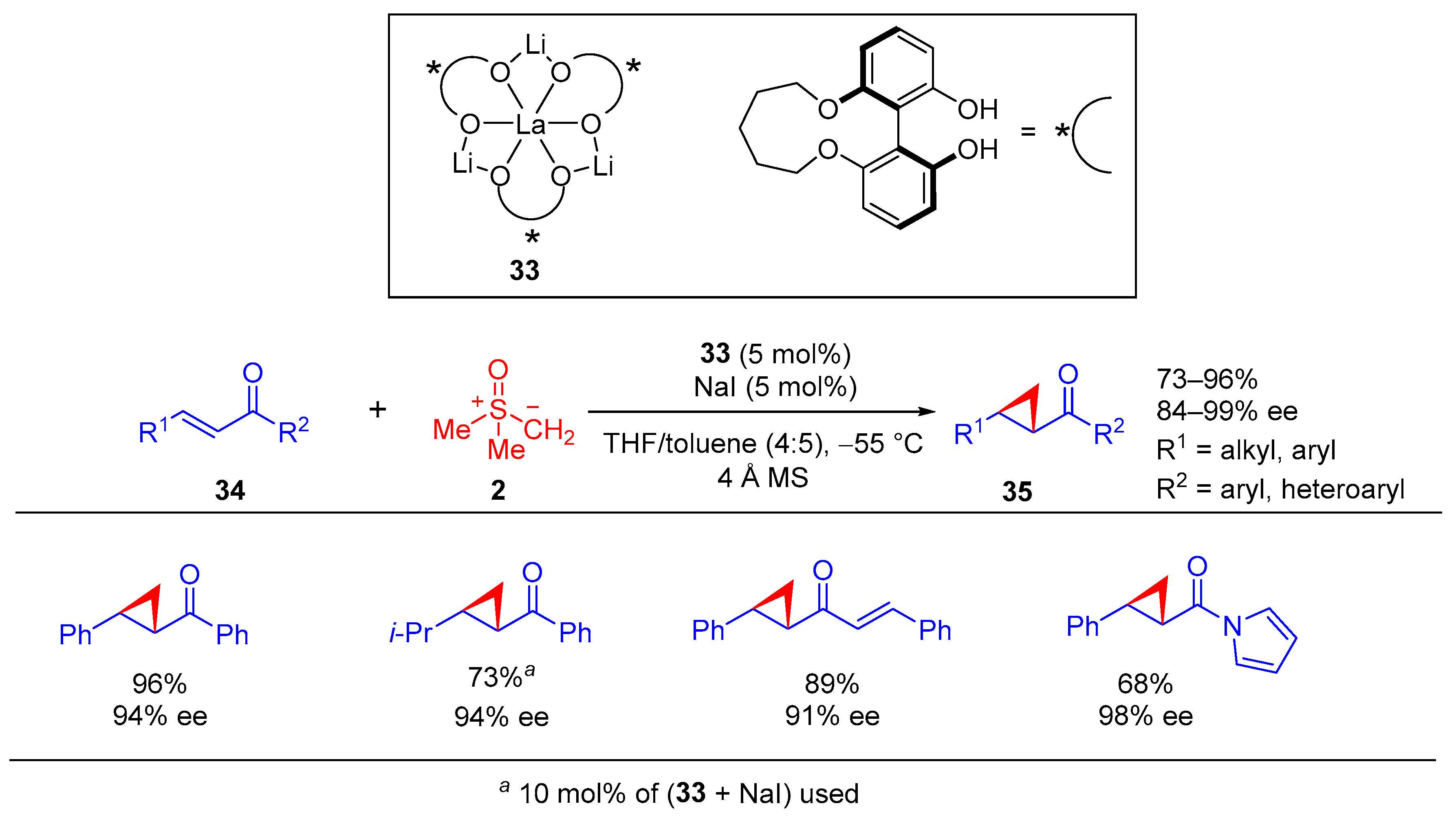
Shibasaki and colleagues introduced La-Li
3-(biphenyldiolate)
3/NaI complex (
33) as the chiral Lewis acid catalyst for catalytic asymmetric cyclopropanation reaction of enones or
α,
β-unsaturated pyrrole amides
34 (
Scheme 22) [
68]. This methodology furnished diverse functionalized cyclopropane derivatives
35 in high yields and with excellent enantioselectivities. In this methodology, the solution of dimethylsulfoxonium methylide
2 was prepared from trimethylsulfoxonium chloride analogue of
1 by treatment with NaH in THF, followed by filtration (× 2). This procedure helped to avoid undesired salt effects for the Lewis acid-catalyzed asymmetric cyclization reaction.
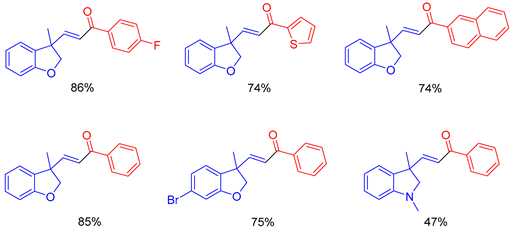
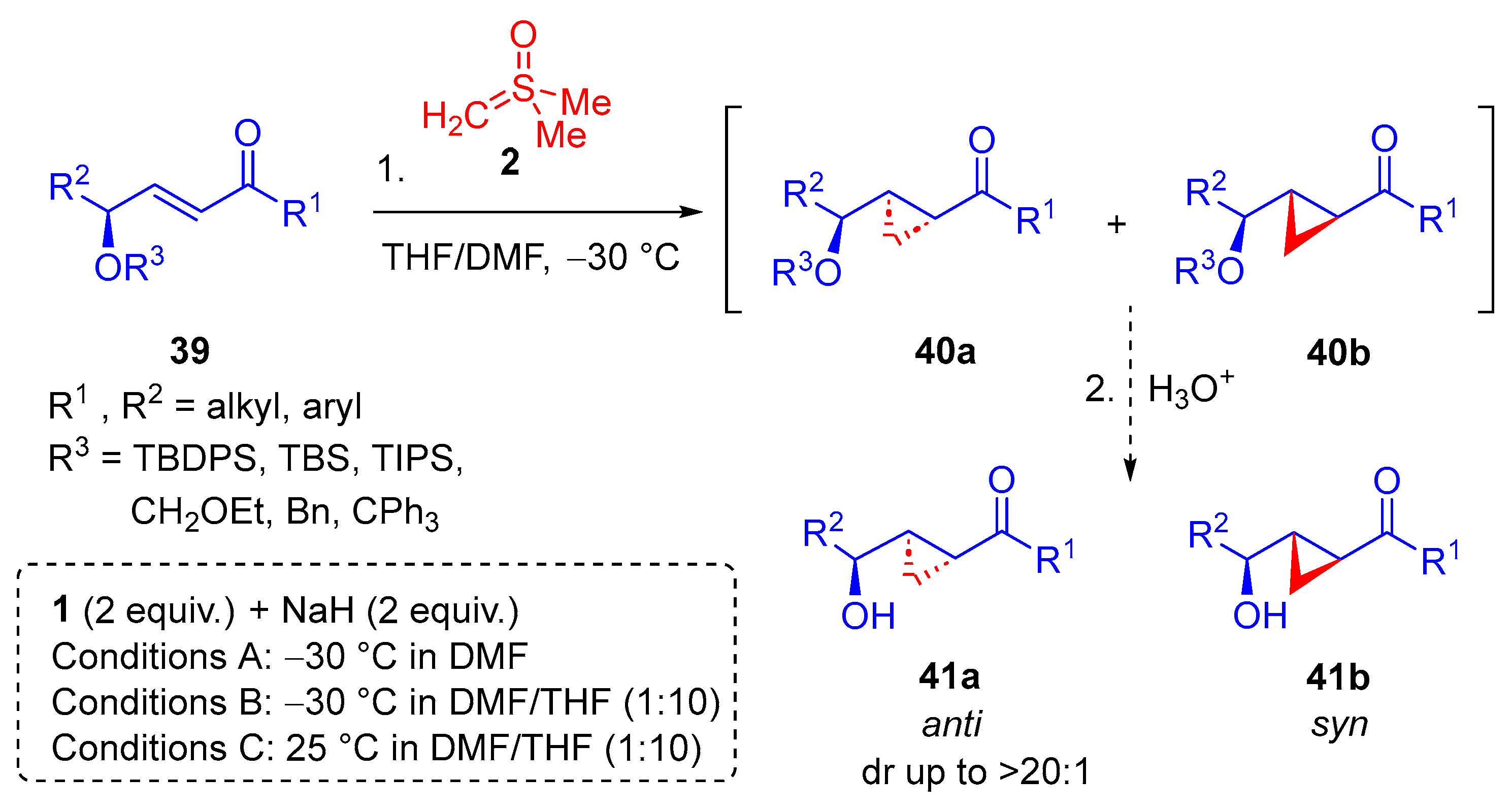
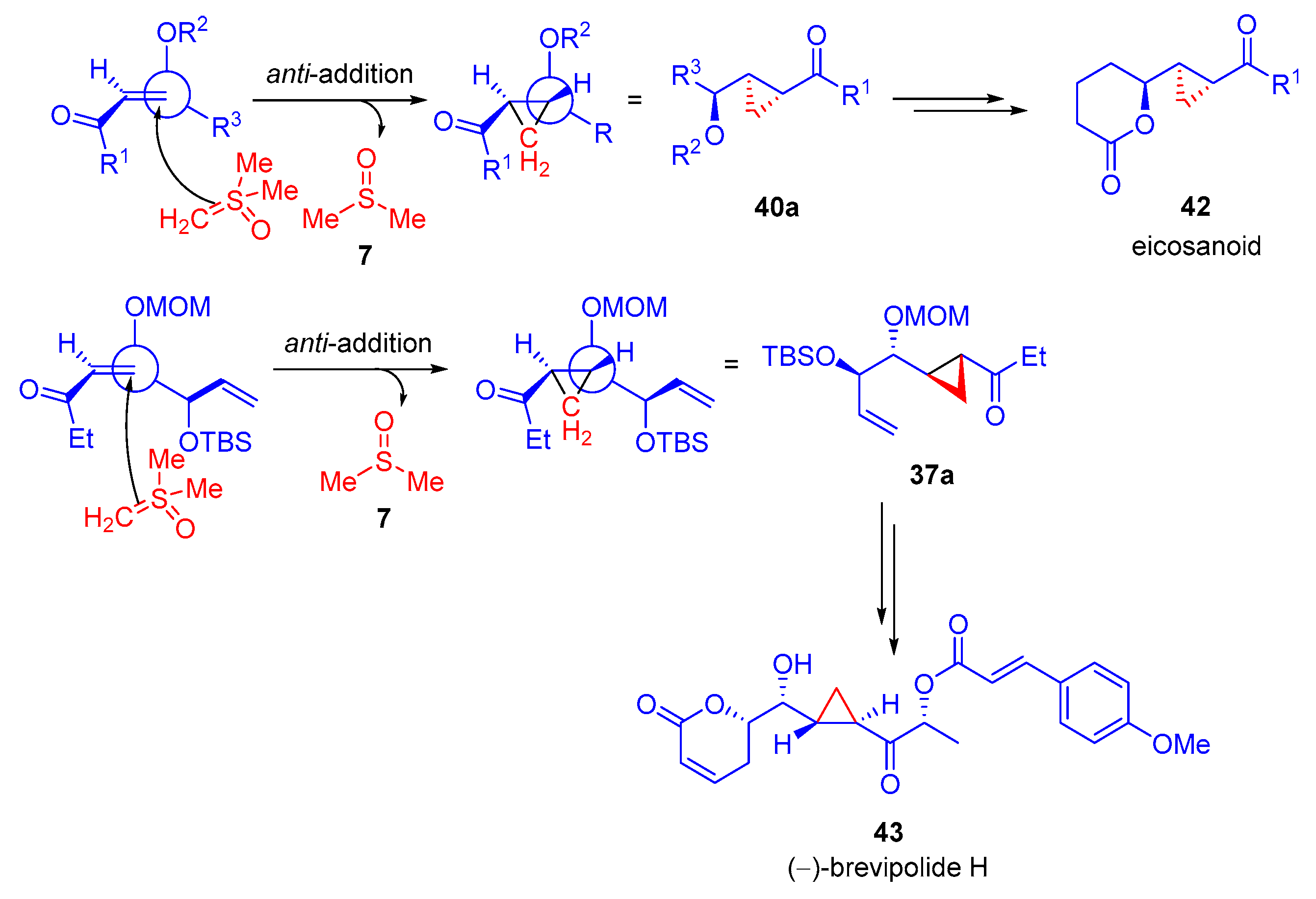
In 2014, Hou and colleagues reported the diastereoselective synthesis of cyclopropanes through the
anti-addition of dimethylsulfoxonium methylide
2 to
α,
β-unsaturated ketones
36 as part of the first formal synthesis of (−)-brevipolide H
43 [69]. The reaction was shown to operate most effectively using a solvent system of THF and DMF (20:1) at −78 °C for 6–8 h. After deprotection,
38a was afforded with high diastereoselectivity (
Scheme 23). In addition to the work carried out in 2014, they reported a diastereoselective
anti-addition of dimethylsulfoxonium methylide
2 to an
α,
β-unsaturated ketone
39 in the formal synthesis of an eicosanoid
42 [70]. A number of enones were probed, and three different sets of reaction conditions were developed to suit substrates, substituted with a particular protecting group (
Scheme 24).
The diastereoselectivity observed in the formal synthesis of both (−)-brevipolide H
43 and an eicosanoid
42 is understood to be as a result of
anti-addition of dimethylsulfoxonium ylide
2 to the
α,
β-unsaturated carbonyl starting material (
36 and
39) (
Scheme 25). This occurs due to the bulky alkoxy group being perpendicular to the alkene, which sterically hinders the methylene transfer trajectory occurring in
syn-addition and promotes
anti-addition from the less sterically hindered side. The favored spatial arrangement of the alkoxy group to the alkene maximizes the overlap of the C-O
σ-bond and alkene
π*-orbital.
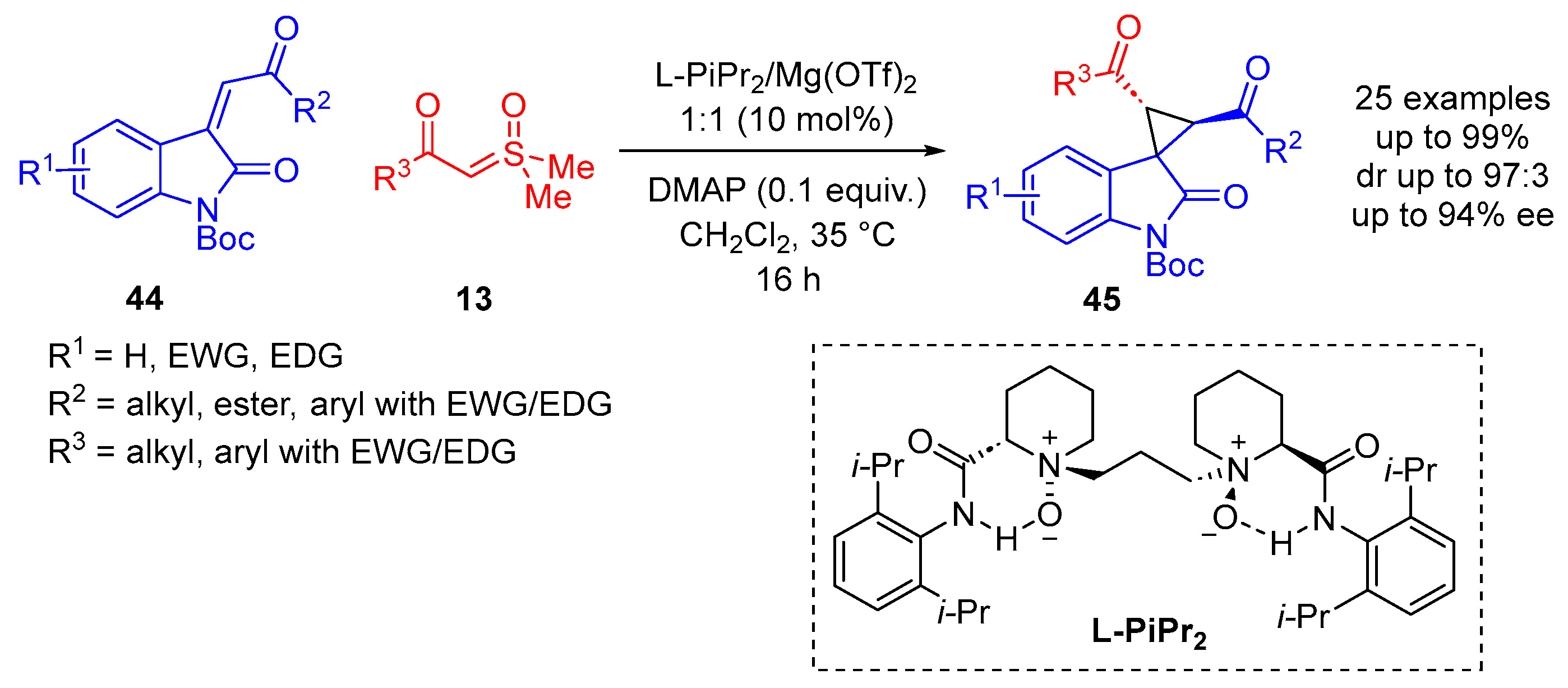
In 2018, Feng and colleagues reported the catalytic asymmetric synthesis of chiral spiro-cyclopropyl oxindoles
45 in good yields of up to 99%, with good diastereoselectivity and high enantioselectivity (
Scheme 26) [
71]. This methodology uses a chiral
N,
N-dioxide/Mg(OTf)
2 complex. Nickel(II) and zinc(II) catalysts were probed when the reaction was initially optimized, both of which provided moderate yields (66% and 69%, respectively) and good diastereoselectivity (dr 93:7 and dr 95:5, respectively). L-PiPr
2 was determined to be the best chiral ligand when utilized with Mg(OTf)
2 in CH
2Cl
2 at 35 °C for 16 h, providing a yield of 99%, diastereoselectivity of 92:8 and enantioselectivity of 91% ee. They reported 25 substrate examples, exploring the scope of the sulfoxonium ylides
13 and 3-phenacylideneoxindoles
44 [
71]. Both electron-donating and electron-withdrawing groups were tolerated when substituted on the oxindole backbone (R
1). Alkyl, ester and aryls bearing electron-donating or electron-withdrawing groups were tolerated when probed as the R
2 and R
3 substituents.
As discussed in the Epoxidation section (
Section 4.1), the developing area of interrupted Johnson–Corey–Chaykovsky reactions has seen success in the context of cyclopropanation reactions [
40,
41,
42]. In 2019, Ramasastry and colleagues developed an interrupted Johnson–Corey–Chaykovsky reaction methodology in which they discovered the unexpected formation of cyclopropanoids through the reaction of dimethylsulfoxonium methylide
2 (1.2 equiv.) with
α,
β-unsaturated aryl ketones
34aa in DMSO for 30 min at room temperature (
Scheme 27) [
40]. It was reported that, depending upon the functional group in the
ortho-position, an unexpected cyclopropanoid product would be formed. Three different
α,
β-unsaturated aryl ketones were probed in reaction with a sulfoxonium ylide. Cyclopropane-fused tetralones
46 were observed to form in the case where a formyl group was placed in the
ortho-position of
α,
β-unsaturated ketone
34, contrary to the expected cyclopropanation of the
α,
β-unsaturated ketone, or indeed, epoxidation of the carbonyl group.
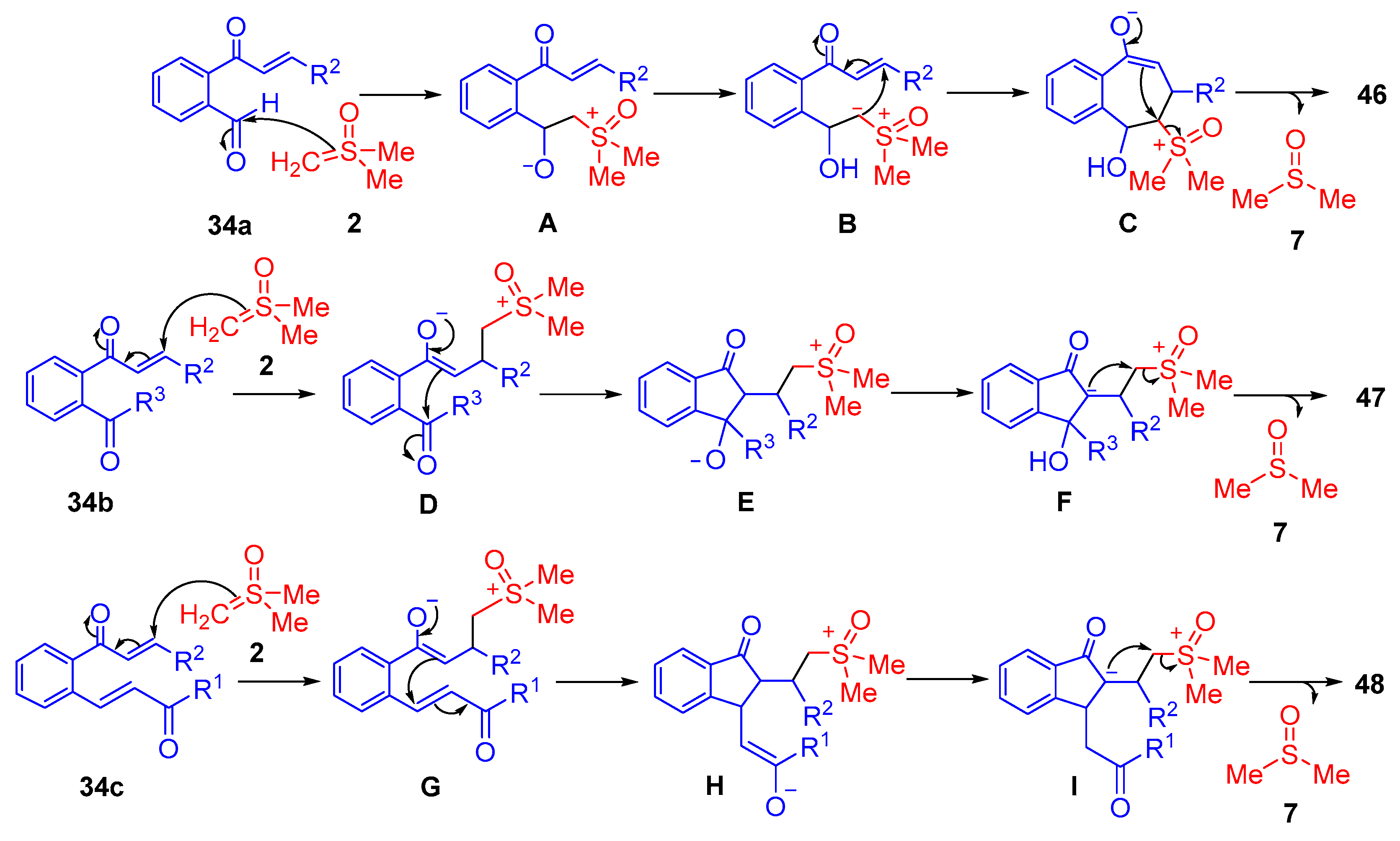
The plausible reaction mechanism starts with an initial 1,2-addition of dimethylsulfoxonium methylide
2 to the carbonyl group of the aldehyde
34a, which is followed by a 1,3-H
+ transfer in
A (
Scheme 28). The ylidic carbanion of intermediate
B carries out a 1,4-addition in intramolecular fashion. The resulting enolate of intermediate
C nucleophilically attacks the carbon adjacent to the sulfoxonium group, which forms a fused cyclopropane ring
46, with concomitant loss of DMSO
7. Overall, 13 examples were reported with good to excellent yields (70-92%) and good diastereoselectivity (dr up to 5:1). When a ketone group was substituted in the
ortho-position in
34b, an indene-type spirocyclopropane
47 was observed to form. The proposed reaction mechanism starts with a 1,4-addition by sulfoxonium ylide
2, and the resulting enolate of intermediate
D carries out a 1,2-addition to the carbonyl of the ketone. A 1,5-H
+ transfer of
E occurs to afford
F, followed by the nucleophilic addition of the carbanion to the carbon adjacent to the sulfoxonium group of
F, resulting in a spiro-cyclopropane
47 (
Scheme 28). In total, 10 examples were reported with good to excellent yields (76–93%) and moderate diastereoselectivity (up to dr 4:1). In the case of unsymmetrical dienones
34c, the reaction proceeds similarly through an initial 1,4-addition by dimethylsulfoxonium methylide
2. An intramolecular cyclization occurs through a second 1,4-addition by the enolate of intermediate
G. Following this, a 1,5-H
+ transfer of
H occurs to afford
I, which allows the resulting carbanion to nucleophilically attack the carbon adjacent to the sulfoxonium group of
I, leading to the formation of an indene-type spirocyclopropane
48 (
Scheme 28). Overall, 10 examples were reported with good to excellent yields (78–91%) and moderate to good diastereoselectivity (dr up to 6:1). The three reactions demonstrated tolerance for alkyl, heteroaryl and aryls bearing EWG/EDG groups substituted in the R
1, R
2 and R
3 positions. The aryl backbone also showed tolerance for EDG/EWG-substituted aryls and heteroaryls.
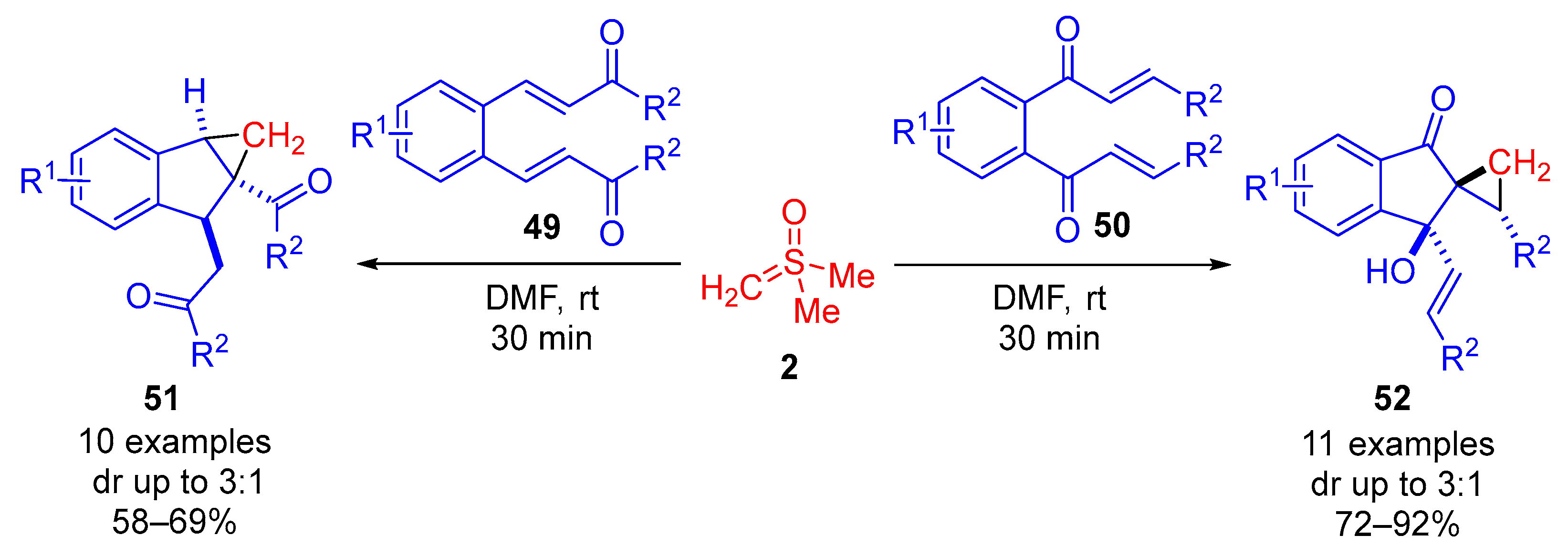
In 2019, Ramasastry and colleagues further explored the reaction between dimethylsulfoxonium methylide
2 and symmetrical aryl dienones (
Scheme 29) [
41]. The paper described the unexpected synthesis of cyclopropanoids through desymmetrization by sulfoxonium ylides. The reaction proceeded optimally with 1.2 equivalents of sulfoxonium ylide in DMF at room temperature for 30 min. When the symmetrical dienone with terminal ketone groups
49 was treated with sulfoxonium ylide
2 under these conditions, 10 examples of cyclopropane-fuse indanes
51 were formed in good reaction yields (58−69%) and with moderate diastereoselectivity (dr up to 3:1). Symmetrical dienones with terminal vinyl groups
50 produced 11 examples of indeno-spirocyclopropane
52 in good to excellent reaction yields (72–92%) and with moderate diastereoselectivity (dr up to 3:1). The paper reported substrate tolerance for halogen-, alkyl- and methoxy-substituted aryls in the R
1 and R
2 positions.
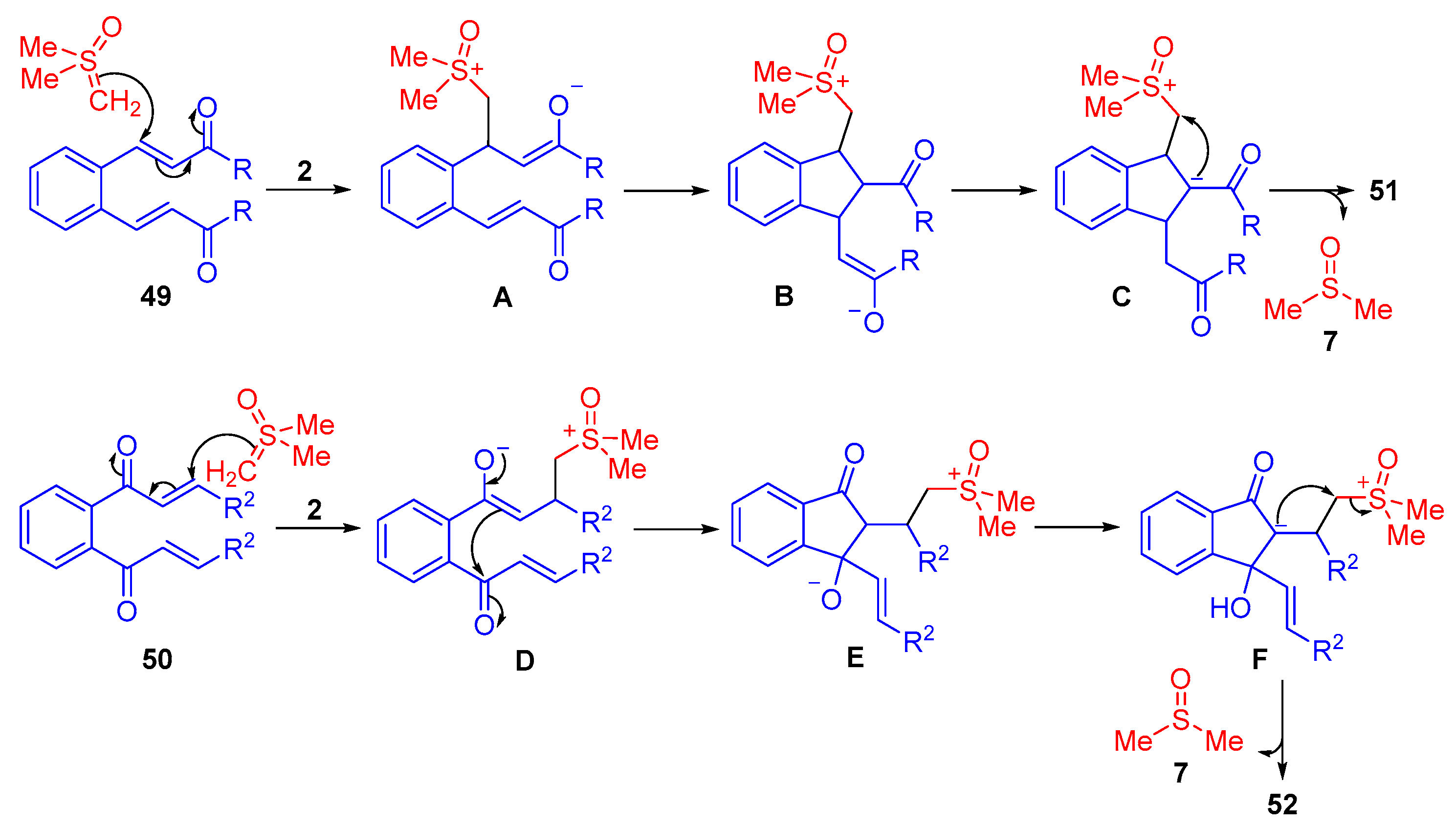
The reaction mechanism for the unexpected synthesis of cyclopropane-fused indanes from symmetrical dienones with terminal ketone groups
49 proceeds with an initial 1,4-addition by the sulfoxonium ylide
2 (
Scheme 30). The enolate of intermediate
A intramolecularly cyclizes through another 1,4-addition. A 1,3-H
+ transfer of
B occurs, which allows the resulting carbanion of
C to displace DMSO
7 and form a cyclopropane-fused indane
51 (
Scheme 30). In the case of terminal vinyl groups
50, the reaction also starts through an initial 1,4-addition by a sulfoxonium ylide
2. A 1,2-addition occurs between the enolate of the resulting intermediate
D to the opposite carbonyl group of the ketone. Following this, a 1,3-H
+ transfer of
E occurs and allows for the displacement of DMSO
7 by the carbanion of
F to form an indene-type spirocyclopropane
52 (
Scheme 30).
In 2023, Ramasastry and colleagues further explored the interrupted Johnson–Corey–Chaykovsky reactions of sulfoxonium ylides
2 with
α,
β-unsaturated aryl ketones bearing a pyridinium salt in the
ortho-position of
53 (
Scheme 31) [
42]. The reaction functioned most optimally in DMF at room temperature for 3 h using 1 equivalent of trimethylsulfoxonium iodide
1 and 1.2 equivalents of NaH to form the sulfoxonium ylide
2. When the starting substrate
53 was placed under these conditions, an unexpected synthesis of vicinal bis-spirocyclic indanones
54, providing three contiguous quaternary carbons, was obtained. The group reported 23 examples of vicinal bis-spirocyclic indanones with moderate to good reaction yields (49–81%) and good diastereoselectivity (dr up to 9:1). The reaction demonstrated substrate tolerance for EDG/EWG substituted aryls and heteroaryls in the R
1 and R
2 positions, alkyl groups in the R
3 position, as well as alkyl groups, EDG-substituted aryls and heteroaryls in the R
4 position.
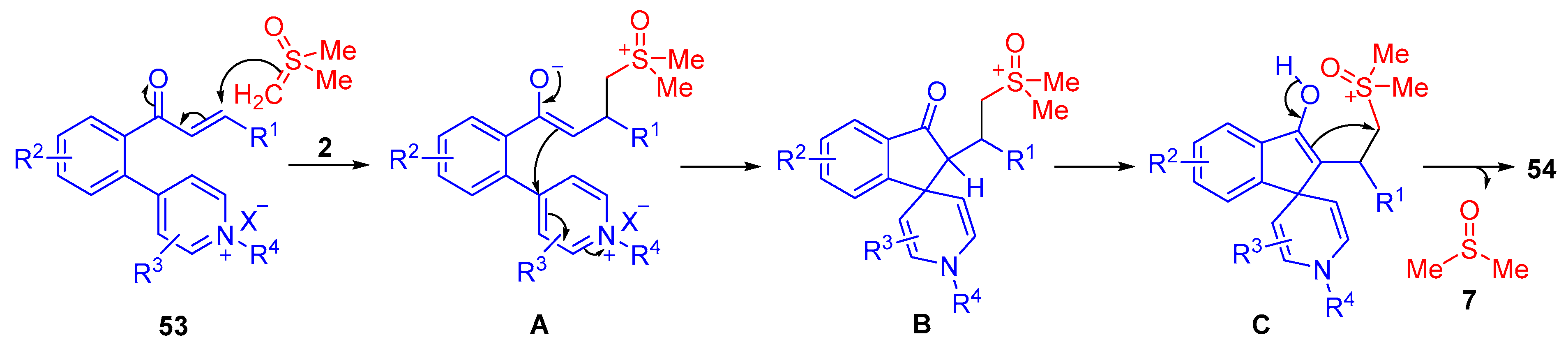
The proposed reaction mechanism starts with an initial 1,4-addition by dimethylsulfoxonium methylide
2, resulting in intermediate
A (
Scheme 32). The enolate of
A derived from
53 intramolecularly cyclizes by nucleophilically attacking C4 of the pyridinium ring, which forms an indanone ring. The keto carbonyl group of
B tautomerizes to form an enol
C. The enol nucleophilically attacks the carbon adjacent to the sulfoxonium group, allowing the leaving group to cleave and form the desired vicinal bis-spirocyclic indanone
54.
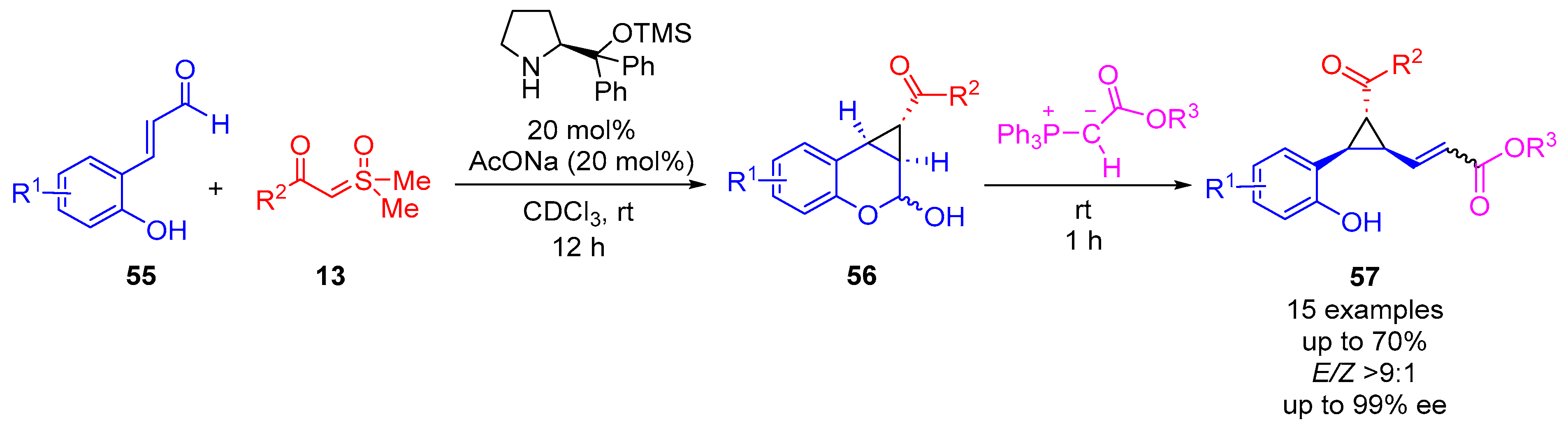
In 2022, Bernardi and colleagues developed a methodology for the enantioselective synthesis of chiral-fused cyclopropane-chromane scaffolds
56, named 1,1a,2,7b-tetrahydrocyclopropa[c]chromenes, through aminocatalysis using sulfoxonium ylides
13 with 2-hydroxycinnamaldehydes
55 (
Scheme 33) [
72]. This method utilized the Jørgensen–Hayashi catalyst in a catalytic amount (20 mol%), along with NaOAc (20 mol%), in deuterated chloroform at room temperature for 12 h. NaOAc, as an additive, was found to facilitate the best combination of enantioselectivity and reaction yield. Benzoic acid and acetic acid were also trialed and performed well, giving good reaction yields and strong enantioselectivity. The cyclopropane ring was formed with perfect diastereoselectivity. The fused cyclopropane–chromane intermediate
56, formed through aminocatalysis, underwent a Wittig olefination at room temperature for 1 h to produce 1,2,3-trisubstituted cyclopropanes
57, exhibiting 15 examples with moderate to good yields (up to 70%), excellent enantioselectivity (up to 99% ee) and good
E-selectivity (
E/Z > 9:1). The reaction showed tolerance for alkyl and activating groups substituted in the
meta- and
para- positions, in addition to the
ortho- hydroxyl group, for R
1. Ester groups and ketone groups were investigated in the R
2 position for the ylide
13, with
α-estersulfoxonium ylides observed to perform better than
α-ketosulfoxonium ylides.
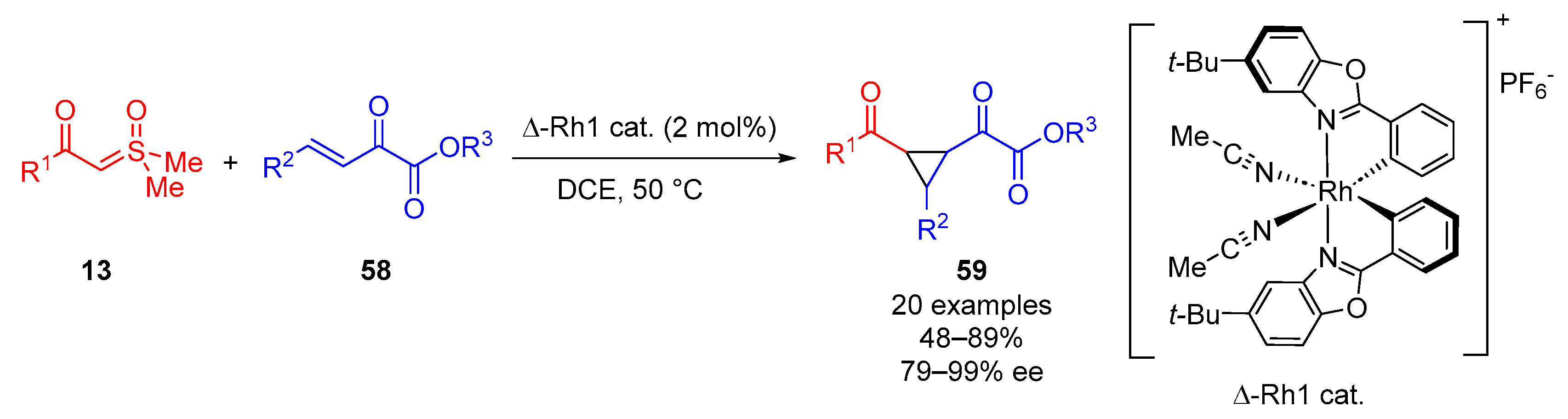
In 2022, Li and colleagues carried out the asymmetric synthesis of cyclopropanes from
α-ketosulfoxonium ylides
13 and
β,
γ-unsaturated ketoesters
58 using a chiral Rh(III) catalyst (
Scheme 34) [
73]. The synthesis of 20 examples of chiral 1,2,3-trisubstituted cyclopropanes
59 was reported to proceed with moderate to excellent yields (48–89%), high enantioselectivity (79–99% ee) and excellent diastereoselectivity (dr > 20:1). Following optimization studies, the most effective rhodium catalyst (Δ-Rh1 cat.) facilitated the best yield, diastereoselectivity and enantioselectivity when the reaction was carried out in 1,2-dichloroethane (75%, dr > 20:1, 97% ee) at 50 °C for 45 h under an argon atmosphere. Toluene facilitated high diastereoselectivity (dr > 20:1) and high enantioselectivity (91% ee), albeit with a moderate reaction yield of 58%. THF facilitated high diastereoselectivity (dr > 20:1) but moderate enantioselectivity (40% ee), and a poor product yield of 10% was obtained. Alkyl, electron-donating/electron-withdrawing group-substituted aryl and heteroaryl groups were tolerated when probed as the R
1 and R
2 substituents.
In 2023, Zhao and colleagues reported the synthesis of cyclopropanes
61 through an Au-catalyzed (2 + 1)-cycloaddition of allene-type tosylamide
60 with sulfoxonium ylides
13 (
Scheme 35) [
74]. A series of
α-ketosulfoxonium ylides were probed in reaction with tosylamide
60, resulting in 20 examples with reaction yields of up to 94% and good diastereoselectivity (up to dr 25:1). The reaction was initially optimized to determine the most suitable Au-catalyst for the synthesis of 1-benzoyl-2-formylcyclopropane. A mixture of PhPAuCl (5 mol%) and AgNTf
2 (5 mol%) in CH
3CN provided the best results when refluxed for 5 h, whereas using Ph
3PAuCl (5 mol%) or AgNTf
2 (5 mol%) singularly resulted in no reaction. CH
3CN was observed to afford the best reaction yield, with toluene, CH
2Cl
2, THF and EtOAc providing moderate yields (38–58%). The substrate scope was investigated through the exploration of 20 different
α-ketosulfoxonium ylides
13, showing tolerance for the alkyl, heteroaryl and aryl groups (as R
1). Both electron-donating and electron-withdrawing group-substituted aryls were tolerated.
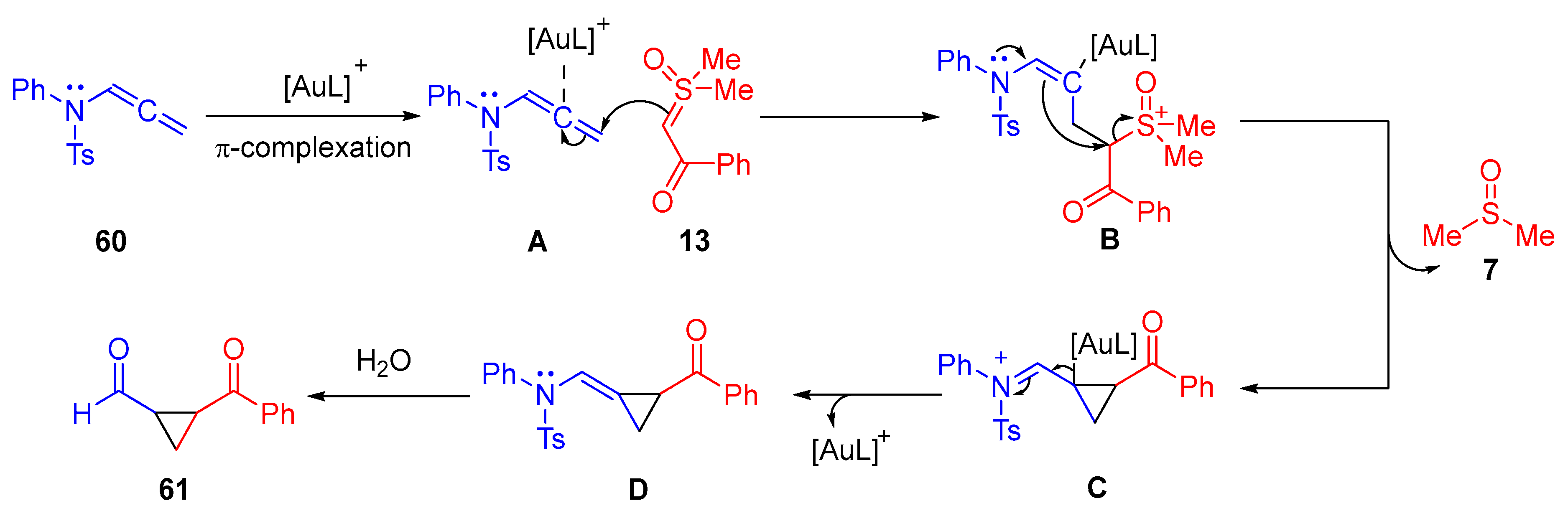
The plausible reaction mechanism starts with an initial
π-complexation of the cationic gold-catalyst to the allene
60 (
Scheme 36). Following this,
α-ketosulfoxonium ylide
13 nucleophilically attacks the terminal end of the Au-complexed allene
A. The resulting Au-substituted enamide intermediate moiety intramolecularly attacks the carbon adjacent to the sulfoxonium group in
B, eliminating DMSO
7 and forming
C. An enamide
D forms as a result of the cationic Au-catalyst being eliminated. Intermediate
D then undergoes acid-catalyzed hydrolysis to form the formylated cyclopropane product
61.
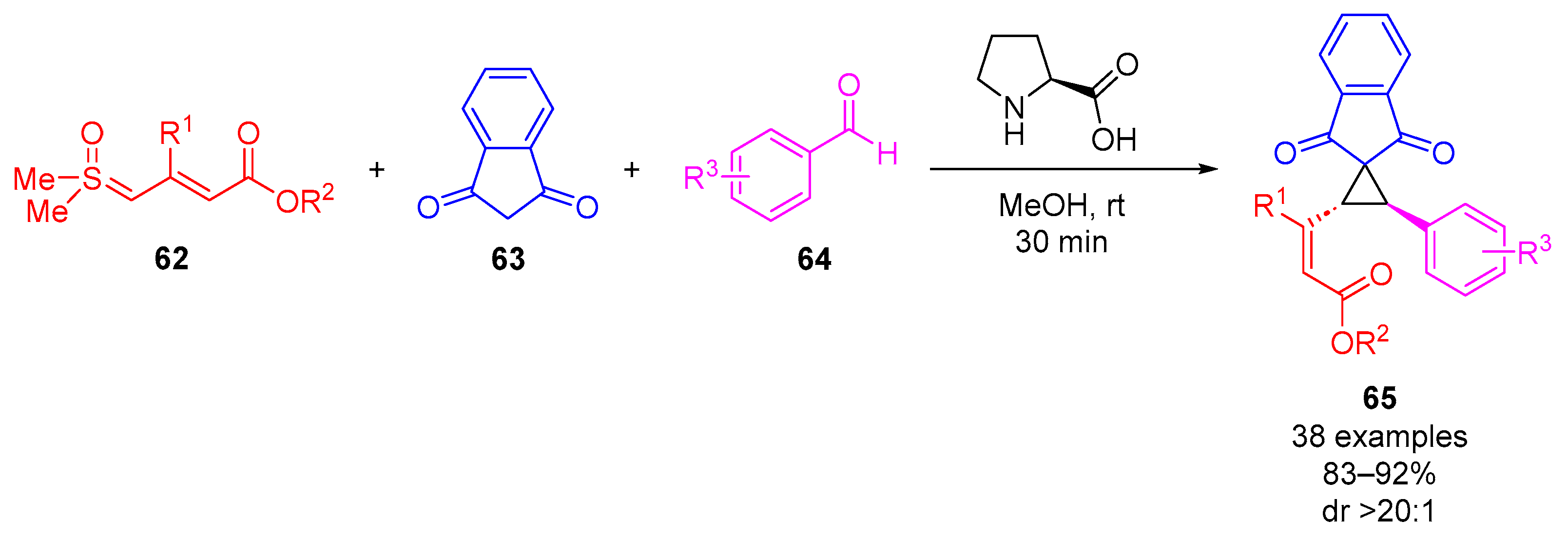
Vaitla and colleagues developed the L-proline-catalyzed diastereoselective reaction sequence involving cyclopropanation using aryl aldehydes
64, indane-1,3-diones
63 and vinyl sulfoxonium ylides
62 (
Scheme 37) [
75]. They reported 38 examples of 1,2,3-trisubstituted cyclopropanes
65 obtained in excellent yields (83–92%) and with excellent diastereoselectivity (dr > 20:1). The cyclopropanation reaction functioned most effectively when carried out in MeOH at room temperature for 30 min using L-proline as catalyst (20 mol%), 1.4 equivalents of sulfoxonium ylide
62, 1.1 equivalents of aldehyde
64 and 1 equivalent of indane-1,3-dione
63. A series of different catalysts were trialed in place of L-proline, and all showed excellent diastereoselectivity (dr > 20:1). Indium(III) triflate, scandium(III) triflate and triethylamine resulted in poor reaction yields of 15%, 22% and 34%, respectively. Tetrafluoroboric acid diethyl ether complex performed effectively to give a reaction yield of 85%. DMSO, CH
3CN and EtOAc were explored as solvents and gave good yields of 80%, 65% and 78%, respectively. In terms of substrate scope, the reaction was tolerant of EDG/EWG-substituted aryl, alkyl and heteroaryl aldehydes as well as EDG/EWG-substituted aryl at the
β-position (R
1) of vinyl sulfoxonium ylides.
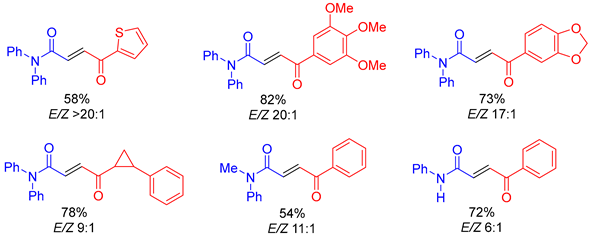
4.4. Olefination
The C=C double bond is a central functional group in organic chemistry due to its importance and application in total synthesis [
76]. Typical methodologies to construct C=C double bond include the Wittig reaction, Peterson, Julia, Tebbe olefinations and the Horner–Wadsworth–Emmons reaction by employing phosphonium ylides/phosphonate anions, silicon-stabilized carbanions, sulfones or metal alkylidenes as reagents [
77,
78,
79,
80,
81]. All these reactions proceed through cyclic intermediates, either a four-membered ring or five-membered spirocyclic intermediate.
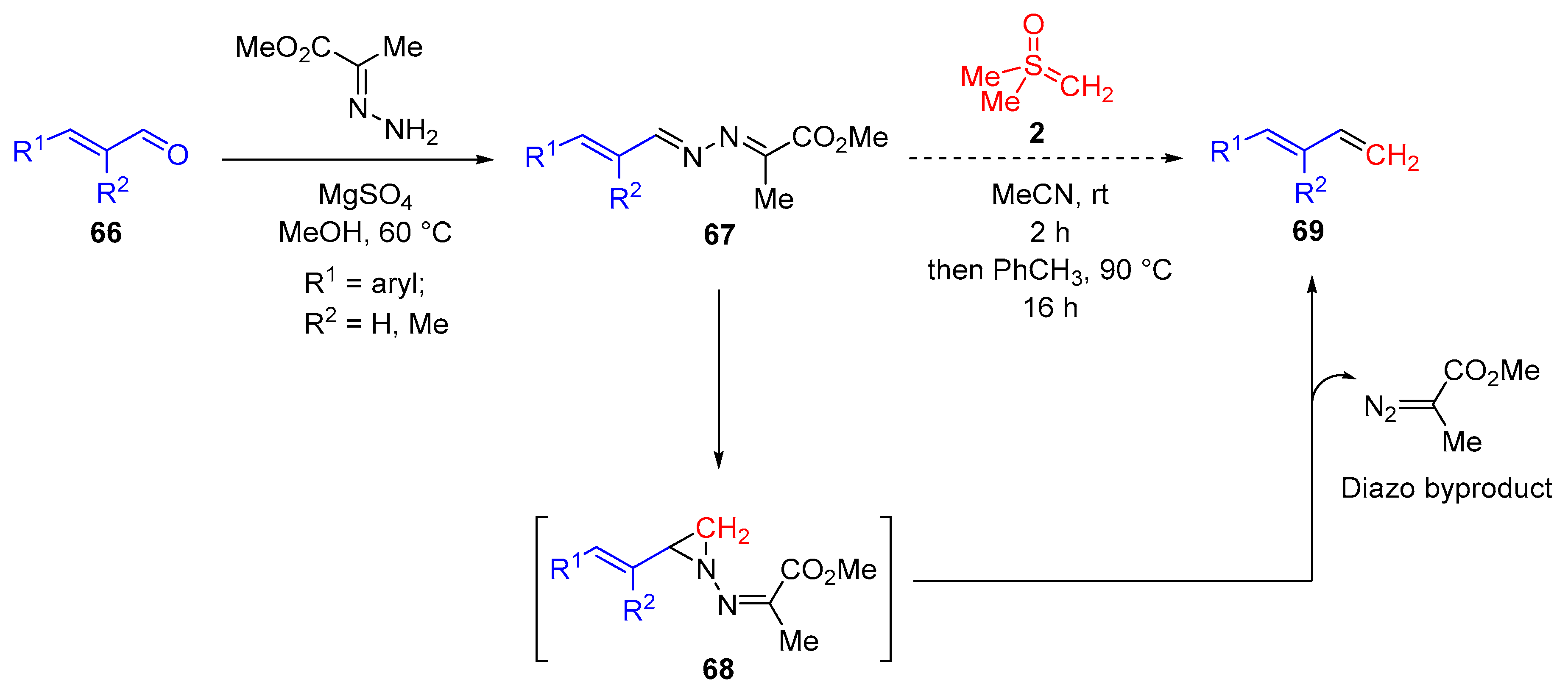
Very recently, Maulide and colleagues reported a three-membered ring approach to carbonyl olefination (
Scheme 38) [
82]. They applied a one-pot procedure to access the key intermediate, a three-membered
N-iminyl aziridine
68, by addition of a sulfoxonium ylide
2 to an in situ-generated
N-iminyl imine (azine)
67 from the corresponding aldehyde
66. Cheletropic cycloreversion or thermal decomposition of the N-iminyl aziridine
68 led to the desired alkene
69 in good to high yields. This methodology tolerated substrates ranging from aromatic aldehydes, aliphatic aldehydes and
α,
β-unsaturated aldehydes to sensitive
α-chiral aldehydes. The
E-isomer selectivity of the alkene products might be explained by involvement of the more stable
trans-aziridine intermediate or non-concerted ring-opening pathways causing isomerization. The procedure was not successful for trisubstituted olefin synthesis because the required aziridine could not be obtained from either a branched sulfoxonium salt or from a ketone. However, the reaction provides a convenient alternative to Wittig-type methylenation in that it avoids the generation of difficult-to-separate triphenylphosphine oxide byproduct.
In addition to this, they reported a methodology for the stereoselective synthesis of 1,2-disubstituted alkenes
70 from
α-diazoesters
18a and
α-ketosulfoxonium ylides
13 through ruthenium catalysis (
Scheme 39) [
83]. It was reported that 22 examples were obtained in moderate to good reaction yields (40–78%), and very good
Z-selectivity (
Z/E up to >13:1) was also demonstrated (
Table 5). The reaction showed tolerance for bulky alkyl ester groups substituted at the R
1 position as well as neutral and EWG-substituted aryls at the R
2 position. The stereoselective cross-olefination proceeds through initial formation of a carbene
A, generated from the loss of nitrogen of an
α-diazoester
18a. A metal carbenoid
B was formed through the combination of the carbene and the [Ru(cymene)
2Cl
2]
2 catalyst. The metal carbenoid coupled with the sulfoxonium ylide
13, allowing the formation of the
Z-olefin product
70.
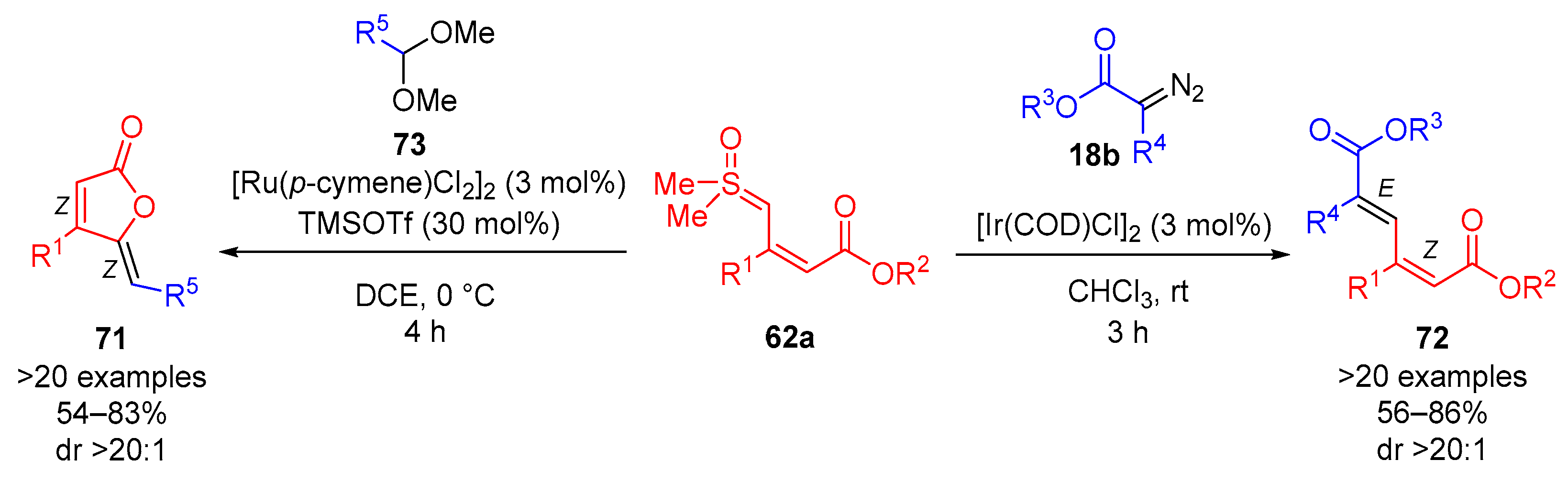
In 2024, Vaitla and colleagues reported a diastereoselective synthesis of conjugated dienoates
72 from
α-diazoesters
18b and vinyl sulfoxonium ylides
62a using iridium-catalysis (
Scheme 40) [
84]. In addition, the paper described a diastereoselective synthesis of ylidene butenolides
71 from acetals
73 and vinyl sulfoxonium ylides
62a using ruthenium-catalysis aided by TMSOTf. The synthesis of conjugated dienoates
72 provided >20 examples with excellent diastereoselectivity (dr > 20:1) and in moderate to very good reaction yields (56–86%). The reaction was observed to function most efficiently in CHCl
3 at room temperature for 5 h, using equimolar amount of sulfoxonium ylide
62a and
α-diazoester
18b and 3.0 mol% of [Ir(COD)Cl]
2. The conjugated dienoates
72 obtained were formed as the
Z,
E-isomer, which was congruent with
cis,
trans-muconic acid. The reaction was tolerant of EDG-substituted and EWG-substituted (R
1, R
5) aryls. This work reported >20 examples of ylidene butanolides
71 formed with excellent diastereoselectivity (dr > 20:1) and in favorable yields (54–83%). The optimal reaction conditions were determined to be DCE as solvent at 0 °C for 4 h, using 3.0 mol% of [Ru(
p-cymene)Cl
2]
2 and 30 mol% of TMSOTf. The diastereoselectivity of the ylidene butenolide
71 synthesis favored the formation of the
Z,
Z-isomer, which is congruent with the structure of rubrolide analogues. This reaction was observed to be tolerant of heteroaryls and EWG- and EDG-substituted (R
1, R
4) aryls.
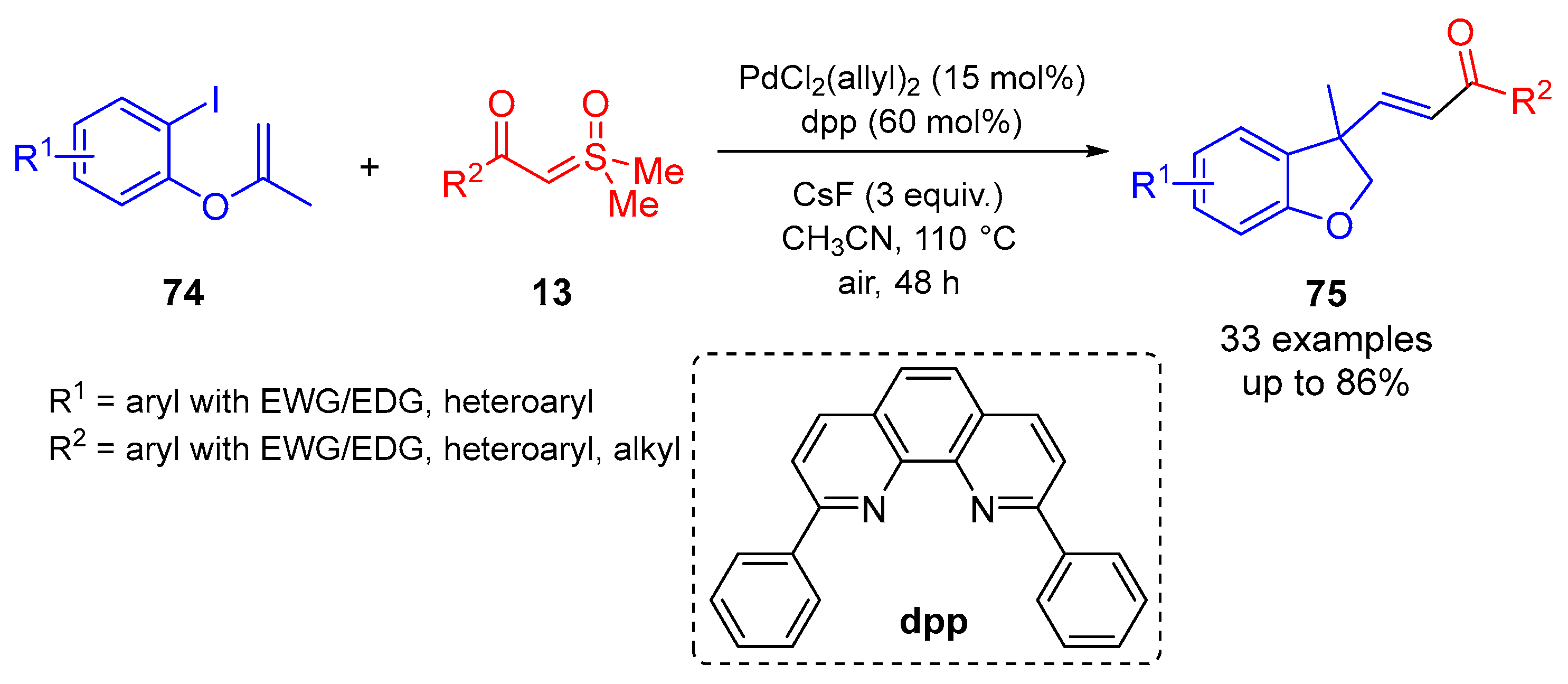
Jiang and colleagues reported a Pd-catalyzed stereoselective synthesis of
α,
β-unsaturated carbonyls from
β-ketosulfoxonium ylides and 1-iodo-2-((methylallyl)oxy)benzene compounds
74, which proved to be favorable across a range of substrates for both starting materials (
Scheme 41 and
Scheme 42,
Table 6) [
85]. The initial reaction optimization study found that the cross-coupling reaction between
β-ketosulfoxonium ylides and 1-iodo-2-((methylallyl)oxy)benzene compounds performed best at 110 °C for 48 h in CH
3CN with the PdCl
2(allyl)
2 catalyst (15 mol%), 2,9-diphenyl-1,10-phenathroline ligand (60 mol%) and 3.0 equiv. of CsF, resulting in an 86% yield of benzofuran-type product
75. Other Pd-based catalysts were probed, including PdCl
2 and PdBr
2, and both resulted in lower yields of 18% and 10%, respectively. Several phosphine-based and chelating ligands were examined, with 2,9-diphenyl-1,10-phenathroline performing most effectively. CH
3CN was determined to produce the best performance when compared to the other solvents investigated, such as DCE, THF and acetone. The reaction was found to perform effectively under an air atmosphere, with no beneficial effect when carried out under a N
2 or O
2 atmosphere. A number of Cs bases were examined, with 3.0 equiv. of CsF providing the best results. This work provides an effective stereoselective olefination methodology which favors the formation of
E-isomers, with moderate to good yields (35–86%) and is tolerant of a wide scope of
β-ketosulfoxonium ylide
13 and 1-iodo-2-((methylallyl)oxy)benzene
74 starting substrates.
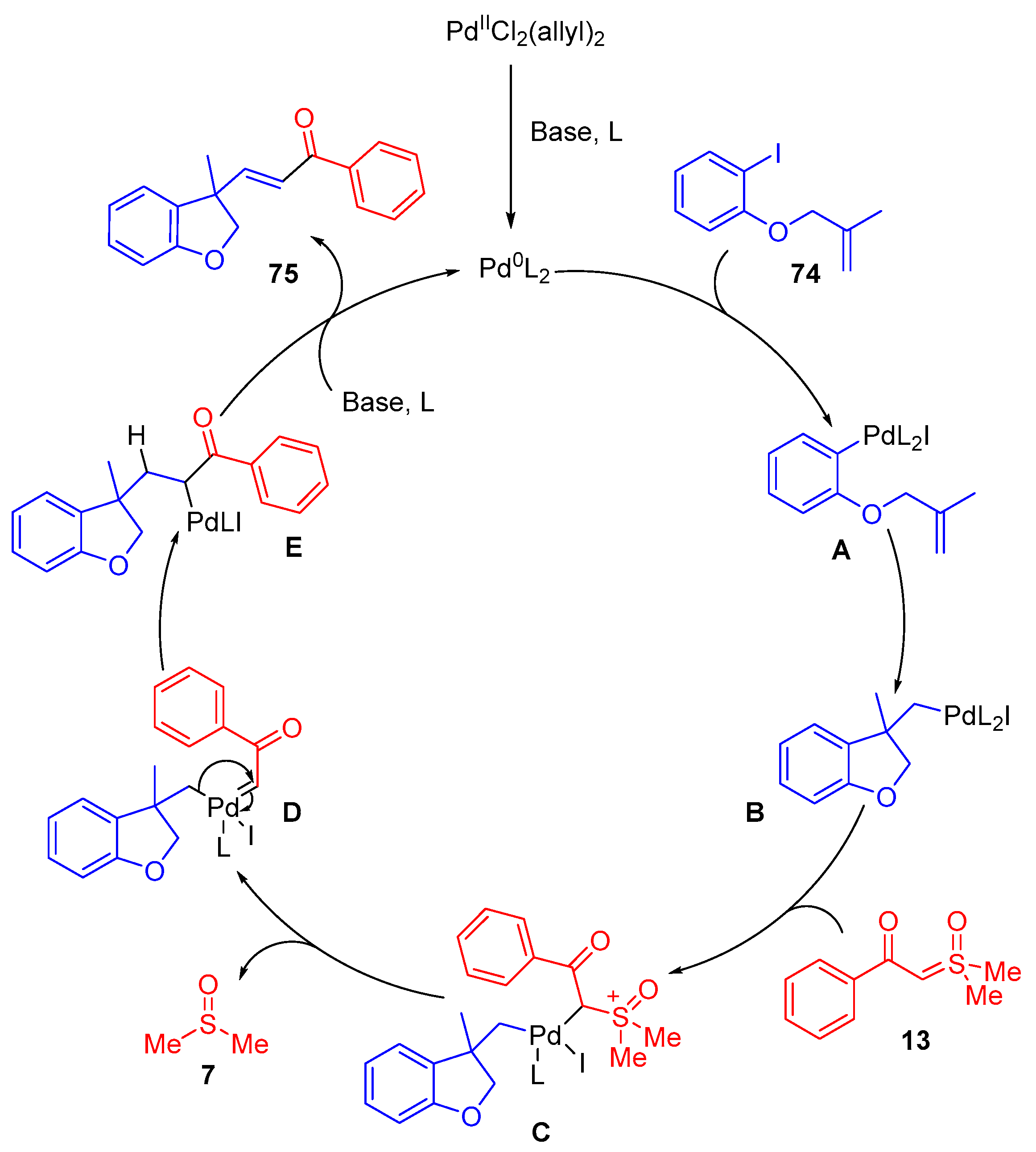
The proposed mechanism proceeds via an initial reduction of the Pd(II)-catalyst using basic CsF and with 2,9-diphenyl-1,10-phenathroline and triphenylphosphine acting as ligands. The Pd(0)-catalyst inserts into the C–I bond of 1-iodo-2-((methylallyl)oxy)benzene 74 in an oxidative addition step. The Pd(II)-complex A undergoes intramolecular cyclization to form the 2,3-dihydrobenzofuranyl ring B. β-ketosulfoxonium ylide 13 associates to the Pd(II)-complex, DMSO 7 is eliminated, and a Pd-carbene D is formed. Following a 1,2-alkyl migration, an alkylpalladium complex E is formed, which undergoes β-hydride elimination to form the α,β-unsaturated carbonyl product 75 with reforming Pd(0)-catalyst.
Liao and colleagues reported an iridium-catalyzed olefination of sulfoxonium ylides
13, providing 80 examples with excellent
E-isomer diastereoselectivity (
E/Z up to >20:1) of disubstituted alkenes
77 in favorable yields of up to 88% (
Scheme 43) [
86]. They reported the reactions of
α-ketosulfoxonium ylides or
α-estersulfoxonium ylides with
α-amidesulfoxonium ylides or
N-heterocyclic
α-amidesulfoxonium ylides. The most optimal conditions employed were
α-amidosulfoxonium ylide
76 (1 equiv.) and
α-carbonylsulfoxonium ylide
13 (1.5 equiv.) in CH
2Cl
2 at −10 °C for 24 h under an N
2 atmosphere. The catalyst employed was [Ir(COD)Cl]
2 (5 mol%). The proposed reaction mechanism starts with the initial formation of an Ir−carbene intermediate
A through the reaction of Ir(I) catalyst with ylide
76, which eliminates DMSO
7 (
Scheme 44). An
α-amidosulfoxonium ylide was observed to form a carbene metalloid faster than an
α-carbonylsulfoxonium ylide. Following this, an
α-carbonylsulfoxonium ylide
13 adds to the carbon adjacent to the Ir catalyst to afford
B. Elimination of DMSO
7 and the Ir catalyst provides
C, and following catalyst dissociation,
E-alkene
77 is obtained (
Scheme 44). In terms of substrate scope, the reaction was shown to be tolerant of heterocyclic, aryl and alkyl groups in the R
1 and R
2 positions. EDG/EWG-substituted aryl, alkyl, vinyl and heteroaryl groups were tolerated in the R
3 position (
Scheme 44,
Table 7).
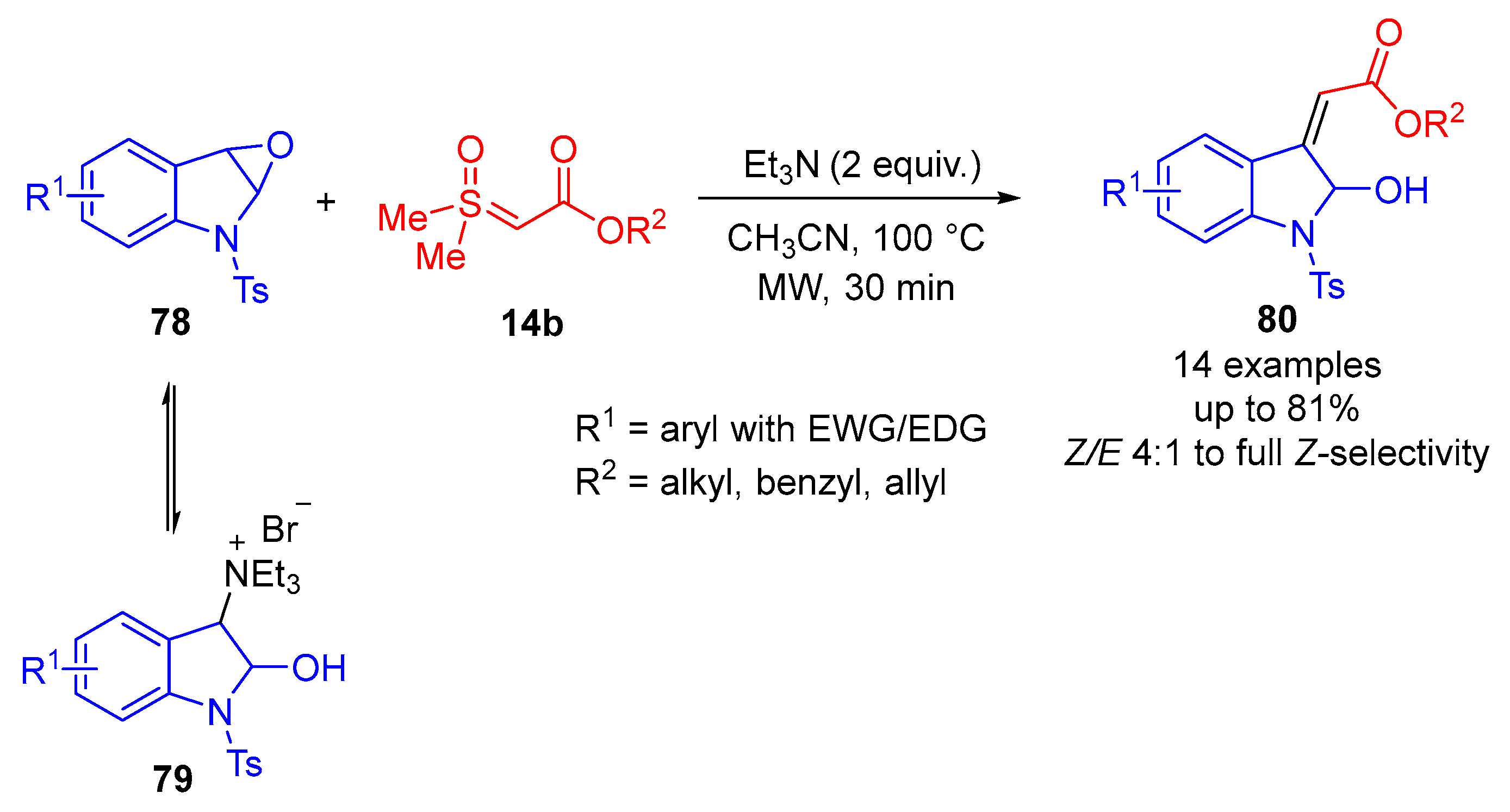
Recently, in 2024, Burtoloso and colleagues reported a selective ring-opening of epoxide-fused indolines
78 by
α-estersulfoxonium ylides
14b (
Scheme 45) [
87]. Epoxide-fused indolines were unstable and were prepared in situ from 2-hydroxyindoline-3-triethylammonium bromide
79 solution as developed by Abe and colleagues [
88]. They detected the formation of an epoxide-fused indoline
78 in solution through NMR studies [
87]. The reaction functioned most effectively using two equivalents of triethylamine in CH
3CN at 100 °C for 30 min under microwave irradiation. When one equivalent of 2-hydroxyindoline-3-triethylammonium bromide
79 and two equivalents of sulfoxonium ylide
14b were treated under these reaction conditions, 14 olefin examples (
80) were formed in favorable yields of up to 81% and with moderate to excellent
Z-selectivity (
Z/E 4:1 to exclusively
Z-isomer formation). The reaction demonstrated tolerance for the activated and deactivated indolines (R
1), as well as the alkyl, benzyl and allyl groups substituted at the R
2 position.
4.5. Insertion Reactions
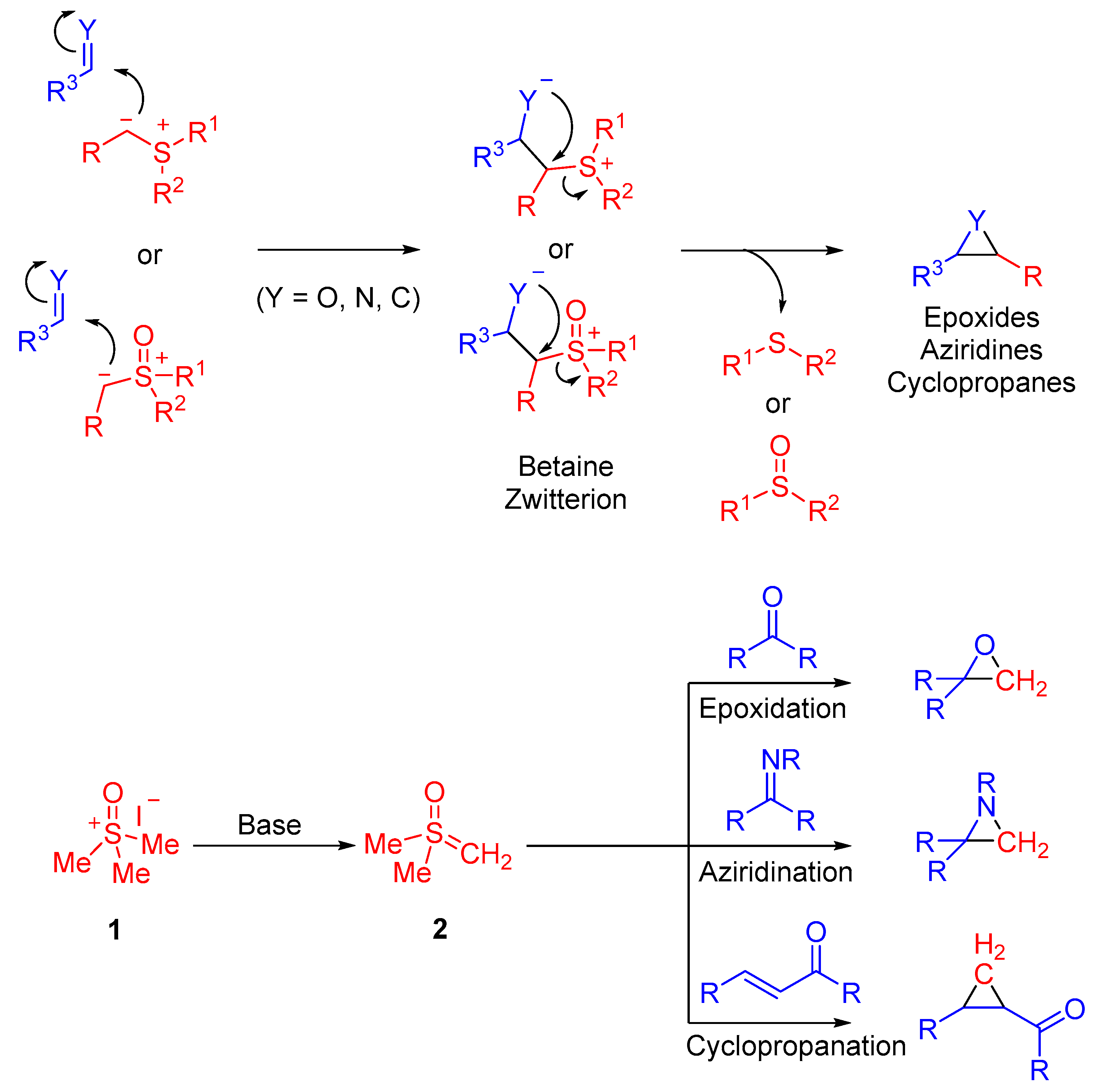
The most commonly observed mechanistic pathway of sulfoxonium ylides is the formal cycloaddition: (2 + 1) (e.g., epoxides, cyclopropanes, aziridines), (3 + 1) (e.g., oxetanes, azetidines), (4 + 1) (e.g., indanones, indolines). This pathway proceeds through the formation of a betaine intermediate occurring from the nucleophilic addition of the carbanion to an electrophilic site. A cyclic product forms as a result of an intramolecular cyclization. Insertion reactions of sulfoxonium ylides have also been observed to proceed with great success in recent years. This typically occurs as a result of protonation of the sulfoxonium ylide carbanion by X–H bonds (e.g., X = S, N or P), which is followed by a nucleophilic addition of X, to form an inserted product.
Mangion and colleagues reported that transition metal complexes, such as RuCl
2(Cp)(PPh
3)
2, [Ir(COD)
2Cl]
2, Pt(COD)Cl
2 and AuCl(SMe
2), could be used to produce the corresponding metal carbenoids from sulfoxonium ylides at room temperature (
Scheme 46) [
89,
90]. These metal carbenoids underwent facile reaction with anilines, Boc- or Cbz-protected aliphatic amines, thiophenols and aliphatic alcohols with good efficiency. This method has applications in both intra- and intermolecular reactions, including a practical ring-expansion strategy for lactams. The intramolecular N–H insertion reactions of the carbamate-containing sulfoxonium ylide
81 were developed to facilitate access to structurally complex aza-heterocyclic products
82 [
89]. The authors also proposed a possible reaction pathway to explain these transformations: catalyst [Ir(COD)Cl]
2 reacts with sulfoxonium ylide
81 to generate the metal carbenoid intermediate
A, and then a subsequent intramolecular N–H insertion generates zwitterionic intermediates
B and
C, which, on autoprotolysis, yields the final aza-heterocyclic product
82.
In 2020, Burtoloso and colleagues reported an enantioselective organocatalytic insertion of
α-aryl-
α-carbonylsulfoxonium ylides
14c and aryl thiols
83 (
Scheme 47) [
91]. Due to the acidic nature of thiols, aryl thiols donate a proton to sulfoxonium ylides and the nucleophilic thiolate attacks the carbon adjacent to the sulfoxonium group, eliminating DMSO and resulting in an inserted product
85. The reported methodology utilizes a chiral thiourea organocatalyst
84 to induce enantiocontrolled addition and is proposed to function using hydrogen-bonding intermolecular interactions to the sulfoxonium ylide. The enantioselectivity of the reaction was explained by a
re-face protonation of the sulfoxonium ylide. The S–H insertion was discovered to perform most efficiently in CHCl
3 at −28 °C for 24–168 h, depending on the substrate, using a bulky thiourea
84. They reported 31 examples of S–H insertions with excellent enantioselectivity (up to 95% ee) and moderate to excellent reaction yields (up to 97%), demonstrating tolerance for electron-donating and electron-withdrawing aryl substituents on the thiol
83 and sulfoxonium ylide
14c [
91].
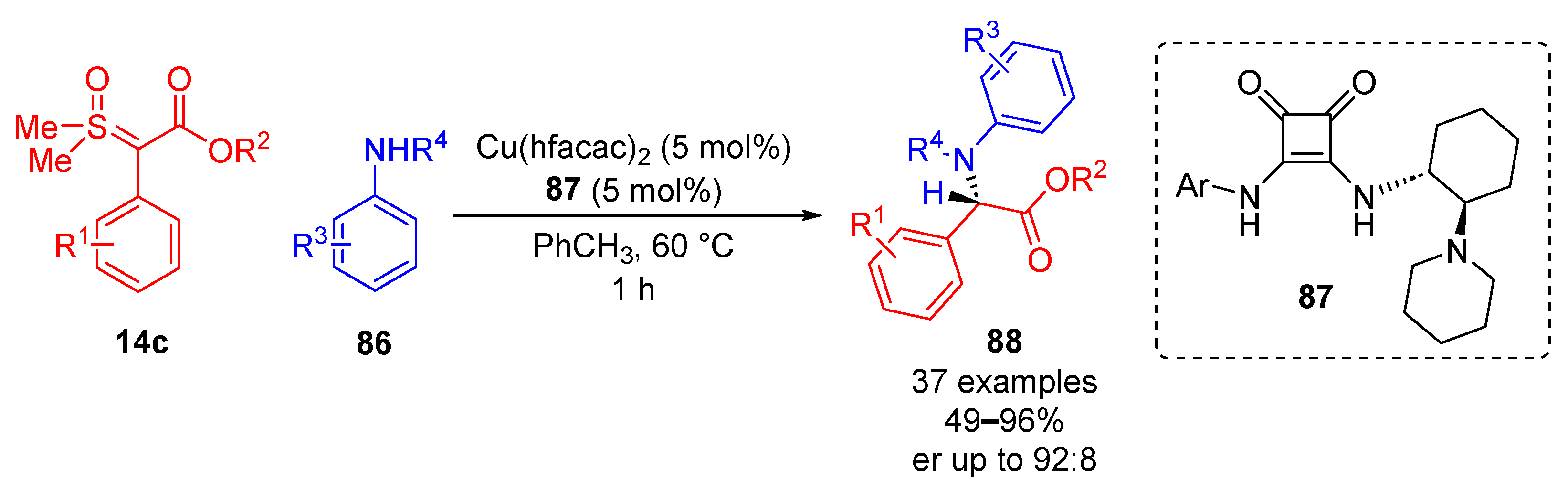
Following this, Burtoloso and colleagues reported the enantioselective N–H insertion of sulfoxonium ylides
14c and anilines
86 using a copper–squaramide catalyst
87, observing 37 substrate examples of the inserted product
88 with reaction yields of up to 96% and good enantiomeric ratios of up to 92:8 (
Scheme 48) [
92]. Anilines and sulfoxonium ylides possessing both EDG and EWG performed equally well when probed, indicating a broad tolerance of substrate types. The reaction conditions which gave the best results were in toluene for 1 h at 60 °C. The most effective catalytic system employed was a copper–squaramide complex, giving a high enantiomeric ratio and reaction yield. A series of phosphoric acids and thiourea catalysts were trialed, and they returned excellent reaction yields (>90%); however, they did not grant desirable enantiocontrol. The plausible reaction mechanism starts with the formation of a copper carbene as a result of the displacement of DMSO. The nucleophilic aniline attacks the central carbon, displacing the copper catalyst. The intermediate tautomerizes to form an enol, which complexes with the squaramide bound to copper and asymmetrically protonates the central carbon, allowing for the synthesis of the inserted product.
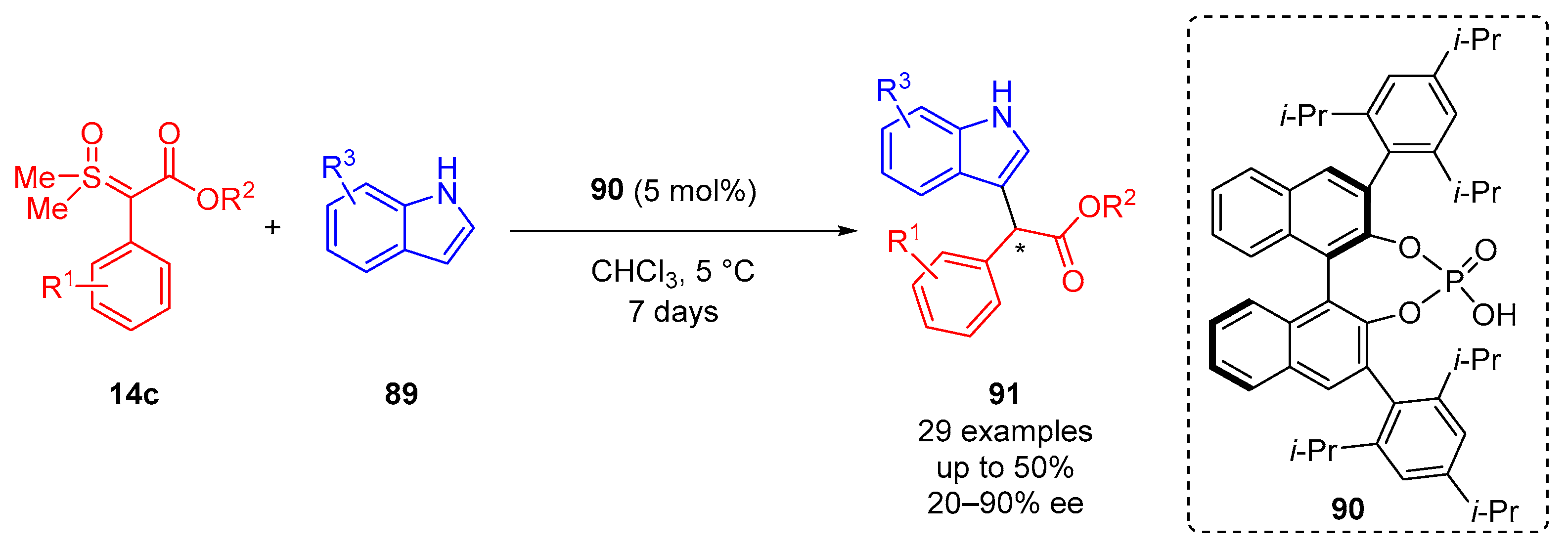
Burtoloso and colleagues additionally reported the enantioselective C–H insertion of indoles
89 and
α-aryl-
α-estersulfoxonium ylides
14c was promoted by phosphoric acid catalysis in 2021 (
Scheme 49) [
93]. In this case, a chiral phosphoric acid
90 (5 mol%) was employed in CHCl
3 at 5 °C for a reaction duration of 7 days. Overall, 29 substrate examples were reported, giving a return of low to high enantiocontrol (20–93% ee) with moderate reaction yields of up to 50%. The proposed reaction mechanism starts with the initial protonation of the sulfoxonium ylide by the chiral phosphoric acid. The cationic sulfoxonium forms an ion pair with the anionic phosphate catalyst. The enantiocontrol stems from the chiral nature of the catalyst, allowing the indole to nucleophilically attack the carbon adjacent to the sulfoxonium group from the less sterically hindered side, displacing DMSO. The phosphoric acid catalyst regenerates by the deprotonation of a
β-hydrogen.
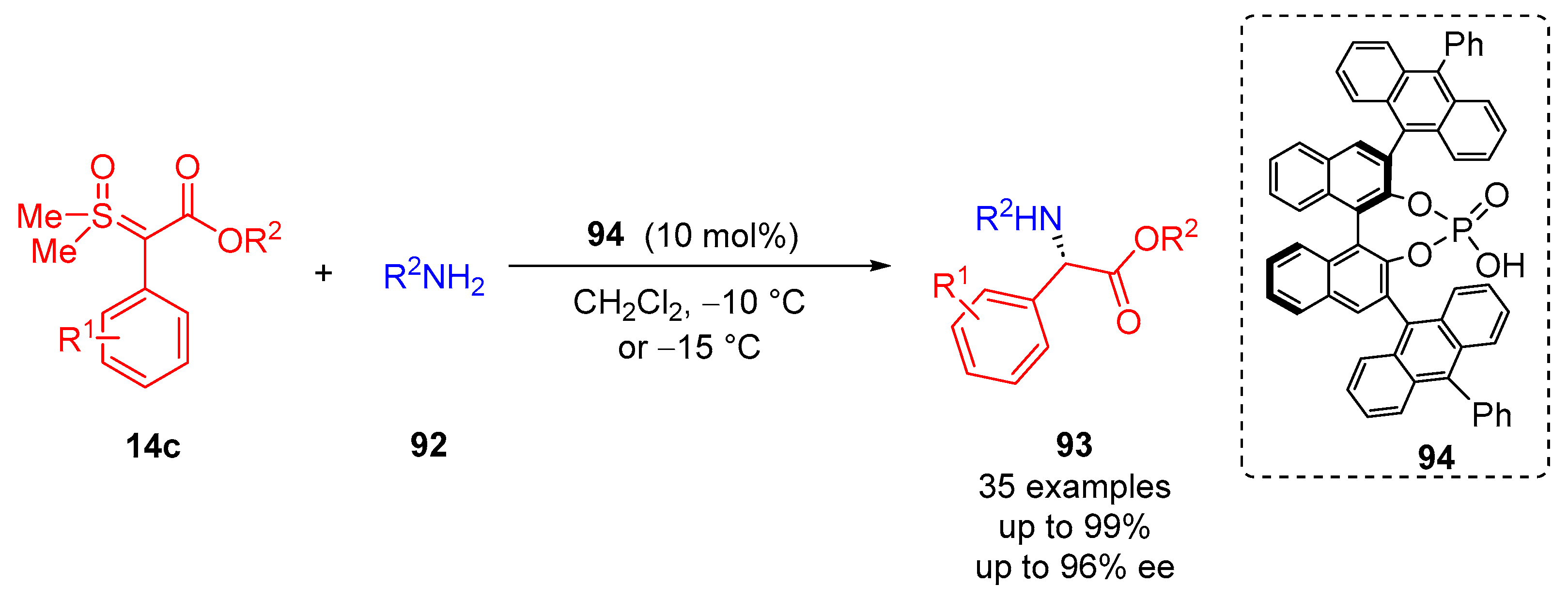
Similarly, Sun and colleagues introduced the asymmetric N–H insertion of amines
92 with sulfoxonium ylides
14c through chiral phosphoric acid catalysis (
94) for the synthesis of
α-amino esters
93 (
Scheme 50)
[94]. In terms of substrate scope, 35 examples were reported with excellent reaction yields of up to 99% and excellent enantioselectivity of up to 96% ee. The reaction allowed broad tolerance for both activated and deactivated
α-aryl-
α-estersulfoxonium ylides, as well as the aryl amines that were explored. The reaction functioned most effectively at colder temperatures (−10 °C or −15 °C depending on the substrate) for long reaction times. CH
2Cl
2 was observed to be the best solvent when compared to CHCl
3, which offered good reaction yields but moderate enantioselectivity. CH
3CN, EtOAc and THF offered high reaction yields (up to 99%) and high enantioselectivity (up to 80% ee) at 25 °C. Moreover, 10 mol% of the chiral phosphoric acid
94 was observed to provide the desired results.
4.6. Miscellaneous Stereoselective Reactions of Sulfoxonium Ylides
γ-Lactones are structural elements that are found as part of many natural products (in about 10% of all natural products), including paraconic acids and numerous bicyclic and tricyclic ring systems (e.g., xanthatin) [
95].
γ-Lactones have a range of biological activities, including strong antifungal, antibiotic, antitumor, antiviral and anti-inflammatory activities, which underscores their potential for pharmaceutical development [
95,
96,
97].
γ-Lactones have also been used as building blocks for the synthesis of complex molecules [
98,
99].
Kerrigan’s group reported direct access to
trans-
γ-lactones
96 via an unprecedented extension to the Johnson−Corey−Chaykovsky reaction, in good yields (up to 93%) and with good diastereoselectivity (up to 92:8) (
Scheme 51) (
Table 8) [
15,
100,
101,
102,
103]. They determined that chiral aminosulfoxonium ylide
95, obtained through treatment of salt with base, was superior to all other onium ylides for
γ-lactone formation. In all cases, tetrafluoroborate (BF
4−) was used as a non-nucleophilic counterion for sulfoxonium salt /ylide
95 in order to minimize side reactions with ketenes (e.g., with iodide as a counterion). This methodology was found to be versatile enough to tolerate ketenes of quite different reactivity, including dimethylketene, alkylarylketenes and even diphenylketene. Furthermore, the level of diastereoselectivity was found to be good for
ortho-substituted aromatic aldehydes, heteroaromatic substituted aldehydes and the
α-branched aliphatic aldehyde, isobutyraldehyde. Generally, the best yields of
γ-lactone were obtained with aromatic aldehydes. The lower yields for the aliphatic aldehyde-derived lactones may be attributed to side reactions involving enolization of starting materials and intermediates. It is speculated that the additive MgCl
2 has a number of roles in the reaction system. Primarily, it activates aliphatic aldehydes, such as isobutyraldehyde, to undergo reaction with the sulfoxonium ylide
95. A second possible effect is that, under certain conditions, it may enable greater organization (through chelation) in the transition state for [3,3]-sigmatropic rearrangement of
F to
G (
Scheme 52).
In the proposed mechanism for the reaction (
Scheme 52), ylide
95 is added to aldehyde to give betaine
A, most likely in equilibrium with oxathietane
A′ [
104,
105,
106]. Reaction of betaine
A with ketene gives rise to enolate
B, which is formed stereoselectively as an
E-enolate (for M = Li) at this point. Deprotonation of
D by a base (e.g., ylide
95) gives sulfurane oxide anion
E, which then undergoes E1cB elimination to give enolate
F [
107]. Enolate
F then undergoes a [3,3]-sigmatropic rearrangement in stereoselective fashion, presumably via a chair transition state, to afford carboxylate
G [108]. Protonation of
G gives
H, which undergoes intramolecular S
N2 (5-
exo-tet) cyclization to provide
trans-
γ-lactone
96.
They reasoned that
γ-lactones could be formed in enantioselective fashion, provided a suitable enantioenriched aminosulfoxonium salt could be identified [
109]. Chiral aminosulfoxonium salt was obtained through a four-step procedure from commercially available methylphenyl sulfoxide [
110,
111,
112,
113]. Enantioenriched aminosulfoxonium (
S)-ylide
97 was subjected to reaction with a variety of aldehydes (both aromatic and aliphatic) and disubstituted ketenes, leading to the formation of
α,
β-substituted
γ-lactones
96 in moderate to very good diastereoselectivity (dr up to 95:5) and with enantiomeric excesses of up to 79% ee (
Table 9). The best levels of enantioselectivity were observed in the reactions using with isobutyraldehyde and various alkylarylketenes.
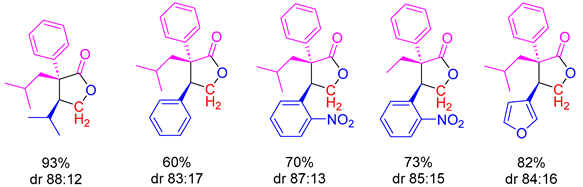
Diastereoselectivity in
γ-lactone forming reaction may be rationalized by a [3,3]-sigmatropic rearrangement of sulfurane oxide
E, involving a six-membered chair-like transition state, where the sterically bulkier substituents (R
1 and R
3) preferentially occupy pseudoequatorial positions (
Scheme 53) [
110,
111,
112,
114]. They propose that the O-enolate and –NMe
2 substituents occupy apical positions at sulfur (in a trigonal bipyramidal geometry), which is consistent with the reported arrangement of cyclic sulfurane oxides bearing electronegative substituents [
105,
107,
115,
116]. High enantioselectivity may be achieved through the rigid transition state depicted in
Scheme 53, where the phenyl substituent blocks the approach by the enolate moiety to the
re face of the
α,
β-unsaturated sulfurane oxide [
117,
118].
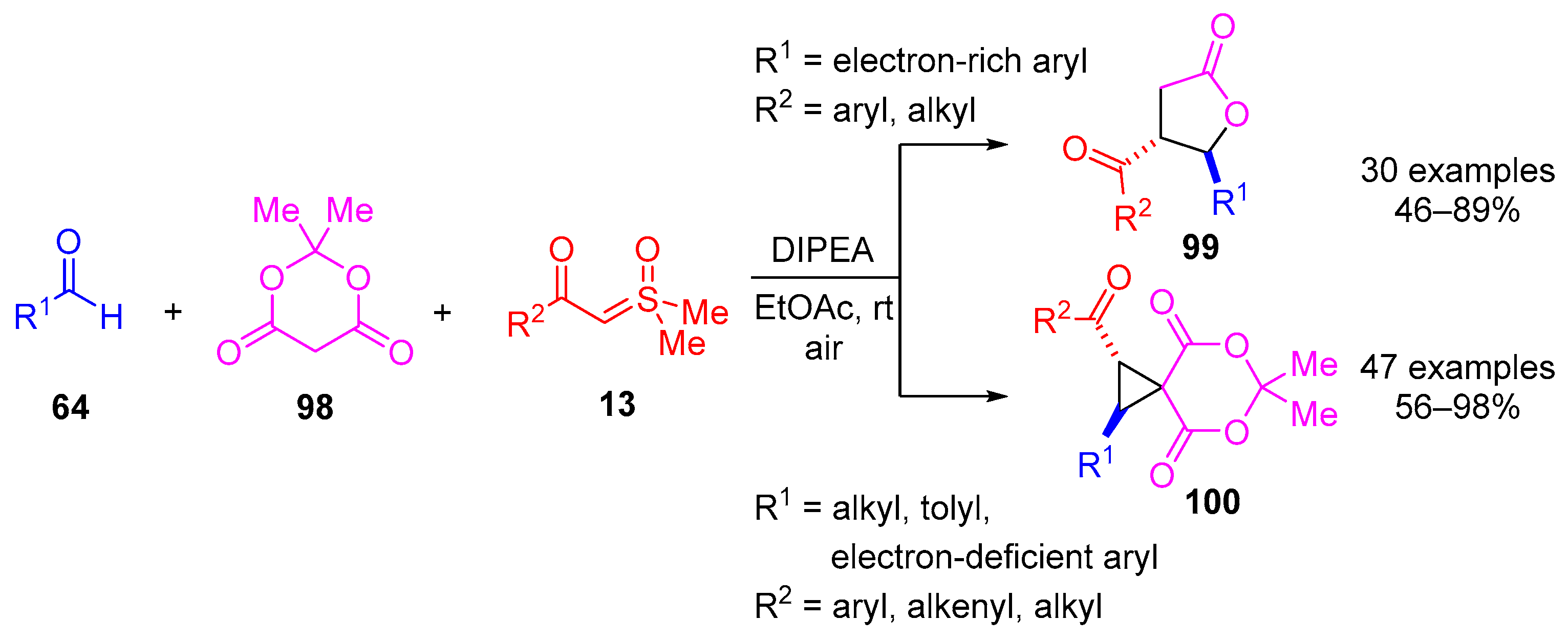
In 2021, Peng and colleagues reported the synthesis of 4,5-disubstituted
γ-butyrolactones
99 from aldehydes
64,
α-carbonylsulfoxonium ylides
13 and Meldrum’s acid
98 using
N,
N-diisopropylethylamine (DIPEA) (
Scheme 54) [
119]. The reaction displayed diastereoselectivity for
trans-
γ-butyrolactones
99, which were formed in moderate to excellent yields (30 examples, 46–98%). They discovered that the reaction provided
trans-spirocyclopropanes
100 from electron-deficient benzaldehydes (or methyl-substituted benzaldehydes) with alkyl or aryl-substituted
β-ketosulfoxonium ylides in moderate to excellent yields (47 examples, 56–98%). However, the formation of
trans-
γ-butyrolactones was realised through the reaction involving electron-rich benzaldehydes and alkyl- or aryl-substituted
β-ketosulfoxonium ylides.
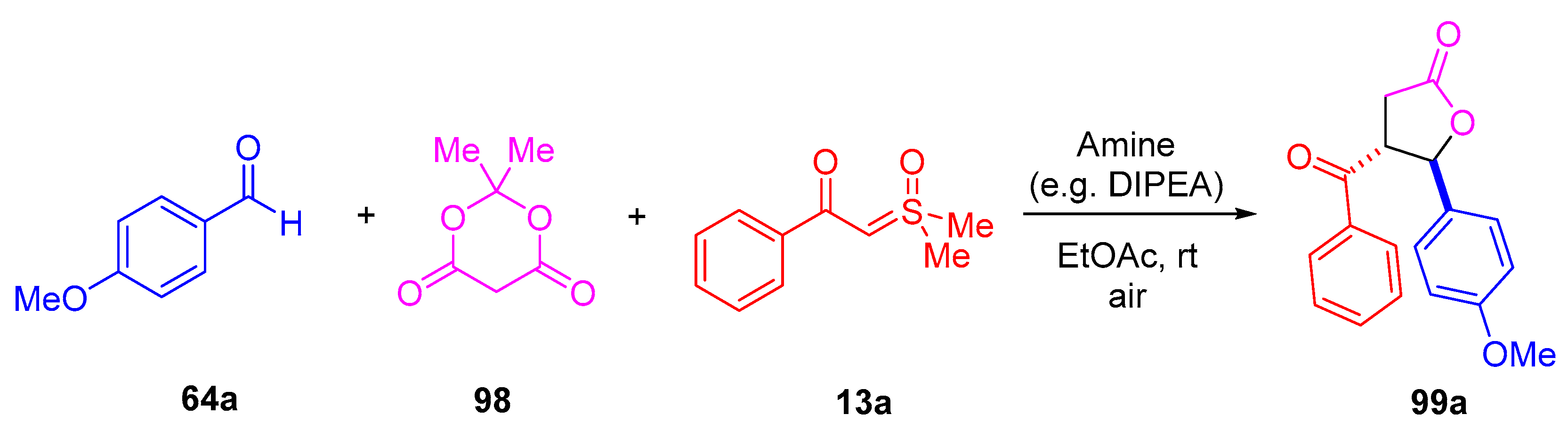
The reaction was optimized by focusing on the synthesis of 4-benzoyl-5-(4-methoxyphenyl)-
γ-butyrolactone
99a from 4-methoxybenzaldehyde
64a,
β-benzoyl-substituted dimethylsulfoxonium methylide
13a and Meldrum’s acid
98 (
Scheme 55). The reaction proceeded well in CH
2Cl
2 (73%) and moderately well when attempted in CCl
4 (32%). However, no reaction was observed when examined in EtOH. The most effective reaction conditions were noted as EtOAc (98%) employing 2 equivalents of DIPEA, 1.5 equivalents of aldehyde
64, 2 equivalents of Meldrum’s acid
98 and 1 equivalent of sulfoxonium ylide
13a. An excellent yield of 94% was observed when triisopropylamine (instead of DIPEA) was tested. Moderate to excellent yields were observed when using triethylamine (32–86%), while 4-dimethylaminopyridine provided a yield of 41%.
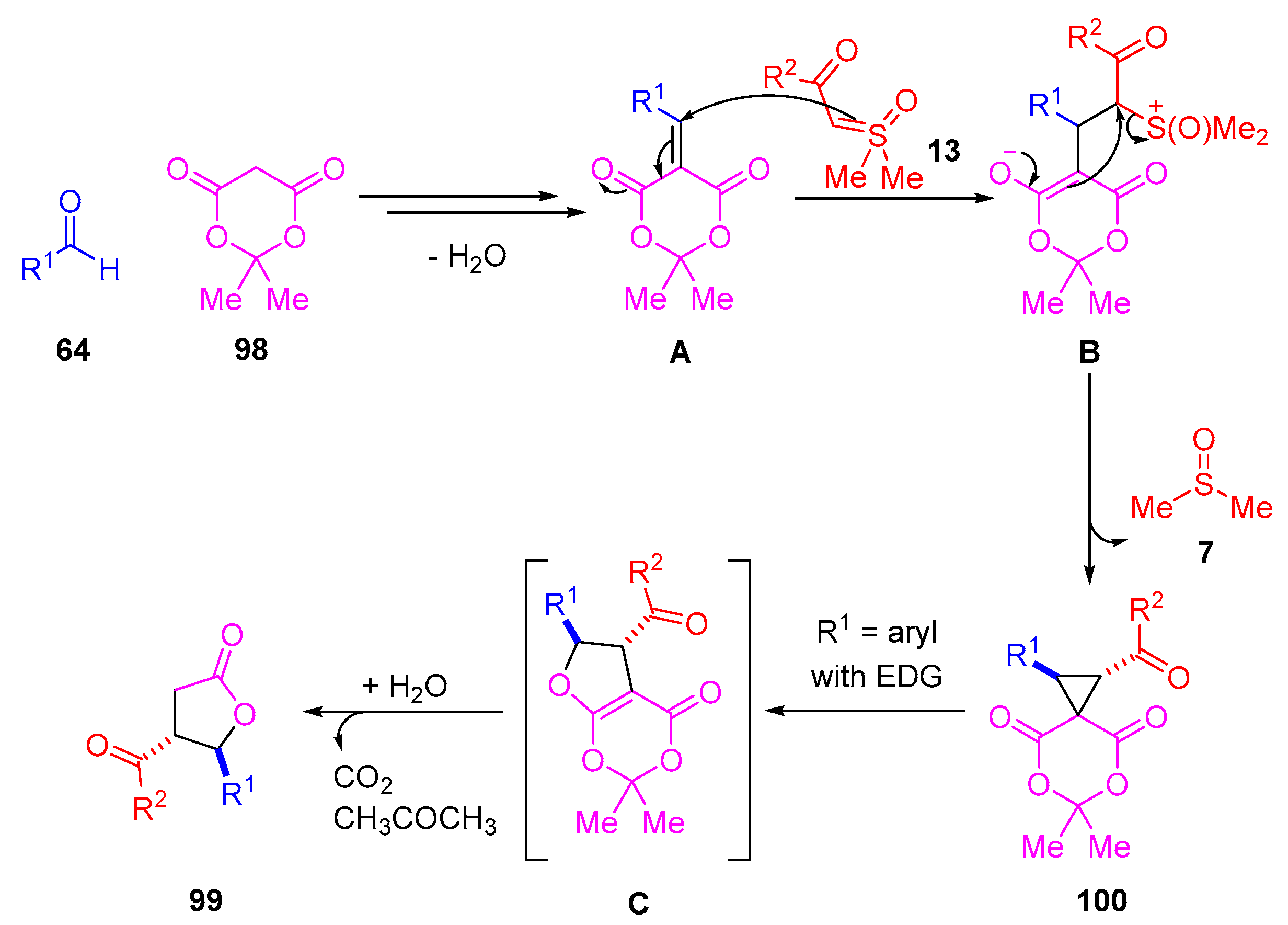
The proposed reaction mechanism for the synthesis of
trans-
γ-butyrolactones and
trans-spirocyclopropane proceeds through a similar pathway (
Scheme 56,
Table 10). The proposed reaction mechanism starts with an initial Knoevenagel condensation of an aldehyde
64 with Meldrum’s acid
98, which forms an alkylidene/arylidene intermediate
A. A sulfoxonium ylide
13 undergoes a 1,4-addition and the resulting enolate of intermediate
B displaces DMSO
7, leading to the formation of cyclopropane product
100. In cases where electron-rich benzaldehydes are examined, the
trans-spirocyclopropane undergoes a Cloke-Wilson rearrangement to form a dihydrofuran intermediate
C [
120,
121,
122]. Aided by water,
C decarboxylates and liberates acetone to form
trans-
γ-butyrolactone
99 (
Scheme 56).
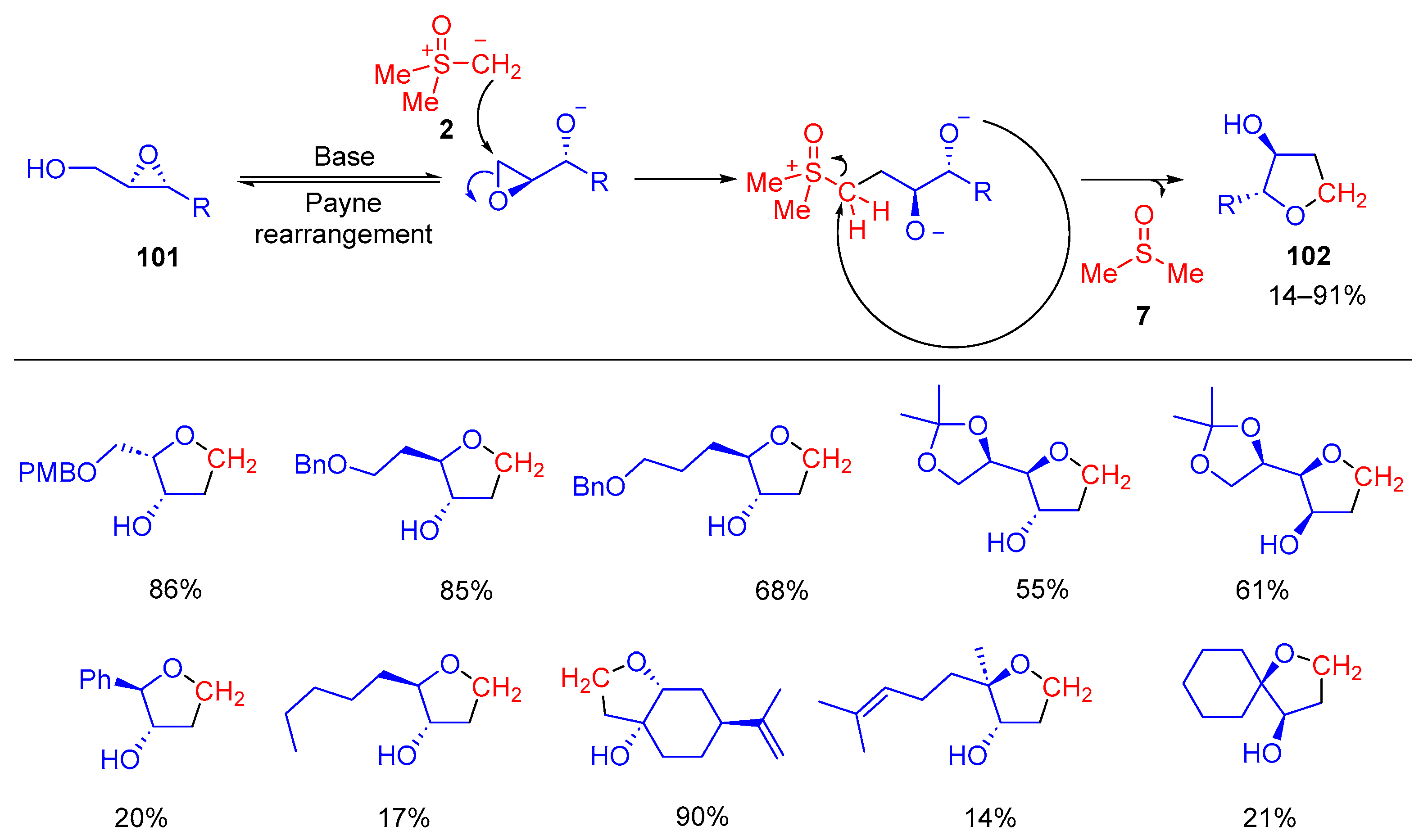
Borhan and co-workers introduced a methodology for regioselective nucleophilic substitution reactions of enantiopure 2,3-epoxy alcohols
101 with dimethylsulfoxonium methylide
2 to yield diastereomerically and/or enantiomerically pure disubstituted tetrahydrofuran rings
102 (
Scheme 57) [
123]. In this methodology, the stereochemistry that is set by the asymmetric epoxidation is translated fully to the final product. This approach provided an efficient access to tetrahydrofuran rings with stereodefined substituents from chiral 2,3-epoxy-alcohols
101 which are relatively simpler than the method via the Sharpless asymmetric epoxidation reaction.
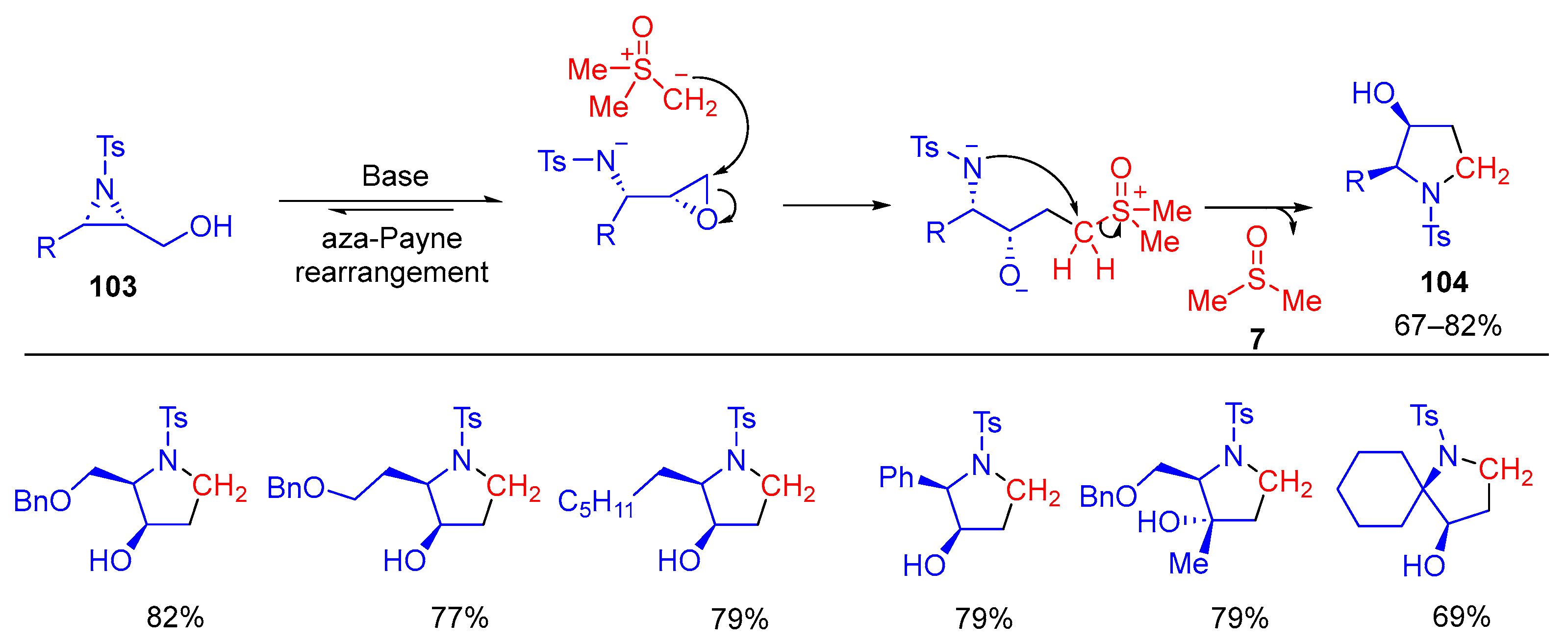
The same group further extended this methodology to the sulfoxonium ylide-based aza-Payne rearrangement of 2,3-aziridin-1-ols
103 for efficient diastereoselective synthesis of substituted pyrrolidines
104 (
Scheme 58)
[124]. The aza-Payne rearrangement under basic reaction conditions favors the formation of epoxy amides. Subsequent nucleophilic attack of the epoxide by the sulfoxonium ylide generates a dianion intermediate, which upon a 5-
exo-tet ring-closure yields the desired pyrrolidine
104, thus completing the relay of the three-membered ring to the five-membered nitrogen-containing ring system. The stereochemistry present in the enantioenriched aziridinols is translated fully to the final products.
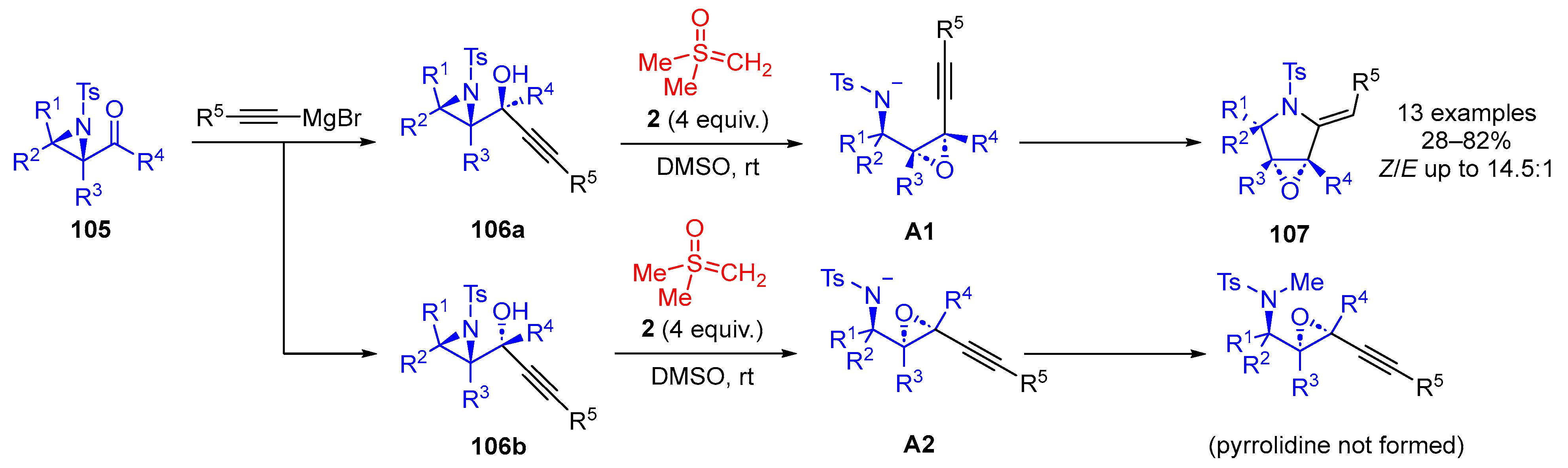
They also reported a sulfoxonium ylide-mediated facile tandem aza-Payne/hydroamination reaction of chiral
syn-aziridinol
106 for diastereoselective synthesis of highly functionalised pyrrolidines
107 (
Scheme 59) [
125]. Addition of alkynyl Grignard reagents to chiral 2,3-aziridinals
105 predominantly yielded
syn-aziridinols
106. Treatment of this chiral
syn-aziridinol
106a with sulfoxonium ylide
2 led initially to an aza-Payne rearrangement, which juxtaposed the amine and the alkyne in a favorable orientation
A1 to complete the hydroamination. The authors also reported that the
anti-aziridinol
106b underwent the aza-Payne rearrangement but could not proceed further with the hydroamination.
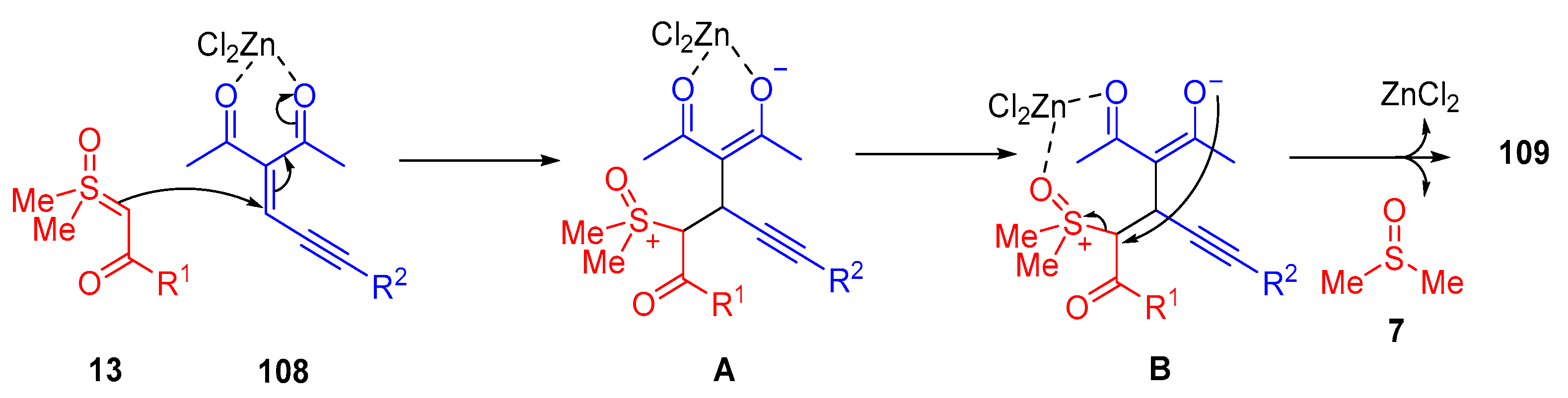
In 2020, Wu and colleagues reported a diastereoselective synthesis of 2,3-dihydrofurans
109 from
α-ketosulfoxonium ylides
13b and enynones
108 induced by Lewis acid-catalysis in ionic liquids (
Scheme 60) [
126]. The proposed mechanism proceeds through the Lewis acid activation of enynone
108, which allows the conjugate addition of
α-ketosulfoxonium ylide
13b to form intermediate
A (
Scheme 61). Cyclization (5-
exo-tet) of the enolate’s O-atom onto the carbon adjacent to the sulfoxonium group of
B, results in the formation of the 2,3-dihydrofuran product
109. They reported 25 product examples with good to excellent yields of up to 92% and excellent diastereoselectivity (dr > 20:1) [
126].
Huang and colleagues reported a metal-free diastereoselective synthesis of trisubstituted
trans-oxazolines
111 from sulfoxonium ylides
13 and enamides
110 by Brønsted acid-catalysis (
Scheme 62) [
127]. The preferred catalyst was perchloric acid (0.2 equivalents) in CH
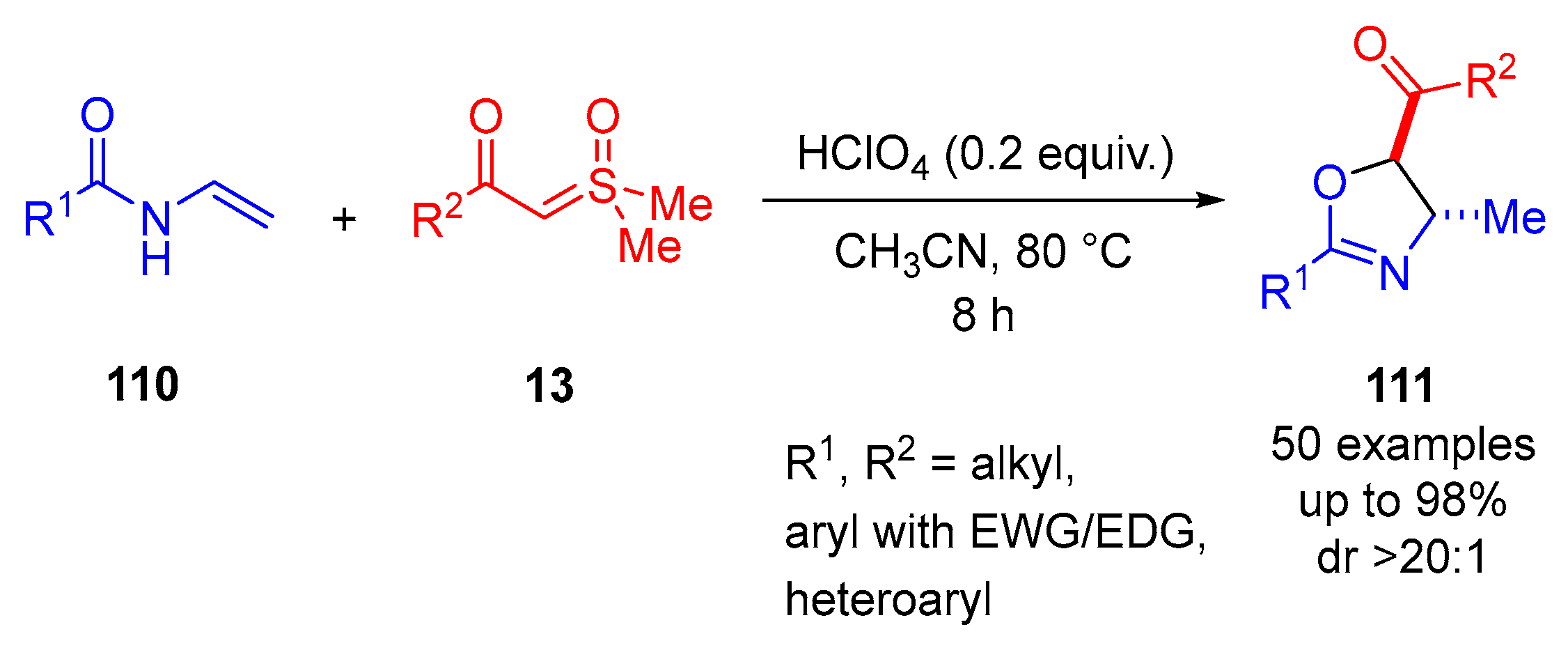
3CN at 80 °C for 8 h. A broad substrate scope was explored and resulted in 50 examples with reaction yields of up 98% and excellent diastereoselectivity (dr >20:1). The reaction demonstrated tolerance for alkyl, heteroaryl and EWG/EDG-substituted aryl groups.
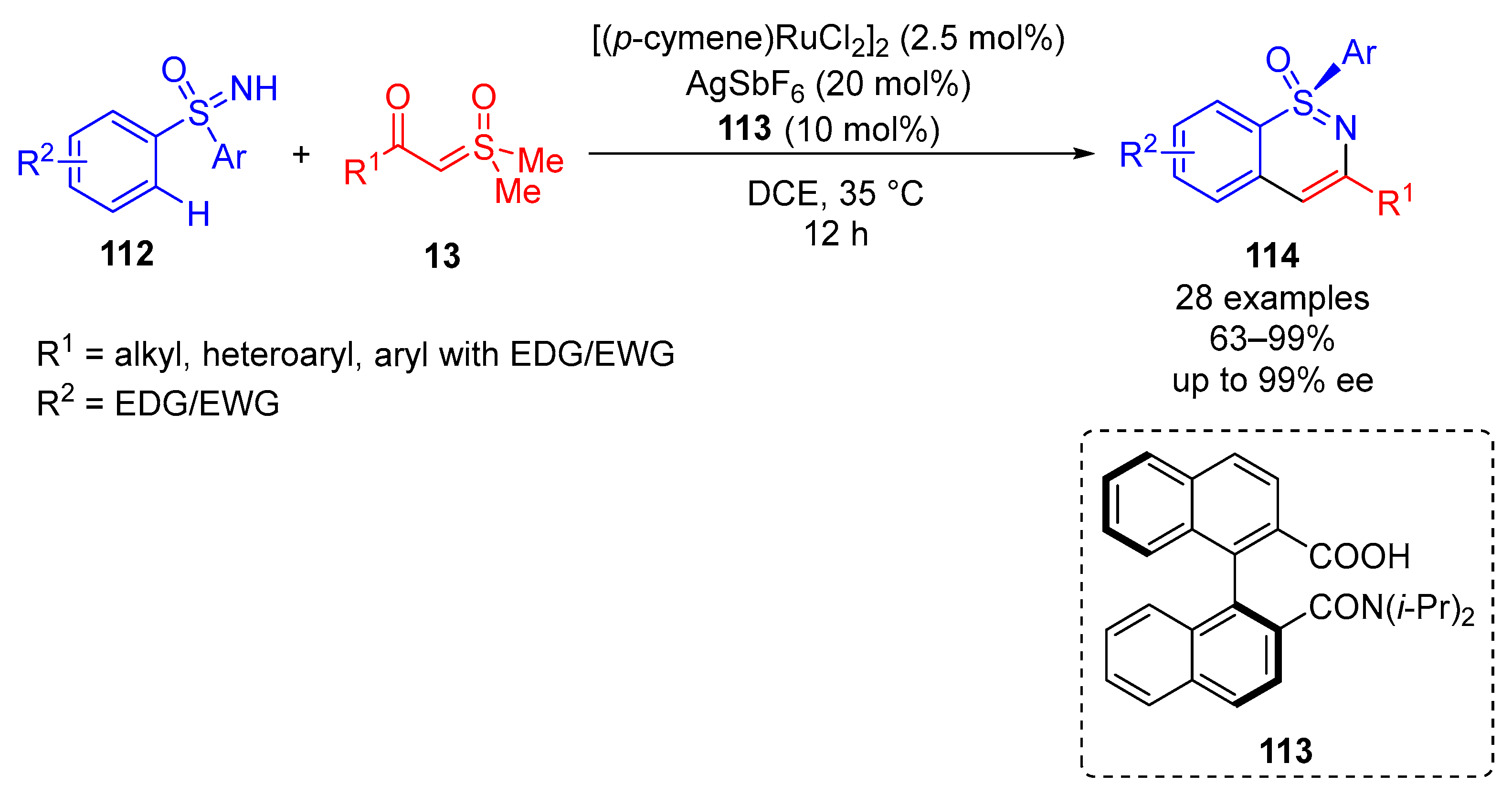
In 2021, Shi and colleagues reported a ruthenium-catalyzed enantioselective synthesis of stereogenic sulfur-containing sulfoximines
114 through the annulation of aryl sulfoximines
112 and
α-ketosulfoxonium ylides
13 (
Scheme 63) [
128]. In total, 28 examples of stereogenic sulfur-containing sulfoximines
114 were presented in good to excellent yields (63–99%) and with excellent enantioselectivity (up to 99% ee). The reaction performed best in DCE, at 35 °C for 12 h when using 2.5 mol% of [(
p-cymene)RuCl
2]
2, 20 mol% of AgSbF
6 and 10 mol% of the chiral binaphthyl monocarboxylic acid ligand
113. The method exhibited substrate tolerance for activated and deactivated aryl sulfoximines
112 and a tolerance for alkyl, heteroaryl and EWG/EDG-substituted aryls in the R
1 position.
In 2023, Sun and colleagues reported an iridium-catalyzed asymmetric [4 + 1] cycloaddition of
α-carbonylsulfoxonium ylides
13 and hydroxyallylanilines
115 resulting in 3-vinylindolines
117 (
Scheme 64) [
129]. The reaction used 3 mol% of [Ir(cod)Cl]
2, three equivalents of TFA and 12 mol% of Carreira’s ligand
116 [
130,
131]. The preferred reactions conditions reported the use of DCE as solvent at 45 °C for 8 h, utilizing molecular sieves (4 Å). A broad substrate scope was tolerated when probed under the optimal conditions, which returned 25 examples of moderate to very good yields (50–80%) with excellent enantio- and diastereoselectivity (up to 99% ee and dr > 19:1). Electron-rich and electron-deficient hydroxylallylanilines were tolerated when EWG/EDG were trialed in the R
2 position, and vinyl, ester, alkyl, heteroaryl and EWG/EDG-substituted aryls returned favorable results when probed in the R
1 position.










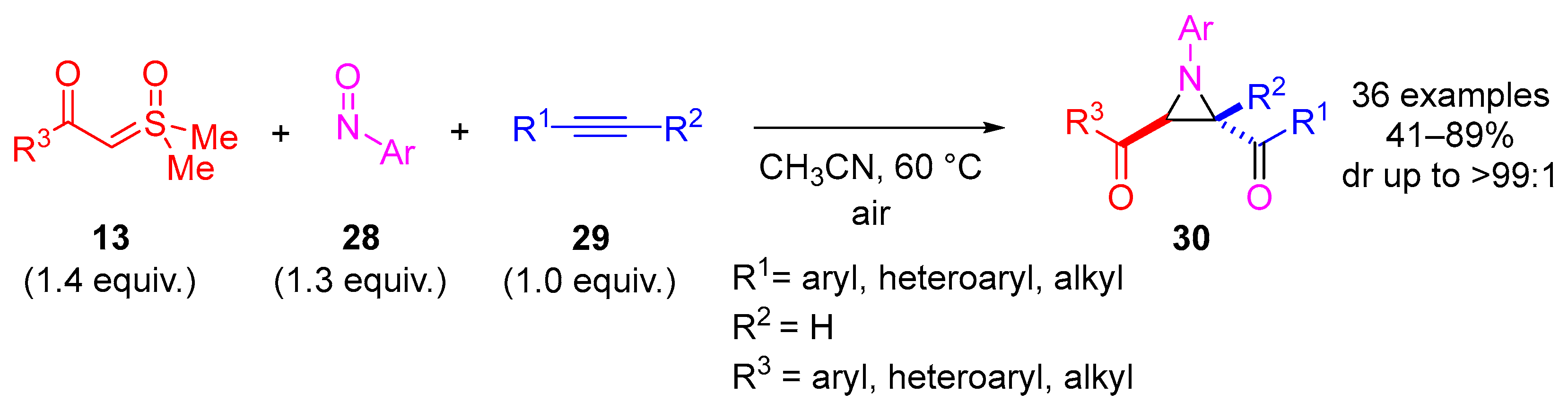


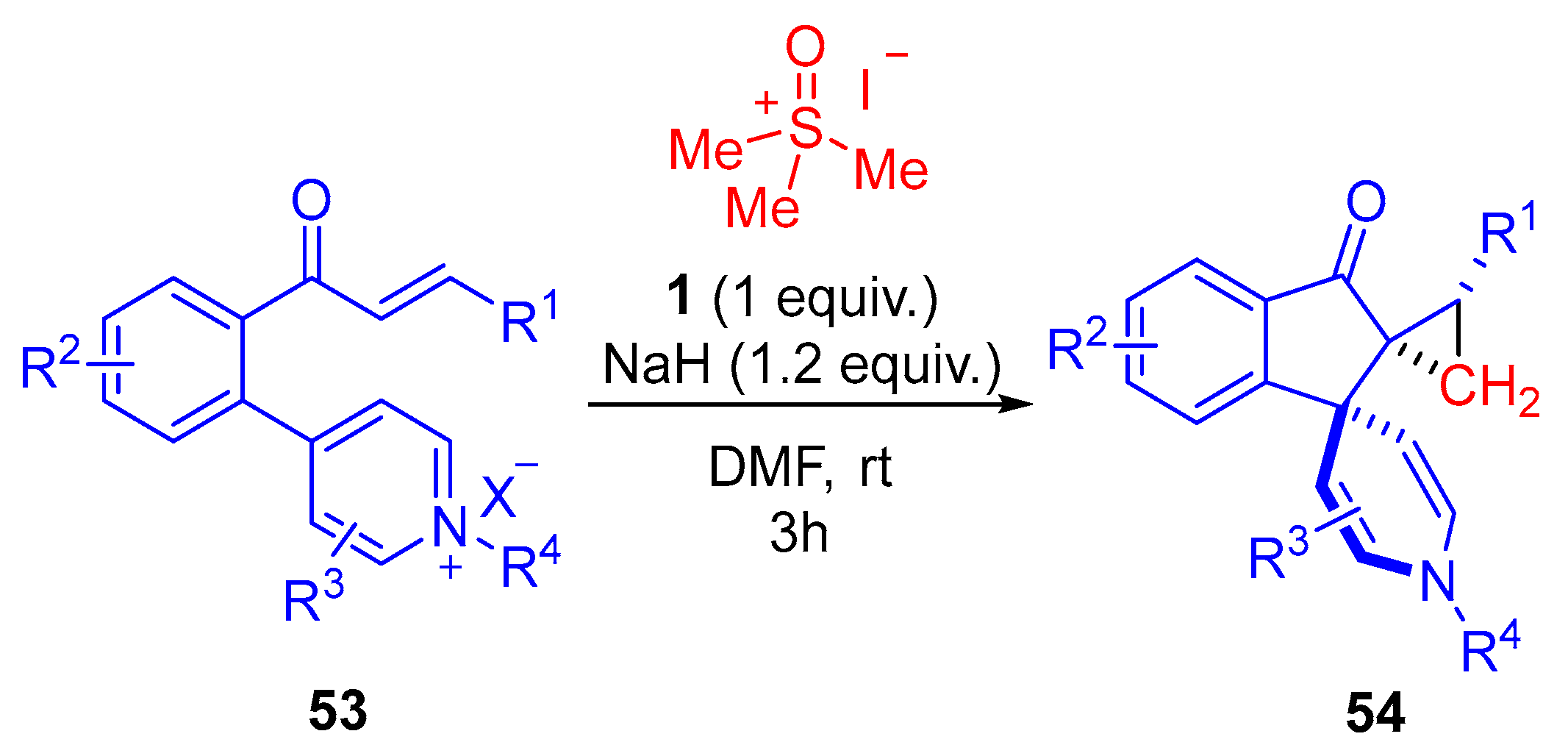
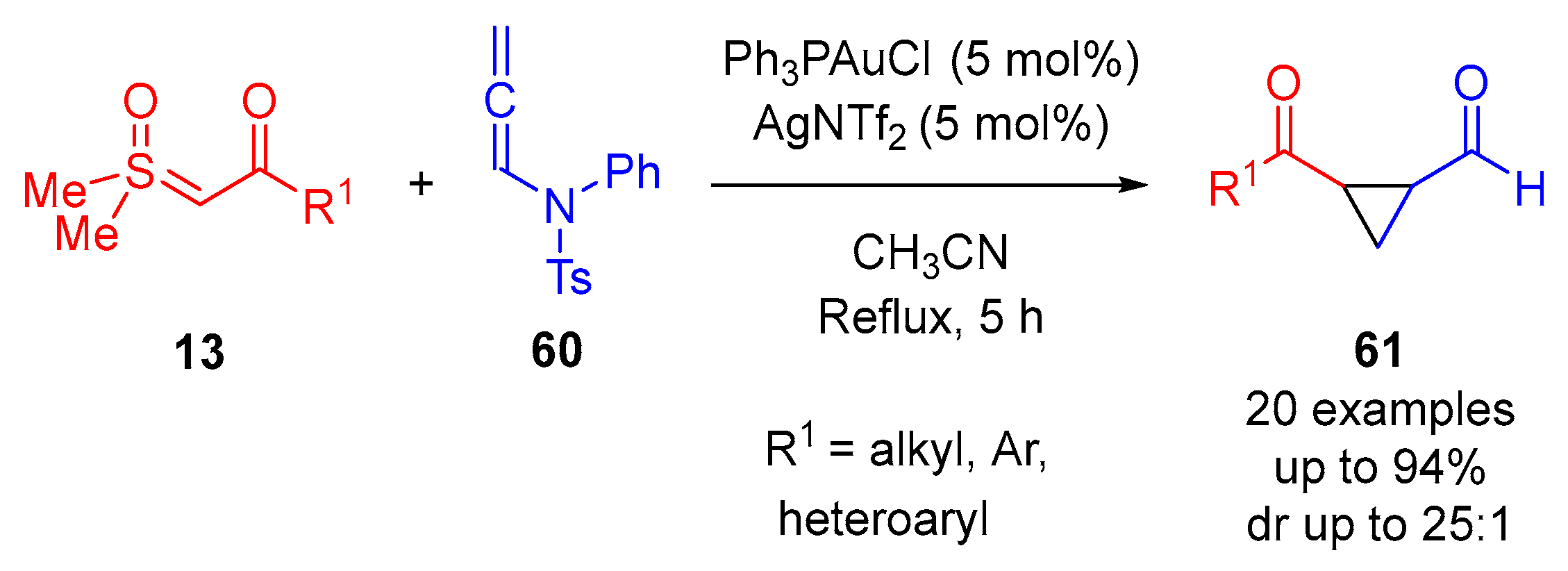

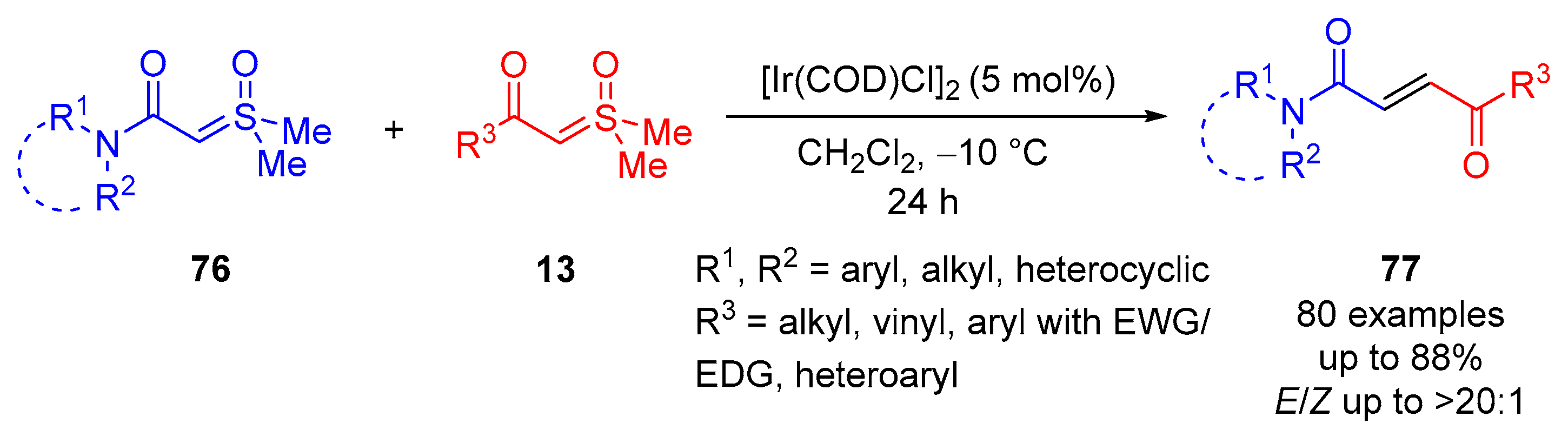



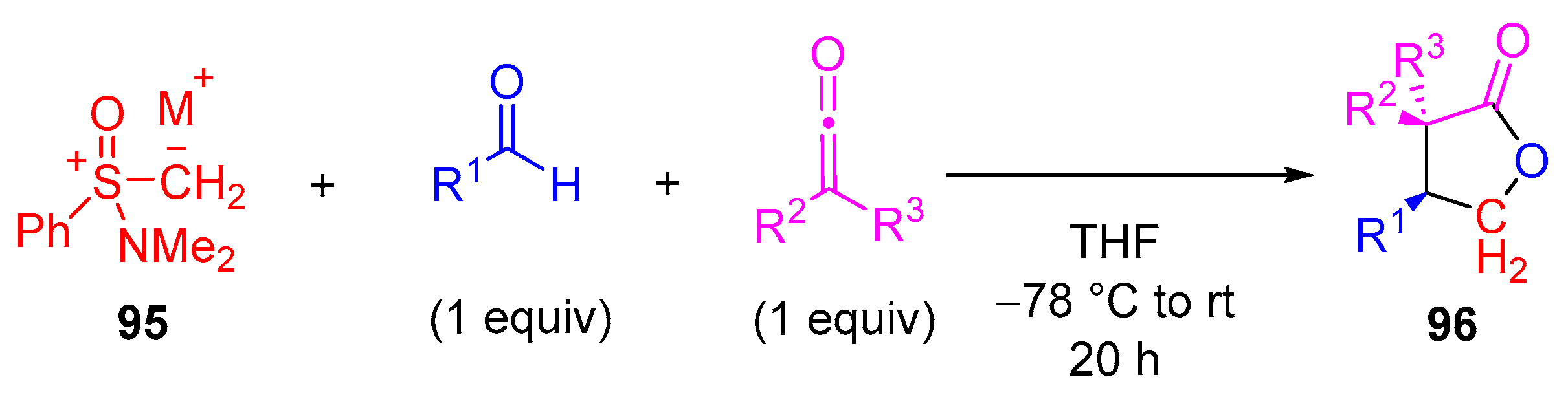



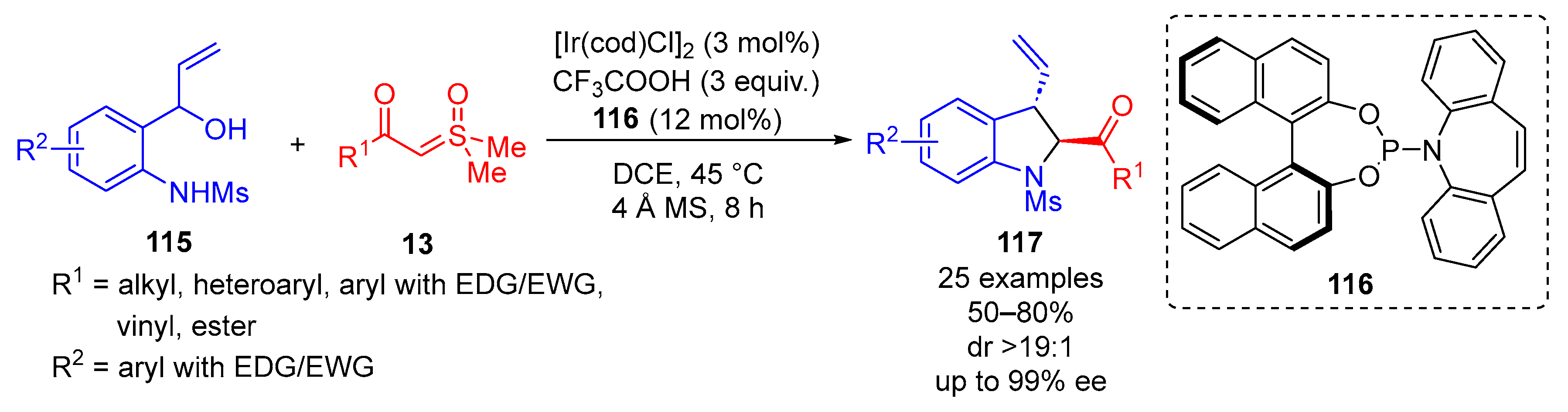

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}