
AQbD-Based UPLC-ELSD Method for Quantifying Medium Chain Triglycerides in Labrafac™ WL 1349 for Nanoemulsion Applications

 , and
, and
Abstract

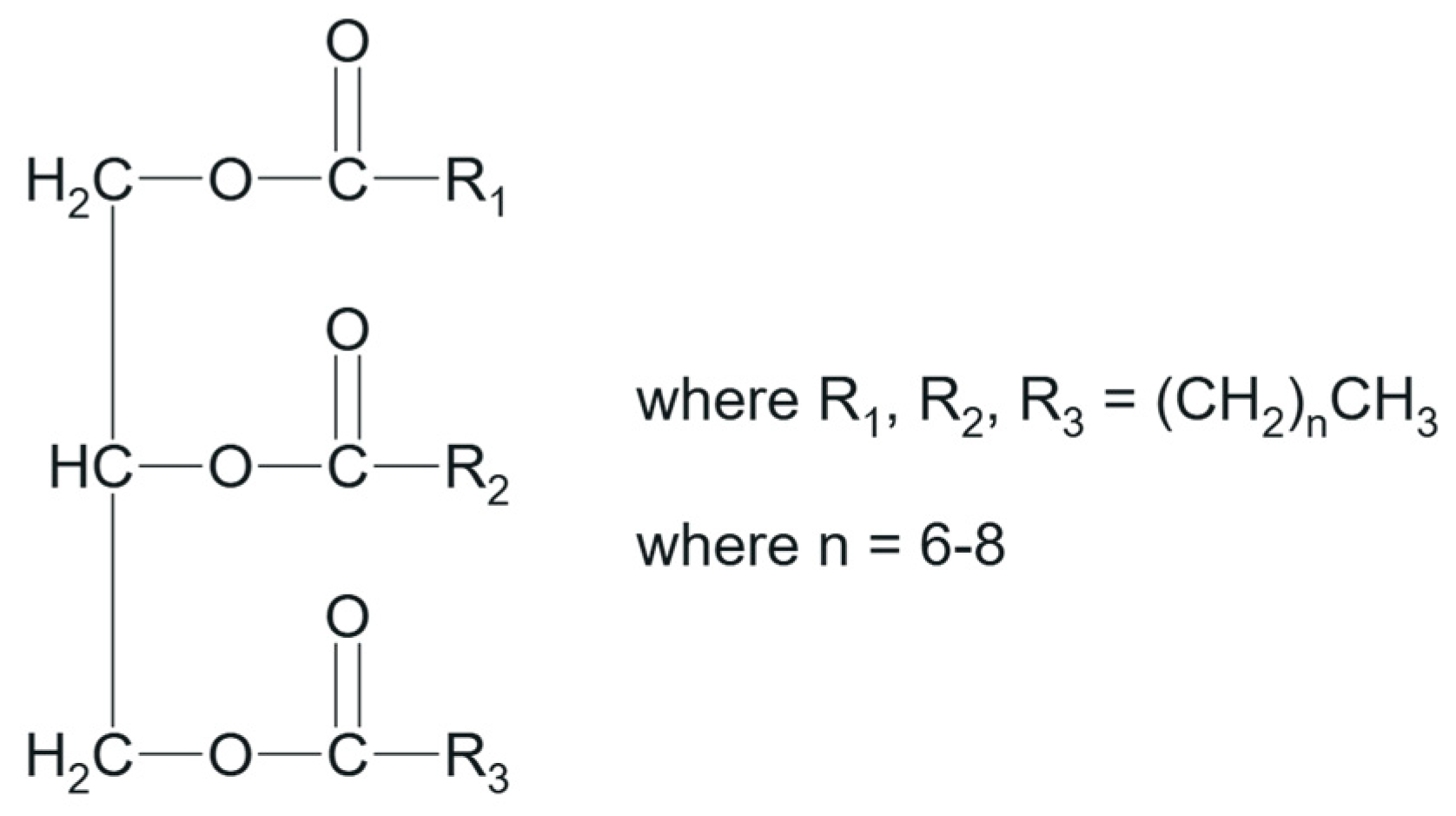
1. Introduction
2. Results and Discussion
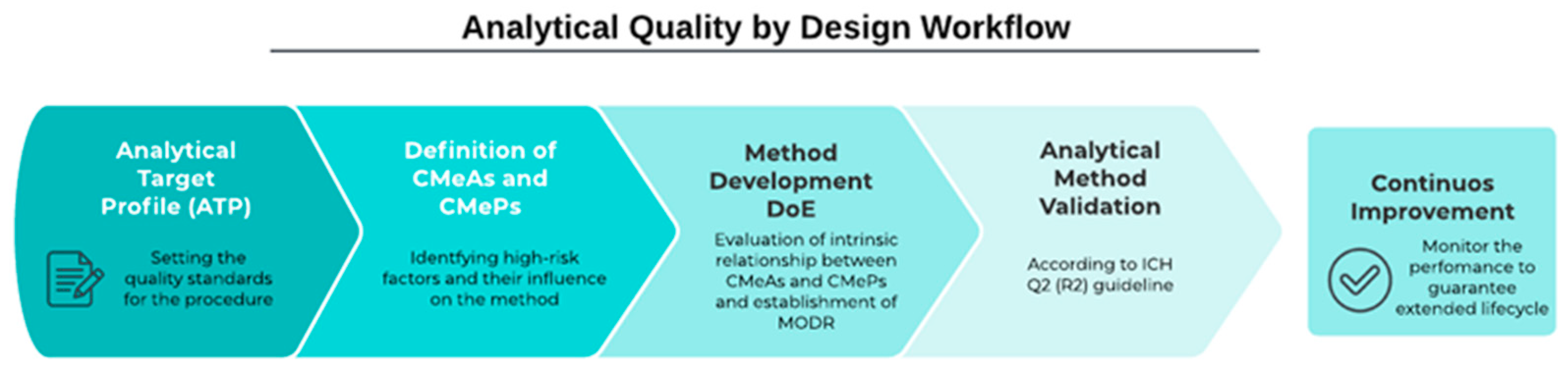
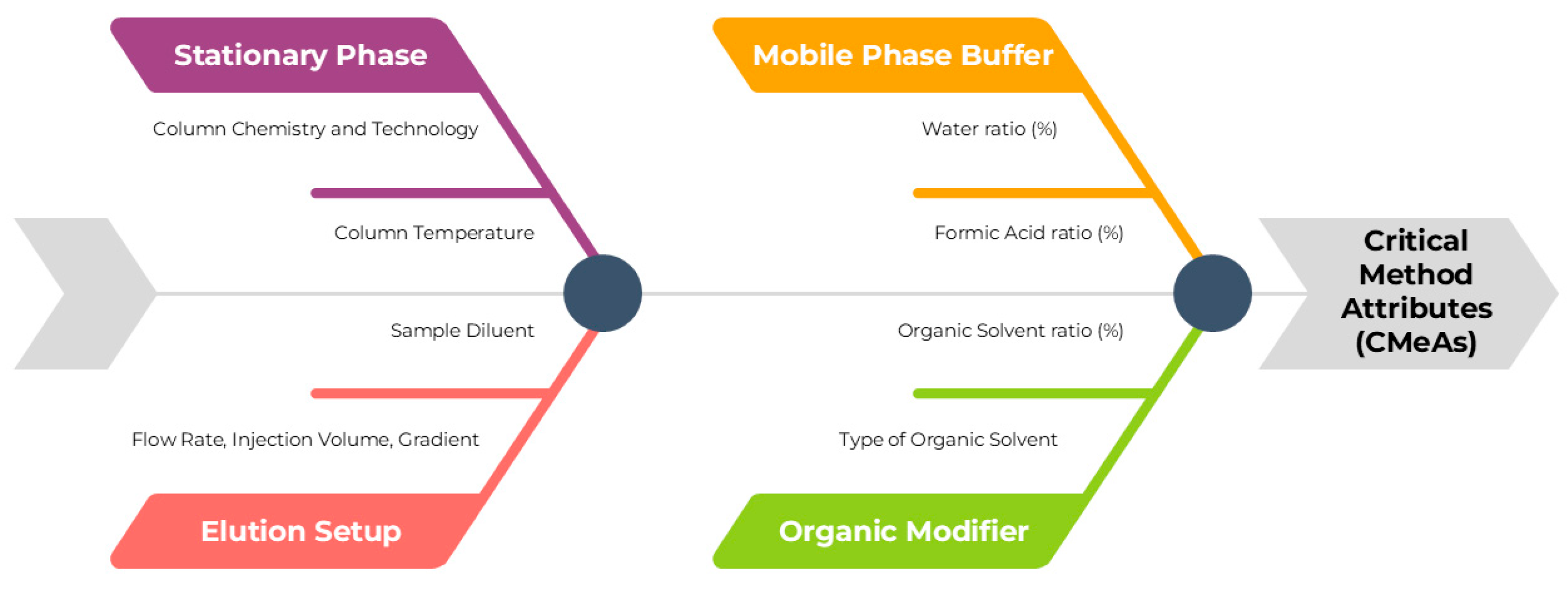
2.1. Method Development by AQbD Principles
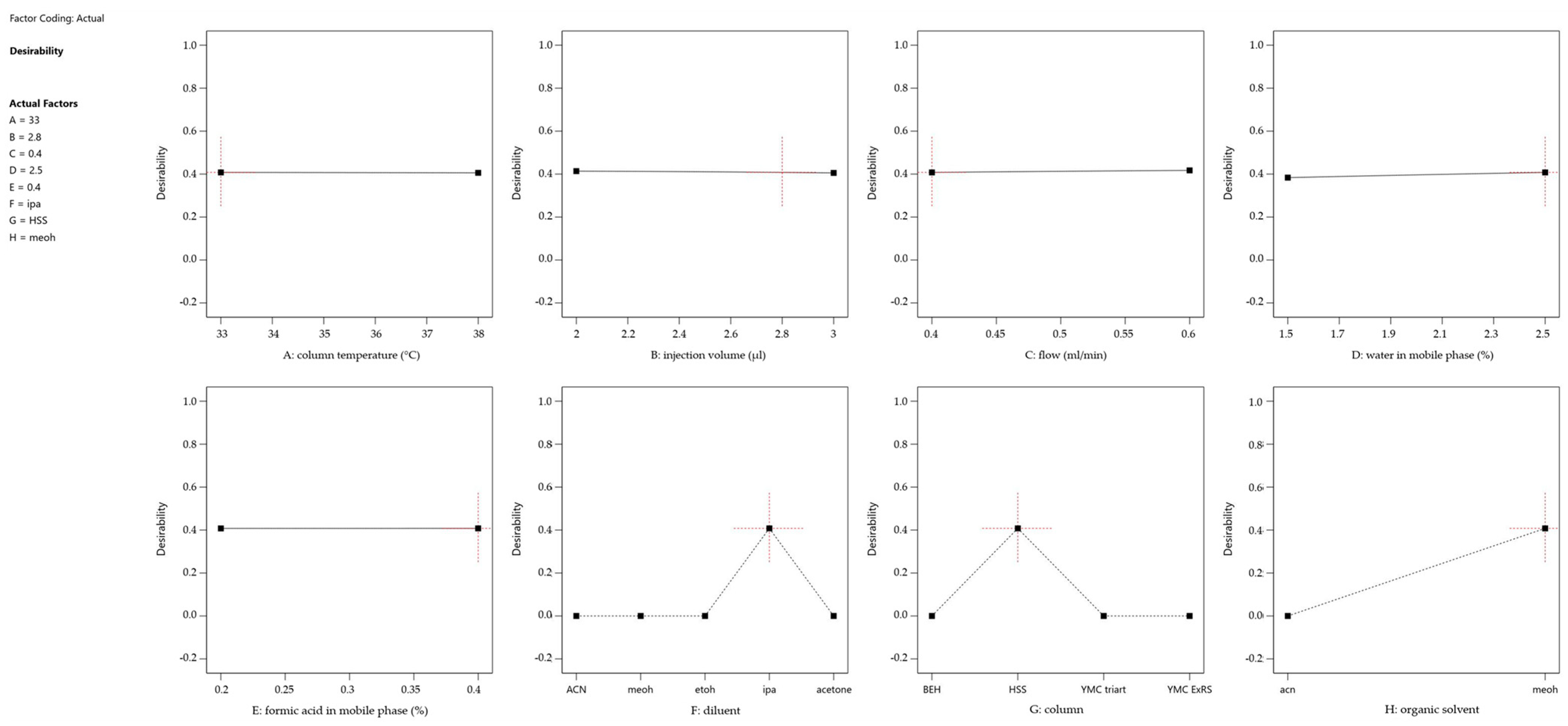
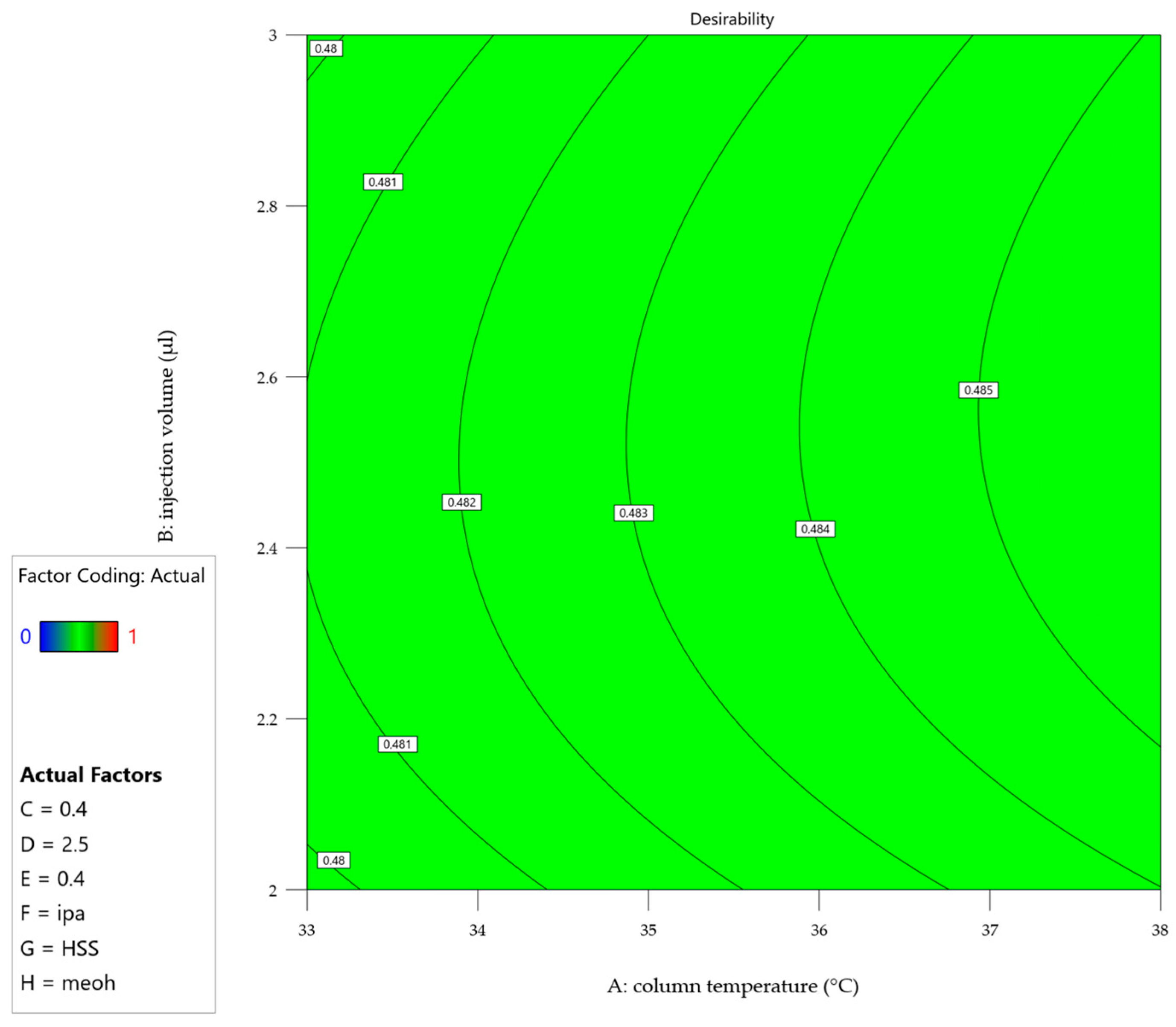
2.2. Statistical Evaluation of the Development DoE and Definition of MODR
2.3. Analytical Method Validation
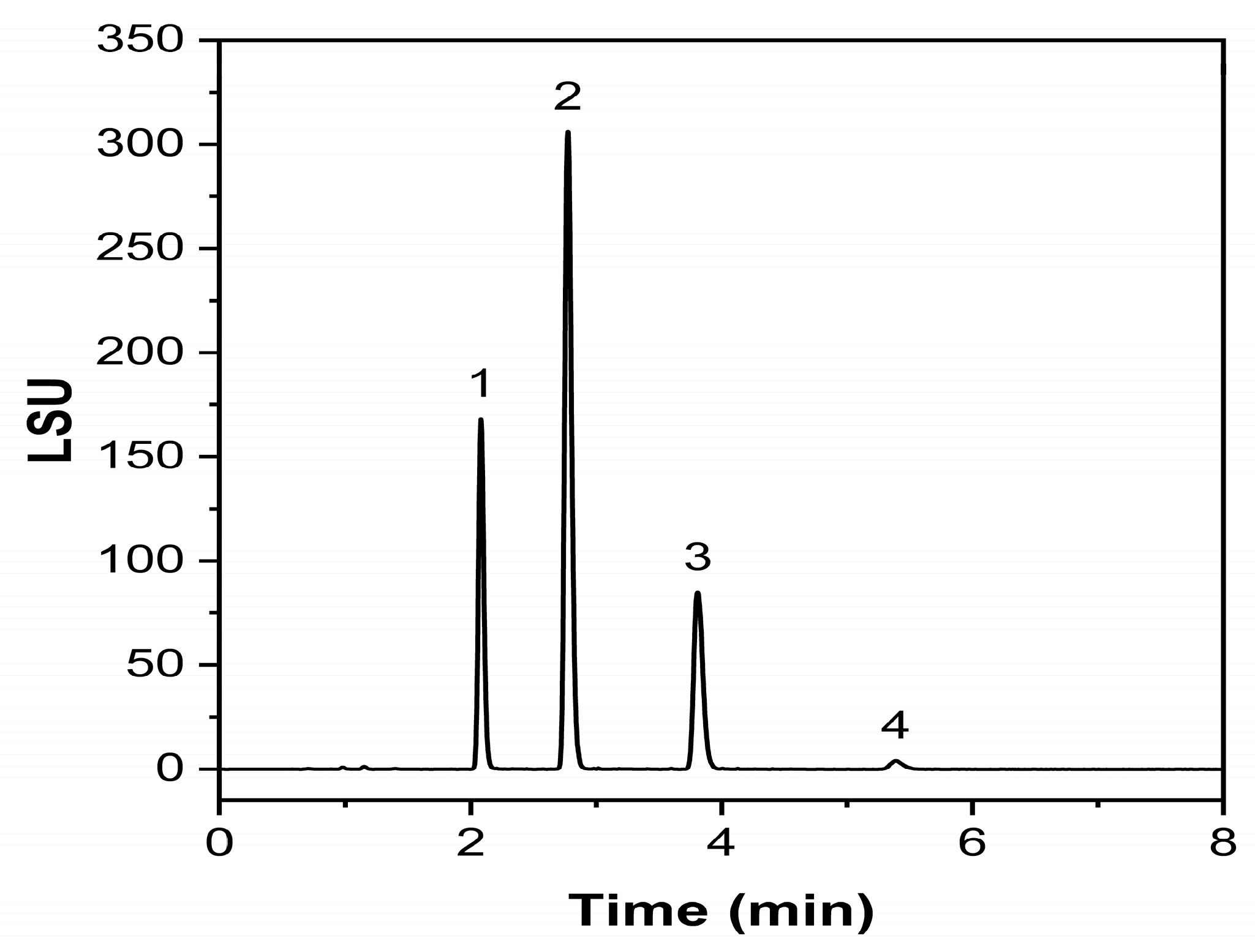
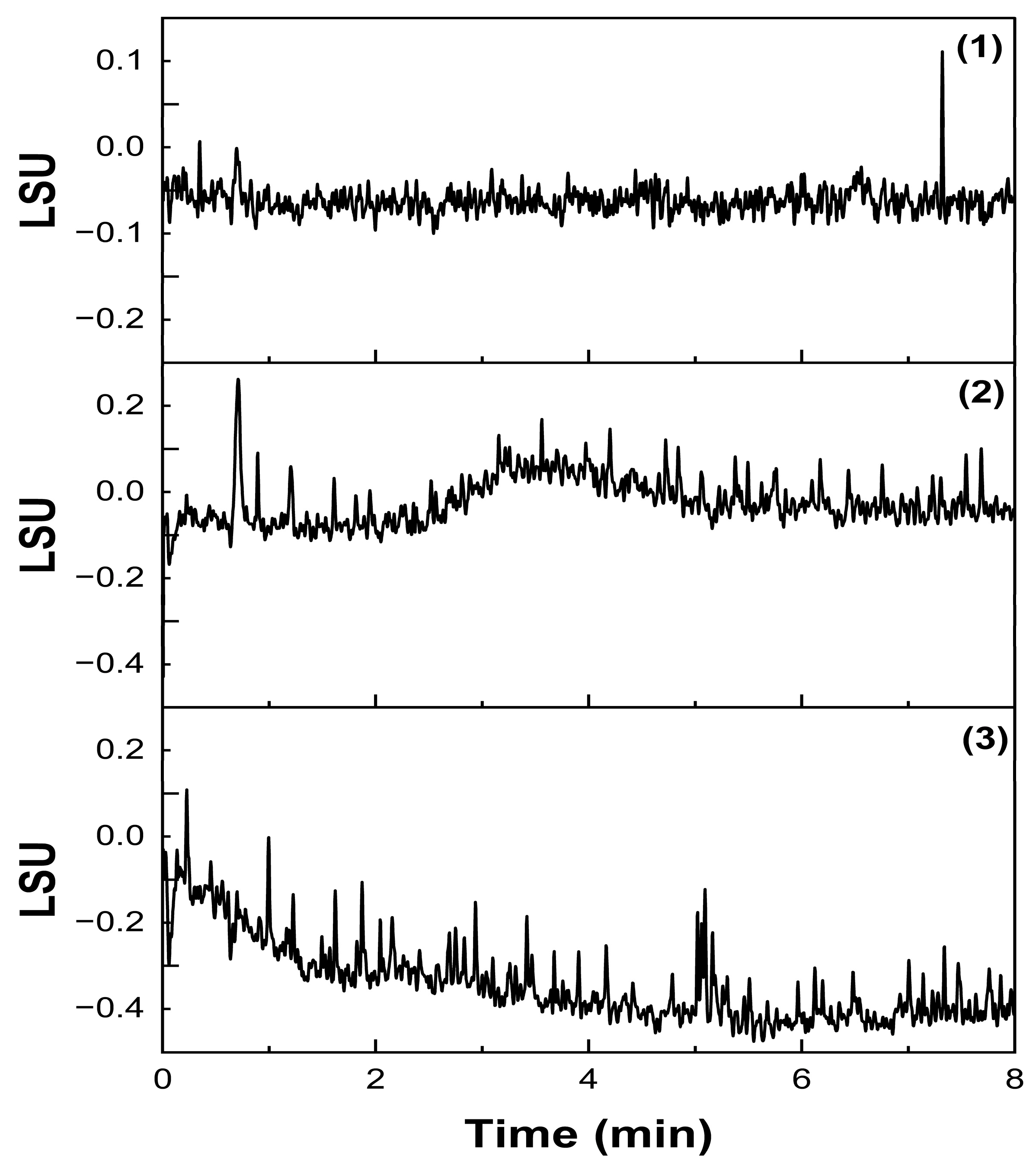
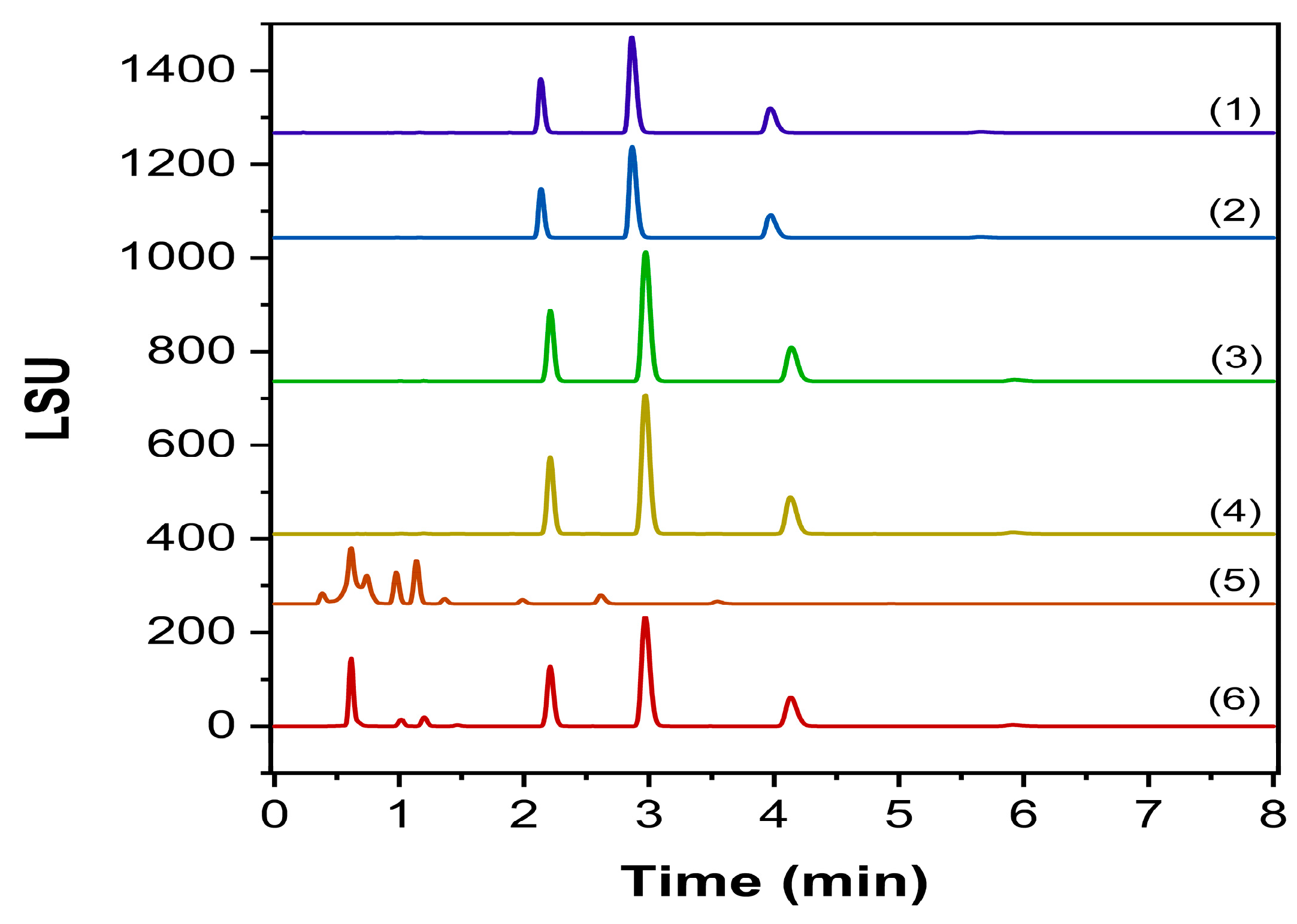
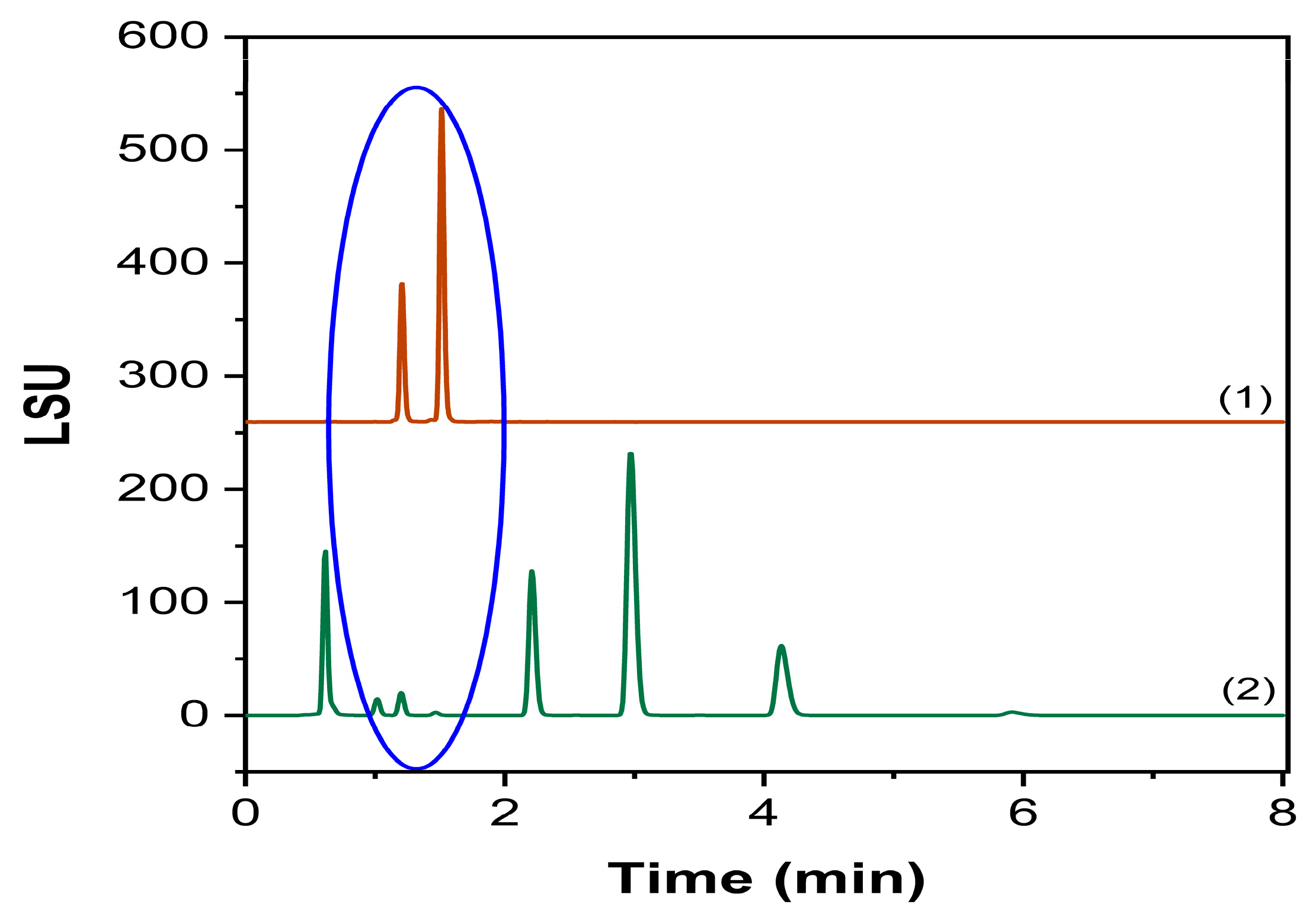
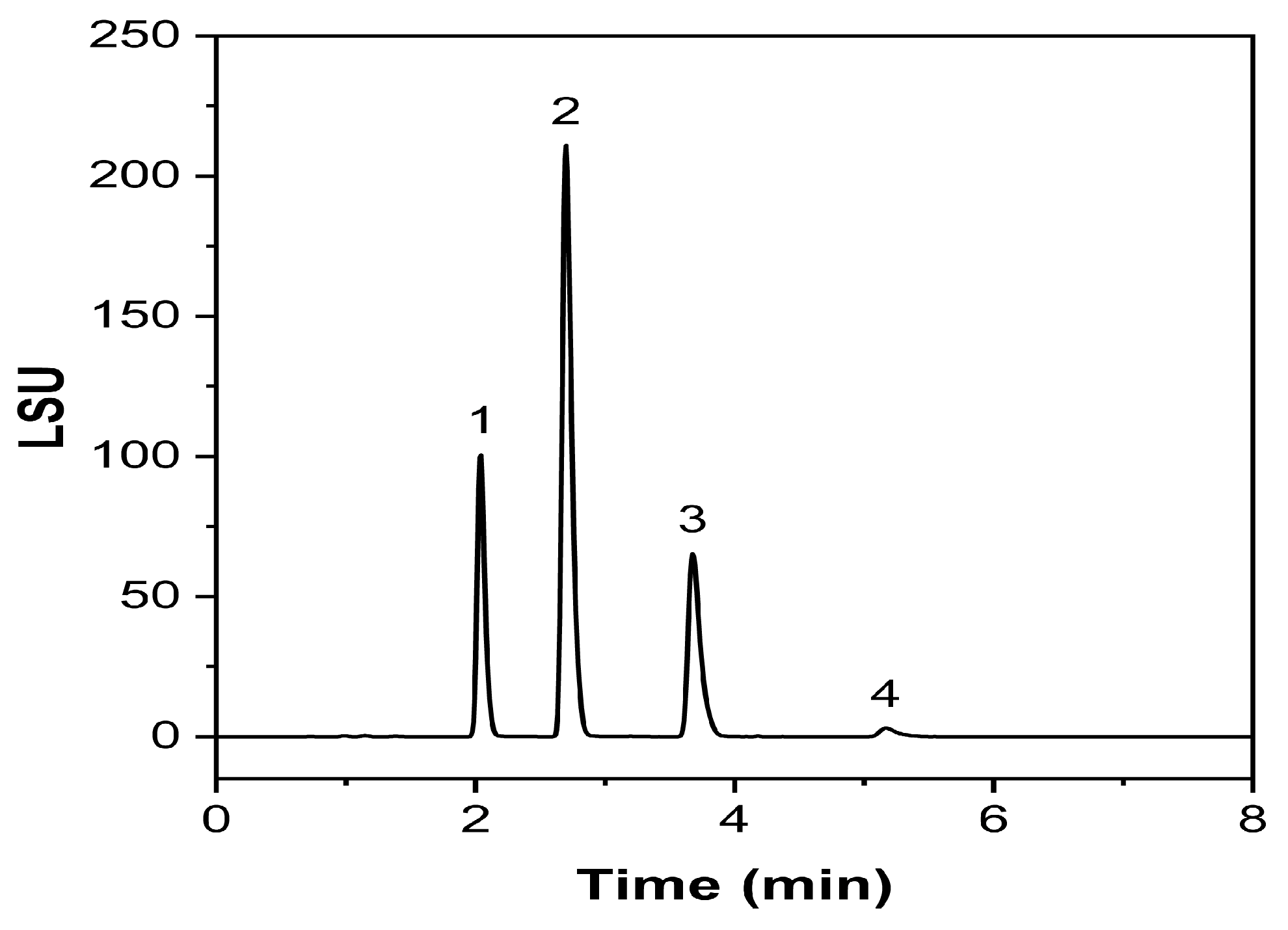
2.3.1. Specificity and Forced Degradation Studies
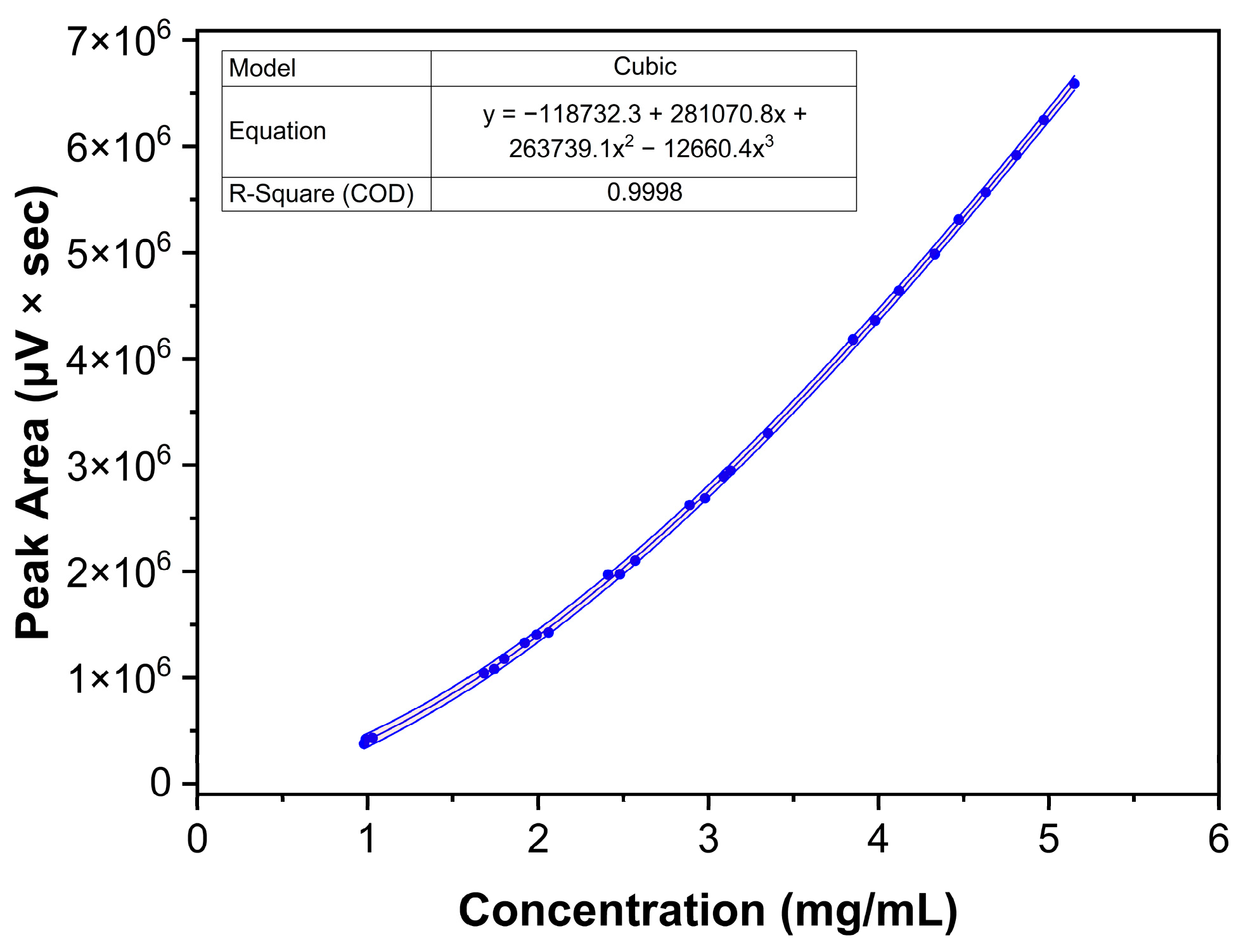
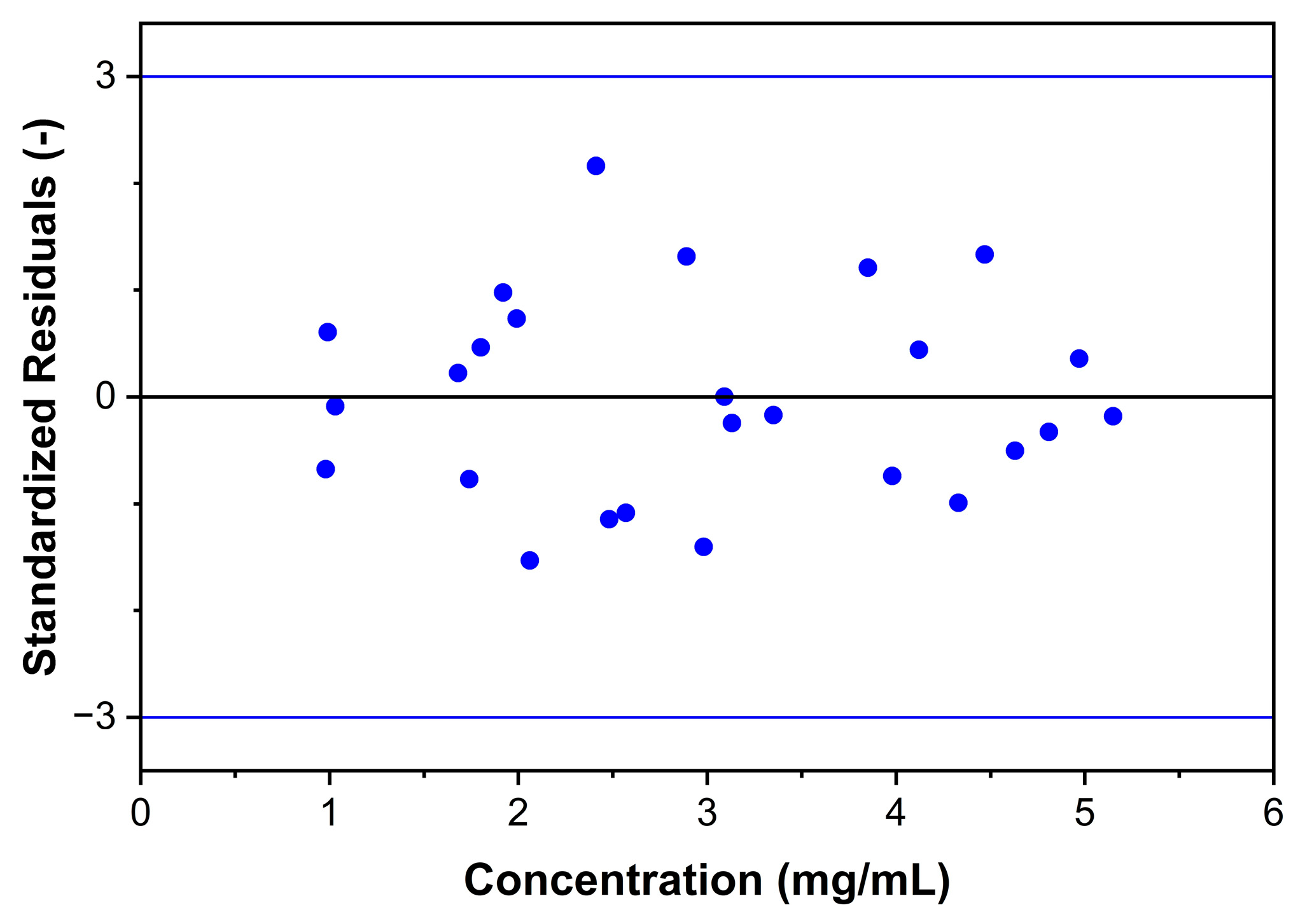
2.3.2. Response, LOD, and LOQ
2.3.3. Accuracy and Precision (Repeatability and Intermediate Precision)
2.3.4. Robustness
2.3.5. Nanopharmaceuticals Applications
2.3.6. Analytical Procedure Lifecycle
3. Materials and Methods
3.1. Reagents and Consumables
3.2. Standards, Samples and Excipients
3.3. Equipment
3.4. Nanoemulsion Matrix Preparation
3.5. Analytical Method Validation
3.5.1. Specificity
3.5.2. Forced Degradation Studies
- Acid hydrolysis: exposure to 1.0 mL of 6N Hydrochloric Acid solution for 5 h;
- Basic hydrolysis: addition of 100 µL of 6N Sodium Hydroxide solution to 1.0 mL of the reference solution and immediate injection;
- Oxidation: exposure to 1.0 mL of 30% w/v H2O2 solution for 5 h;
- Thermolysis: exposure to heat (100 °C) in a dedicated oven for 4 h.
3.5.3. Response, LOD, and LOQ
3.5.4. Accuracy
3.5.5. Precision (Repeatability and Intermediate Precision)
3.5.6. Robustness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| API | Active Pharmaceutical Ingredient |
| AQbD | Analytical Quality by Design |
| ATP | Analytical Target Profile |
| CMeAS | Critical Method Attributes |
| CMePS | Critical Method Parameters |
| DoE | Design of Experiments |
| ELSD | Evaporative Light Scattering Detection |
| GC | Gas Chromatography |
| HPLC | High-Performance Liquid Chromatography |
| HLB | Hydrophilic-lipophilic Balance |
| ICH | International Council of Harmonization |
| LOD | Limit of Detection |
| LOQ | Limit of Quantification |
| MCTs | Medium Chain Triglycerides |
| MODR | Method Operable Design Region |
| NE | Nanoemulsion |
| PDA | Photo Diode Array |
| QbD | Quality by Design |
| QRM | Quality Risk Management |
| RSD | Relative Standard Deviation |
| RT | Retention Time |
| RP | Reversed-phase |
| USP | United States Pharmacopoeia |
| UPLC | Ultra High-Performance Liquid Chromatography |
| UV | Ultraviolet |
References
- Williams, H.D.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of In Vitro Digestion Profiles Across a Range of Representative Formulations. J. Pharm. Sci. 2012, 101, 3360–3380. [Google Scholar] [CrossRef]
- Tetyczka, C.; Griesbacher, M.; Absenger-Novak, M.; Fröhlich, E.; Roblegg, E. Development of nanostructured lipid carriers for intraoral delivery of Domperidone. Int. J. Pharm. 2017, 526, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Kotta, S.; Khan, A.W.; Pramod, K.; Ansari, S.H.; Sharma, R.K.; Ali, J. Exploring oral nanoemulsions for bioavailability enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2012, 9, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Free, M.L. Introduction to Surfactants. Surfactants Precis. Clean. Remov. Contam. Micro Nanoscale 2022, 1–53. [Google Scholar] [CrossRef]
- Göke, K.; Lorenz, T.; Repanas, A.; Schneider, F.; Steiner, D.; Baumann, K.; Bunjes, H.; Dietzel, A.; Finke, J.H.; Glasmacher, B.; et al. Novel strategies for the formulation and processing of poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2018, 126, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, H. Mitigation of Plant Abiotic Stress by Microorganisms; Elsevier: Tamil Nadu, India, 2022; pp. 273–297. [Google Scholar]
- Çinar, K. A Review on Nanoemulsions: Preparation Methods and Stability. Trak. Univ. J. Eng. Sci. 2017, 18, 73–83. [Google Scholar]
- Badnjevic, A. CMBEBIH 2017; Springer Nature: Dordrecht, The Netherlands, 2017. [Google Scholar]
- Hebbi, V.; Chattopadhyay, S.; Rathore, A.S. High performance liquid chromatography (HPLC) based direct and simultaneous estimation of excipients in biopharmaceutical products. J. Chromatogr. B 2019, 1117, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Karlowicz-Bodalska, K.; Szura, D.; Ozimek, L.; Musial, W. Application of HPLC with ELSD Detection for the Assessment of Azelaic Acid Impurities in Liposomal Formulation. Sci. World J. 2013, 2013, 564962. [Google Scholar] [CrossRef] [PubMed]
- Mousli, Y.; Brachet, M.; Chain, J.L.; Ferey, L. A rapid and quantitative reversed-phase HPLC-DAD/ELSD method for lipids involved in nanoparticle formulations. J. Pharm. Biomed. Anal. 2022, 220, 115011. [Google Scholar] [CrossRef] [PubMed]
- Lutzke, B.; Braughler, J. An improved method for the identification and quantitation of biological lipids by HPLC using laser light-scattering detection. J. Lipid Res. 1990, 31, 2127–2130. [Google Scholar] [CrossRef] [PubMed]
- Mounts, T.L.; Abidl, S.L.; Rennick, K.A. HPLC analysis of phospholipids by evaporative laser light-scattering detection. J. Am. Oil Chem. Soc. 1992, 69, 438–442. [Google Scholar] [CrossRef]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2020, 192, 113642. [Google Scholar] [CrossRef] [PubMed]
- EDQM Triglycerides, Medium-Chain. European Pharmacopoeia; European Directorate for the Quality of Medicines & HealthCare: Strasbourg, France, 2020; pp. 4096–4097. [Google Scholar]
- Plante, M.; Bailey, B.; Acworth, I.; Clark, D. Analysis of Lipids by HPLC-CAD. 2011. Available online: https://hplc.eu/Downloads/HALO_Lipids.pdf (accessed on 10 September 2024).
- Graham, C. Analysis of Foods Using HPLC with Evaporative Light Scattering Detection. Available online: https://www.agilent.com/cs/library/applications/5990-9456EN.pdf (accessed on 10 September 2024).
- DeLaney, K.; Han, D.; Birdsall, R.E.; Qing Yu, Y. Optimized ELSD Workflow for Improved Detection of Lipid Nanoparticle Components. Available online: https://www.waters.com/nextgen/us/en/library/application-notes/2022/optimized-elsd-workflow-for-improved-detection-of-lipid-nanoparticle-components.html?srsltid=AfmBOorgq4-8_Tsduhp9Ib99Ojes4KlEfexjh4KEuMmoiTYHEk9l2dH0 (accessed on 10 September 2024).
- Wang, S.; Liu, C.; Chen, Y.; Zhang, Z.; Zhu, A.; Qian, F. A high-sensitivity HPLC-ELSD method for HPMC-AS quantification and its application in elucidating the release mechanism of HPMC-AS based amorphous solid dispersions. Eur. J. Pharm. Sci. 2018, 122, 303–310. [Google Scholar] [CrossRef]
- Rowe, J.C.; Sheskey, E.Q. Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Pharmacopeia, U.S. Mono- and Di-Glycerides Monograph. Available online: https://online.uspnf.com/uspnf/document/1_GUID-EDFBCC40-7304-4BAE-9D27-239F4A6FA6F5_4_en-US?source=QuickSearch&highlight=mono (accessed on 17 October 2024).
- Cernosek, T.; Dalphin, M.; Jain, N.; Richter, N.; Beard, S.; Wang, J.; Osborne, J.; Stone, T.; Mellal, M.; Behrens, S.; et al. Analytical Quality by Design as applied to the development of a SEC-HPLC platform procedure for the determination of monoclonal antibody purity without mobile phase additives. J. Pharm. Biomed. Anal. 2024, 246, 116220. [Google Scholar] [CrossRef] [PubMed]
- Urich, J.A.A.; Marko, V.; Boehm, K.; García, R.A.L.; Jeremic, D.; Paudel, A. Development and Validation of a Stability-Indicating UPLC Method for the Determination of Hexoprenaline in Injectable Dosage Form Using AQbD Principles. Molecules 2021, 26, 6597. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, R. QbD: A Global Implementation Perspective The EU Perspective. In Proceedings of the The Siena Conference on Product and Process Optimization, Siena, Italy, 6 October 2008. [Google Scholar]
- ICH Expert Working Group. Pharmaceutical Development Q8(R2); ICH: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Verch, T.; Campa, C.; Chéry, C.C.; Frenkel, R.; Graul, T.; Jaya, N.; Nakhle, B.; Springall, J.; Starkey, J.; Wypych, J.; et al. Analytical Quality by Design, Life Cycle Management, and Method Control. AAPS J. 2022, 24, 34. [Google Scholar] [CrossRef]
- Alhakeem, M.A.; Ghica, M.V.; Pîrvu, C.D.; Anuța, V.; Popa, L. Analytical Quality by Design with the Lifecycle Approach: A Modern Epitome for Analytical Method Development. Acta Medica Marisiensis 2019, 65, 37–44. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use. ICH Q2(R2) Validation of Analytical Procedures; ICH: Amsterdam, The Netherlands, 2022; Volume 2, pp. 1–34. [Google Scholar]
- Ganorkar, S.B.; Shirkhedkar, A.A. Design of experiments in liquid chromatography (HPLC) analysis of pharmaceuticals: Analytics, applications, implications and future prospects. Rev. Anal. Chem. 2017, 36, 20160025. [Google Scholar] [CrossRef]
- ICH International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH Harmonised Guideline General Considerations for Clinical Studies E8(R1); ICH: Amsterdam, The Netherlands, 2021; Volume 1, pp. 1–29. [Google Scholar]
- Ramalingam, P.; Jahnavi, B. QbD Considerations for Analytical Development; Elsevier Inc.: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Watson, S. Applying QbD Principles to Analytical Development. 2017. Available online: https://www.pharm-int.com/2021/08/16/applying-qbd-principles-to-analytical-development/ (accessed on 19 October 2024).
- USP-NF 〈1220〉 Analytical Procedure Life Cycle. Available online: https://online.uspnf.com/uspnf/document/1_GUID-35D7E47E-65E5-49B7-B4CC-4D96FA230821_2_en-US?source=Activity (accessed on 22 October 2024).
- Volta e Sousa, L.V.; Gonçalves, R.; Menezes, J.C.; Ramos, A. Analytical Method Lifecycle Management in Pharmaceutical Industry: A Review. Aaps Pharmscitech 2021, 22, 128. [Google Scholar] [CrossRef]
- Douville, V.; Lodi, A.; Miller, J.; Nicolas, A.; Clarot, I.; Prilleux, B.; Megoulas, N.; Koupparis, M. Evaporative light scattering detection (ELSD): A tool for improved quality control of drug substances. Pharmeur. Sci. Notes 2006, 2006, 9–15. [Google Scholar] [PubMed]
- Food and Drug Administration Affairs. O.O.R.Q. ORA Lab Manual Vol. II–Methods, Method Verification and Validation (ORA- LAB.5.4.5). Food Drug Adm. Off. Regul. Aff. Qual. 2023, 2, 1–34. [Google Scholar]
- Ponphaiboon, J.; Limmatvapirat, S.; Chaidedgumjorn, A.; Limmatvapirat, C. Optimization and comparison of GC-FID and HPLC-ELSD methods for determination of lauric acid, mono-, di-, and trilaurins in modified coconut oil. J. Chromatogr. B 2018, 1099, 110–116. [Google Scholar] [CrossRef]
- Fauland, A.; Köfeler, H.; Trötzmüller, M.; Knopf, A.; Hartler, J.; Eberl, A.; Chitraju, C.; Lankmayr, E.; Spener, F. A comprehensive method for lipid profiling by liquid chromatography-ion cyclotron resonance mass spectrometry. J. Lipid Res. 2011, 52, 2314–2322. [Google Scholar] [CrossRef] [PubMed]
- Loeser, E. Peculiarities of Mobile Phases Containing Formic Acid. Chromatographia 2009, 69, 807–811. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, S.; Zheng, M.; Peng, J.; He, X.; Han, Y.; Zhu, J.; Xiao, Q.; Lv, R.; Lin, R. Development of the HPLC–ELSD method for the determination of phytochelatins and glutathione in Perilla frutescens under cadmium stress conditions. R. Soc. Open Sci. 2018, 5, 171659. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, M. The Role of Temperature and Column Thermostatting in Liquid Chromatography. 2016. Available online: https://tools.thermofisher.com/content/sfs/brochures/WP-71499-LC-Temperature-Column-Thermostatting-WP71499-EN.pdf (accessed on 28 October 2024).
- Lesellier, E.; Valarché, A.; West, C.; Dreux, M. Effects of selected parameters on the response of the evaporative light scattering detector in supercritical fluid chromatography. J. Chromatogr. A 2012, 1250, 220–226. [Google Scholar] [CrossRef]
- Mccarthy, S.M.; Gilar, M. UPLC Separation of Oligonucleotides: Effect of Increased Flow Rate and Faster Run Time. Available online: https://www.waters.com/nextgen/au/en/library/application-notes/2010/uplc-separation-oligonucleotides-effect-increased-flow-rate-faster-run-time.html?srsltid=AfmBOoptiY7NJUkIq7Zlv_cp5mucMN_rNXZ7_4RnUHIZpsGjrxvpLpgV (accessed on 10 September 2024).
- Shapiro, S.S.; Wilk, M.B. An Analysis of Variance Test for Normality (Complete Samples). Biometrika 1965, 52, 591. [Google Scholar] [CrossRef]
- Nakmode, D.; Bhavana, V.; Thakor, P.; Madan, J.; Singh, P.K.; Singh, S.B.; Rosenholm, J.M.; Bansal, K.K.; Mehra, N.K. Fundamental Aspects of Lipid-Based Excipients in Lipid-Based Product Development. Pharmaceutics 2022, 14, 831. [Google Scholar] [CrossRef]
- Zenebon, O.; Sadocco, N.; Tiglea, P. Métodos Físico-Químicos Para Análise de Alimentos, 4th ed.; Instituto Adolfo Lutz: São Paulo, Brazil, 2008. [Google Scholar]
- Urich, J.A.A.; Marko, V.; Boehm, K.; Garcia, R.A.L.; Fedorko, A.; Salar-Behzadi, S.; Jeremic, D. Stability-Indicating UPLC-PDA-QDa Methodology for Carvedilol and Felodipine in Fixed-Dose Combinations Using AQbD Principles. Sci. Pharm. 2024, 92, 22. [Google Scholar] [CrossRef]
- Raposo, F.; Ibelli-Bianco, C. Performance parameters for analytical method validation: Controversies and discrepancies among numerous guidelines. TrAC Trends Anal. Chem. 2020, 129, 115913. [Google Scholar] [CrossRef]
- Sut, S.; Franceschi, C.; Peron, G.; Poloniato, G.; Dall’acqua, S. Development and Validation of an HPLC-ELSD Method for the Quantification of 1-Triacontanol in Solid and Liquid Samples. Molecules 2018, 23, 2775. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Singh, U.; Pandey, C.M.; Mishra, P.; Pandey, G. Application of student’s t-test, analysis of variance, and covariance. Ann. Card. Anaesth. 2019, 22, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.K.; Jensen, J.S.; Diaz, A.R. An Investigation into Detector Limitations Using Evaporative Light-Scattering Detectors For Pharmaceutical Applications. J. Chromatogr. Sci. 2004, 42, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Parr, M.K.; Schmidt, A.H. Life cycle management of analytical methods. J. Pharm. Biomed. Anal. 2018, 147, 506–517. [Google Scholar] [CrossRef]
- Armoškaitė, V.; Jakštas, V.; Ivanauskas, L.; Ražukas, A.; Ramanauskienė, K.; Briedis, V. Application of HPLC-ELSD for the Quantification of 5-Aminolevulinic Acid after Penetration into Human Skin Ex Vivo. Anal. Lett. 2013, 46, 717–733. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ATP Element | Target | Requirement Reference |
|---|---|---|
| Number of Peaks | =4 | - |
| Peak Tailing | <2 | [28,36] |
| Peak Height | Above LOQ | [28,36] |
| Capacity factor (k’) | >2 | [28,36] |
| Resolution | >2 | [28,36] |
| Analytical Column | Organic Modifier (%) | Aqueous Phase (%) | Formic Acid (%) | Sample Diluent | Flow (mL/min) | Injection Volume (μL) | Column Temperature (°C) |
|---|---|---|---|---|---|---|---|
| (A) (B) (C) (D) | Acetonitrile Methanol | 0–4 | 0–0.5 | Acetonitrile Methanol Acetone 2-Propanol | 0.3–0.7 | 1–5 | 25–38 |
| Validation Conditions | |
|---|---|
| Flow Rate | 0.41 mL/min |
| Injection Volume | 2.8 µL |
| Formic Acid Ratio | 0.4% |
| Mobile Phase | Water/Methanol (2.5 : 97.5) |
| Column | Acquity HSS T3 |
| Column Temperature | 33 °C |
| Sample Diluent | 2-Propanol |
| Stress Condition | Recovery (%) | Average (%) | SD |
|---|---|---|---|
| Acidic hydrolysis | 88.1 88.3 86.2 | 87.5 | 1.3 |
| Basic hydrolysis | 10.2 9.8 9.6 | 9.9 | 0.3 |
| Oxidation | 100.6 96.1 99.4 | 98.7 | 2.3 |
| Thermolysis | 98.4 98.6 98.4 | 98.4 | 0.1 |
| Component | LOD (µg/mL) | LOQ (µg/mL) |
|---|---|---|
| Tricaprylin | 1.48 | 4.94 |
| 1,2-caprate-3-caprylate | 2.29 | 7.64 |
| 1,2-caprylate-3-caprate | 4.14 | 13.81 |
| Tricaprin | 168.91 | 563.06 |
| Percent of Target (%) | Recovery (%) | Average (%) | RSD (%) |
|---|---|---|---|
| 70% | 98.4 100.2 100.9 | 99.8 | 1.3 |
| 100% | 99.5 99.3 99.0 | 99.3 | 0.2 |
| 130% | 98.8 100.6 102.1 | 100.5 | 1.6 |
| Parameter | Analyst I | Analyst II |
|---|---|---|
| Mean | 97.32 | 99.65 |
| SD | 2.25 | 0.31 |
| RSD | 2.30 | 0.30 |
| SEM | 0.03 | 0.04 |
| n | 6 | 6 |
| p-value | 0.065 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaggero, A.; Marko, V.; Jeremic, D.; Tetyczka, C.; Caisse, P.; Afonso Urich, J.A. AQbD-Based UPLC-ELSD Method for Quantifying Medium Chain Triglycerides in Labrafac™ WL 1349 for Nanoemulsion Applications. Molecules 2025, 30, 486. https://doi.org/10.3390/molecules30030486
Gaggero A, Marko V, Jeremic D, Tetyczka C, Caisse P, Afonso Urich JA. AQbD-Based UPLC-ELSD Method for Quantifying Medium Chain Triglycerides in Labrafac™ WL 1349 for Nanoemulsion Applications. Molecules. 2025; 30(3):486. https://doi.org/10.3390/molecules30030486
Chicago/Turabian StyleGaggero, Alessio, Viktoria Marko, Dalibor Jeremic, Carolin Tetyczka, Philippe Caisse, and Jesús Alberto Afonso Urich. 2025. "AQbD-Based UPLC-ELSD Method for Quantifying Medium Chain Triglycerides in Labrafac™ WL 1349 for Nanoemulsion Applications" Molecules 30, no. 3: 486. https://doi.org/10.3390/molecules30030486
APA StyleGaggero, A., Marko, V., Jeremic, D., Tetyczka, C., Caisse, P., & Afonso Urich, J. A. (2025). AQbD-Based UPLC-ELSD Method for Quantifying Medium Chain Triglycerides in Labrafac™ WL 1349 for Nanoemulsion Applications. Molecules, 30(3), 486. https://doi.org/10.3390/molecules30030486