
Domino Synthesis of 1,2,5-Trisubstituted 1H-Indole-3-carboxylic Esters Using a [3+2] Strategy


Abstract
1. Introduction
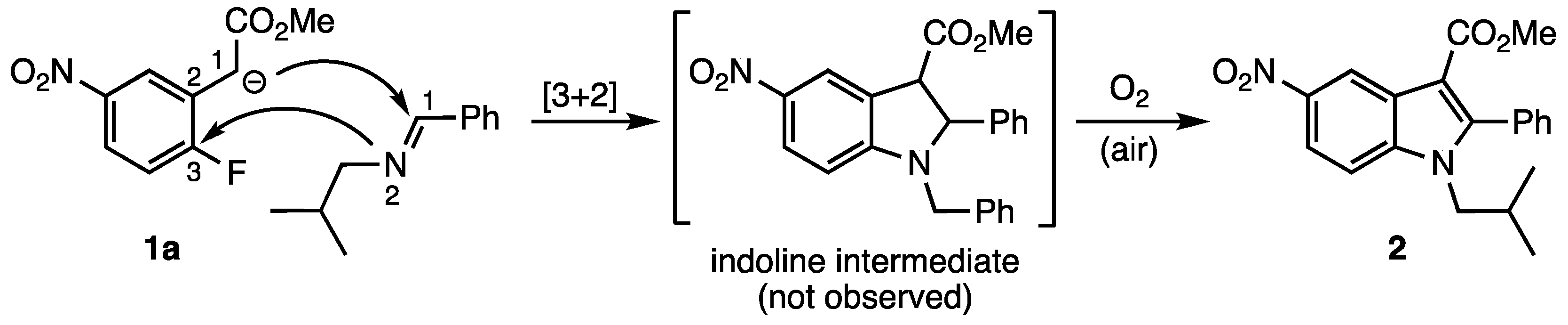
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Representative Procedure for Preparing 1,2,5-Trisubstituted 1H-Indole-3-carboxylate Esters
3.2.1. Methyl 1-Isobutyl-5-nitro-2-phenyl-1H-indole-3-carboxylate (2)
3.2.2. Methyl 1-Benzyl-2-methyl-5-nitro-1H-indole-3-carboxylate (3)
3.2.3. Methyl 2-Methyl-1-(4-methylbenzyl)-5-nitro-1H-indole-3-carboxylate (4)
3.2.4. Methyl 1-(4-Chlorobenzyl)-1-methyl-5-nitro-1H-indole-3-carboxylate (5)
3.2.5. Methyl 1-(2-Chlorobenzyl)-2-methyl-5-nitro-1H-indole-3-carboxylate (6)
3.2.6. Methyl 1-Benzyl-5-nitro-2-phenyl-1H-indole-3-carboxylate (7)
3.2.7. Methyl 5-Nitro-1-phenethyl-2-phenyl-1H-indole-3-carboxylate (8)
3.2.8. Methyl 1-(4-Methylbenzyl)-5-nitro-2-phenyl-1H-indole-3-carboxylate (9)
3.2.9. Methyl 1-Benzyl-2-(4-fluorophenyl)-5-nitro-1H-indole-3-carboxylate (10)
3.2.10. Methyl 1-Benzyl-2-(4-chlorophenyl)-5-nitro-1H-indole-3-carboxylate (11)
3.2.11. Methyl 1-Benzyl-2-(2-chlorophenyl)-5-nitro-1H-indole-3-carboxylate (12)
3.2.12. Methyl 1-Benzyl-2-(3-methoxyphenyl)-5-nitro-1H-indole-3-carboxylate (13)
3.2.13. Methyl 2-(4-Methylphenyl)-5-nitro-1-phenethyl-1H-indole-3-carboxylate (14)
3.2.14. Methyl 2-(4-Fluorophenyl)-5-nitro-1-phenethyl-1H-indole-3-carboxylate (15)
3.2.15. Methyl 2-(4-Methoxyphenyl)-5-nitro-1-phenethyl-1H-indole-3-carboxylate (16)
3.2.16. Methyl 1-Benzyl-5-cyano-2-phenyl-1H-indole-3-carboxylate (17)
3.2.17. Methyl 5-Cyano-1-phenethyl-2-phenyl-1H-indole-3-carboxylate (18)
3.2.18. Methyl 5-Cyano-2-(4-methylphenyl)-1-phenethyl-1H-indole-3-carboxylate (19)
3.2.19. Methyl 5-Cyano-2-(4-fluorophenyl)-1-phenethyl-1H-indole-3-carboxylate (20)
3.2.20. Methyl 2-(4-Chlorophenyl)-5-cyano-1-phenethyl-1H-indole-3-carboxylate (21)
3.2.21. Methyl 5-Cyano-2-(4-methoxyphenyl)-1-phenethyl-1H-indole-3-carboxyl-ate (22)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katritzky, A.R.; Ramsden, C.A.; Joule, J.A.; Zhdankin, V.V. Handbook of Heterocyclic Chemistry, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 801–815. [Google Scholar]
- Organic Chemistry Portal. Available online: https://www.organic-chemistry.org/synthesis/heterocycles/benzo-fused/indoles.shtm (accessed on 20 November 2024).
- Inman, M.; Moody, C.J. Indole synthesis—Something old, something new. Chem. Sci. 2013, 4, 29–41. [Google Scholar] [CrossRef]
- Kudzma, L.V. Synthesis of substituted indoles and carbazoles from 2-fluorophenyl imines. Synthesis 2003, 2003, 1661–1666. [Google Scholar] [CrossRef]
- Bunce, R.A.; Randall, M.H.; Applegate, K.G. 2-Alkylindole-3-carboxylic esters by a tandem reduction-addition-elimination reaction. Org. Prep. Proced. Int. 2002, 34, 493–499. [Google Scholar] [CrossRef]
- Li, J.J. Batcho-Leimgruber indole synthesis. In Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, 5th ed.; Springer International: Cham, Switzerland, 2014; pp. 34–35. [Google Scholar] [CrossRef]
- Ametsetor, E.; Farthing, S.; Bunce, R.A. Domino aza-Michael-SNAr-heteroaromatization route to C5-substituted N-alkyl-1H-indole-3-carboxylic esters. Molecules 2022, 27, 6998. [Google Scholar] [CrossRef]
- Bunce, R.A.; Schammerhorn, J.E.; Sigle, J. Substituted 4-oxo-1,2,3,4-tetrahydroquinoline-3-carboxylic esters by a tandem imine addition-SNAr reaction. J. Heterocycl. Chem. 2013, 50, 373–380. [Google Scholar] [CrossRef]
- Dumitrascu, F.; Illies, M.A. Recent advances in the Nenitzescu indole synthesis (1990–2019). In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 133, pp. 65–157. [Google Scholar] [CrossRef]
- Trost, B.M. The atom economy: A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef]
- Trost, B.M. Atom economy: A challenge for enhanced synthetic efficiency. In Handbook of Green Chemistry; Wiley Online Library: Hoboken, NJ, USA, 2012; pp. 1–33. [Google Scholar]
- Ali, M.A.; Punniyamurthy, T. Domino ligand-free copper-catalyzed synthesis of polysubstituted indoles. Synlett 2011, 2011, 623–626. [Google Scholar] [CrossRef]
- Li, Y.; Peng, J.; Chen, X.; Mo, B.; Li, X.; Sun, P.; Chen, C. Copper-catalyzed synthesis of multisubstituted indoles through tandem Ullmann-type C–N bond formation and cross dehydrogenative coupling reactions. J. Org. Chem. 2018, 83, 5288–5294. [Google Scholar] [CrossRef]
- Kim, J.H.; Li, S. Palladium-catalyzed intramolecular trapping of Blaise reaction intermediate for tandem one-pot synthesis of indole derivatives. Org. Lett. 2011, 13, 1350–1353. [Google Scholar] [CrossRef]
- Vaswani, R.G.; Albrecht, B.K.; Audia, J.E.; Cote, A.; Dakin, L.A.; Duplessis, M.; Gehling, V.S.; Harmanage, J.-C.; Hewitt, M.C.; Leblanc, Y.; et al. A practical synthesis of indoles via a Pd-catalyzed C–N ring formation. Org. Lett. 2014, 16, 4114–4117. [Google Scholar] [CrossRef]
- Liu, B.; Song, C.; Sun, C.; Zhou, S.; Zhu, J. Rhodium(III)-catalyzed indole synthesis using N–N bond as an internal oxidant. J. Am. Chem. Soc. 2013, 135, 16625–16631. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Yu, S.; Li, X. Rh(III)-catalyzed synthesis of N-unprotected indoles from imidamides and diazo ketoesters via C–H activation and C–C/C–N bond cleavage. Org. Lett. 2016, 18, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-Q.; Lei, T.; Song, Z.-Q.; Yang, X.-L.; Wu, C.-J.; Jiang, X.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Visible light promoted synthesis of indoles by single photosensitizer under aerobic conditions. Org. Lett. 2017, 19, 3251–3254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Doyle, M.P. Lewis acid catalyzed indole synthesis via intramolecular nucleophilic attack of phenyl-diazoacetate to iminium ions. J. Org. Chem. 2009, 74, 9222–9224. [Google Scholar] [CrossRef]
- Levesque, P.; Fournier, P.-A. Synthesis of substituted indole from 2-aminobenzaldehyde through [1,2]-aryl shift. J. Org. Chem. 2010, 75, 7033–7036. [Google Scholar] [CrossRef]
- Du, Y.; Liu, R.; Linn, G.; Zhao, K. Synthesis of N-substituted indole derivatives via PIFA-mediated intramolecular cyclization. Org. Lett. 2006, 8, 5919–5922. [Google Scholar] [CrossRef]
- Bugaenko, D.I.; Dubrovina, A.A.; Yurovskaya, M.A.; Karchava, A.V. Synthesis of indoles via electron-catalyzed intramolecular C–N bond formation. Org. Lett. 2018, 20, 7358–7362. [Google Scholar] [CrossRef]
- McAusland, D.; Seo, S.; Pintori, D.G.; Finlayson, J.; Greaney, M.F. The benzyne Fischer-indole reaction. Org. Lett. 2011, 13, 3667–3669. [Google Scholar] [CrossRef]
- Zeng, W.; Han, C.; Mohammed, S.; Li, S.; Song, Y.; Sun, F.; Du, Y. Indole-containing pharmaceuticals: Targets, pharmacological activities and SAR studies. RSC Med. Chem. 2024, 15, 788–808. [Google Scholar] [CrossRef]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef]
- Kumar, S.; Ritika. A brief review of the biological potential of indole derivatives. Future J. Pharm. Sci. 2020, 6, 121. [Google Scholar] [CrossRef]
- Kang, Y.; Shi, Y.; Xu, S. Arbidol: The current demand, strategies and antiviral mechanisms. Immun. Inflamm. Dis. 2023, 11, e984. [Google Scholar] [CrossRef] [PubMed]
- Munjal, A.; Allam, A.E. Indomethacin; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK555936/ (accessed on 26 November 2024). [PubMed]
- Fabian, C.J.; Nye, L.; Powers, K.R.; Nydegger, J.L.; Kreutzjans, A.L.; Phillips, T.A.; Metheny, T.; Winblad, O.; Zalles, C.M.; Hagan, C.R.; et al. Effect of Bazedoxifene and conjugated estrogen (Duavee) on breast cancer risk biomarkers in high-risk women: A pilot study. Cancer Prev. Res. 2019, 12, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Chen, X.; Fu, S.; Zhang, R.; Pan, L.; Cao, Y.; Wu, A.; Xiao, H.; Lin, H.-J.; Lo, H.-W.; et al. Basedoxifene is a novel IL-6/GP130 inhibitor for treating negative breast cancer. Breast Cancer Res. Treat. 2019, 175, 553–566. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Expt No.a | Methyl (2-Fluoro-phenyl)acetate Substrate | Aldehyde | Amine | Product | Yield (%) |
| 3.2.1 | 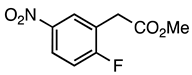 1a |  |  |  | 75 |
| 3.2.2 | 1a | 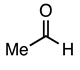 | 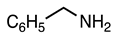 |  | 64 |
| 3.2.3 | 1a |  |  |  | 68 |
| 3.2.4 | 1a |  |  | 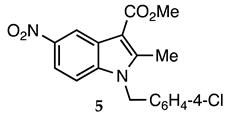 | 85 |
| 3.2.5 | 1a |  |  |  | 60 |
| 3.2.6 | 1a |  |  |  | 89 |
| 3.2.7 | 1a | 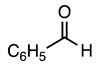 | 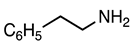 |  | 91 |
| 3.2.8 | 1a | 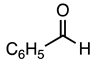 |  | 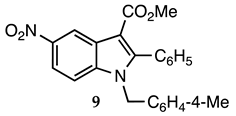 | 81 |
| 3.2.9 | 1a |  | 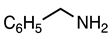 |  | 64 |
| 3.2.10 | 1a | 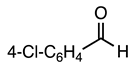 |  | 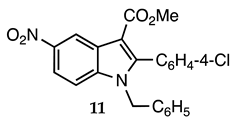 | 85 |
| 3.2.11 | 1a | 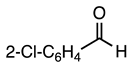 |  |  | 59 |
| 3.2.12 | 1a |  |  |  | 61 |
| 3.2.13 | 1a |  |  | 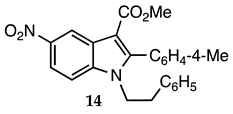 | 90 |
| 3.2.14 | 1a |  |  | 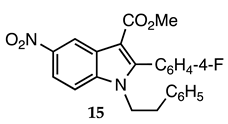 | 64 |
| 3.2.15 | 1a |  |  |  | 85 |
| 3.2.16 |  1b |  |  | 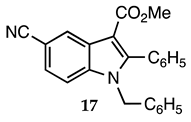 | 82 |
| 3.2.17 | 1b |  |  | 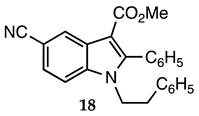 | 96 |
| 3.2.18 | 1b |  | 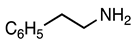 |  | 85 |
| 3.2.19 | 1b |  |  |  | 78 |
| 3.2.20 | 1b | 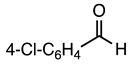 | 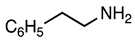 | 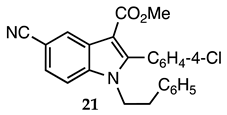 | 78 |
| 3.2.21 | 1b |  |  |  | 80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maji, S.; Fobi, K.; Ametsetor, E.; Bunce, R.A. Domino Synthesis of 1,2,5-Trisubstituted 1H-Indole-3-carboxylic Esters Using a [3+2] Strategy. Molecules 2025, 30, 444. https://doi.org/10.3390/molecules30030444
Maji S, Fobi K, Ametsetor E, Bunce RA. Domino Synthesis of 1,2,5-Trisubstituted 1H-Indole-3-carboxylic Esters Using a [3+2] Strategy. Molecules. 2025; 30(3):444. https://doi.org/10.3390/molecules30030444
Chicago/Turabian StyleMaji, Siddhartha, Kwabena Fobi, Ebenezer Ametsetor, and Richard A. Bunce. 2025. "Domino Synthesis of 1,2,5-Trisubstituted 1H-Indole-3-carboxylic Esters Using a [3+2] Strategy" Molecules 30, no. 3: 444. https://doi.org/10.3390/molecules30030444
APA StyleMaji, S., Fobi, K., Ametsetor, E., & Bunce, R. A. (2025). Domino Synthesis of 1,2,5-Trisubstituted 1H-Indole-3-carboxylic Esters Using a [3+2] Strategy. Molecules, 30(3), 444. https://doi.org/10.3390/molecules30030444