

New Compounds with Enhanced Biological Activity Through the Strategic Introduction of Silylated Groups into Hydroxystearic Acids

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
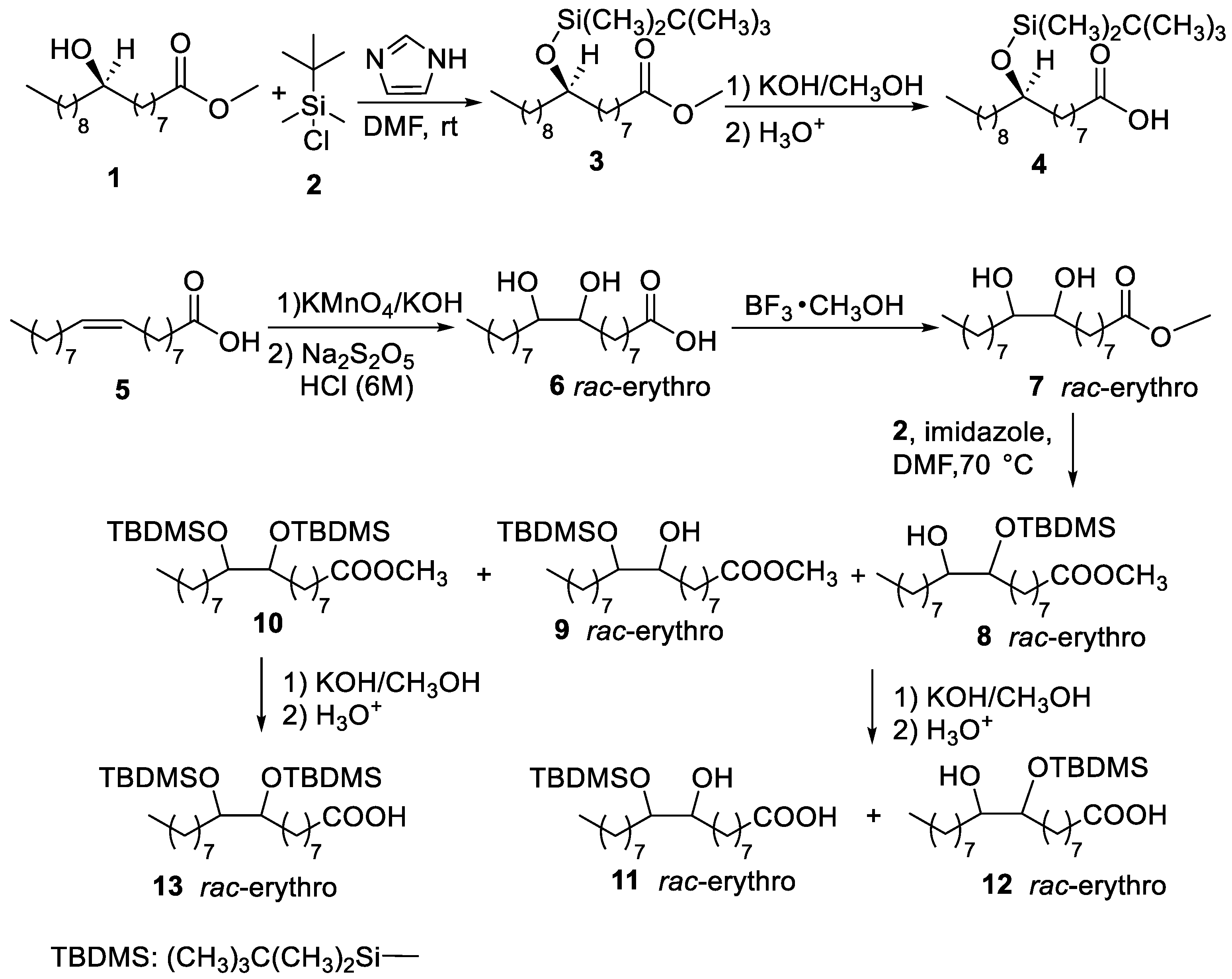
2.1. Synthesis of O-Silylated Hydroxystearic Acids
2.2. Biological Studies
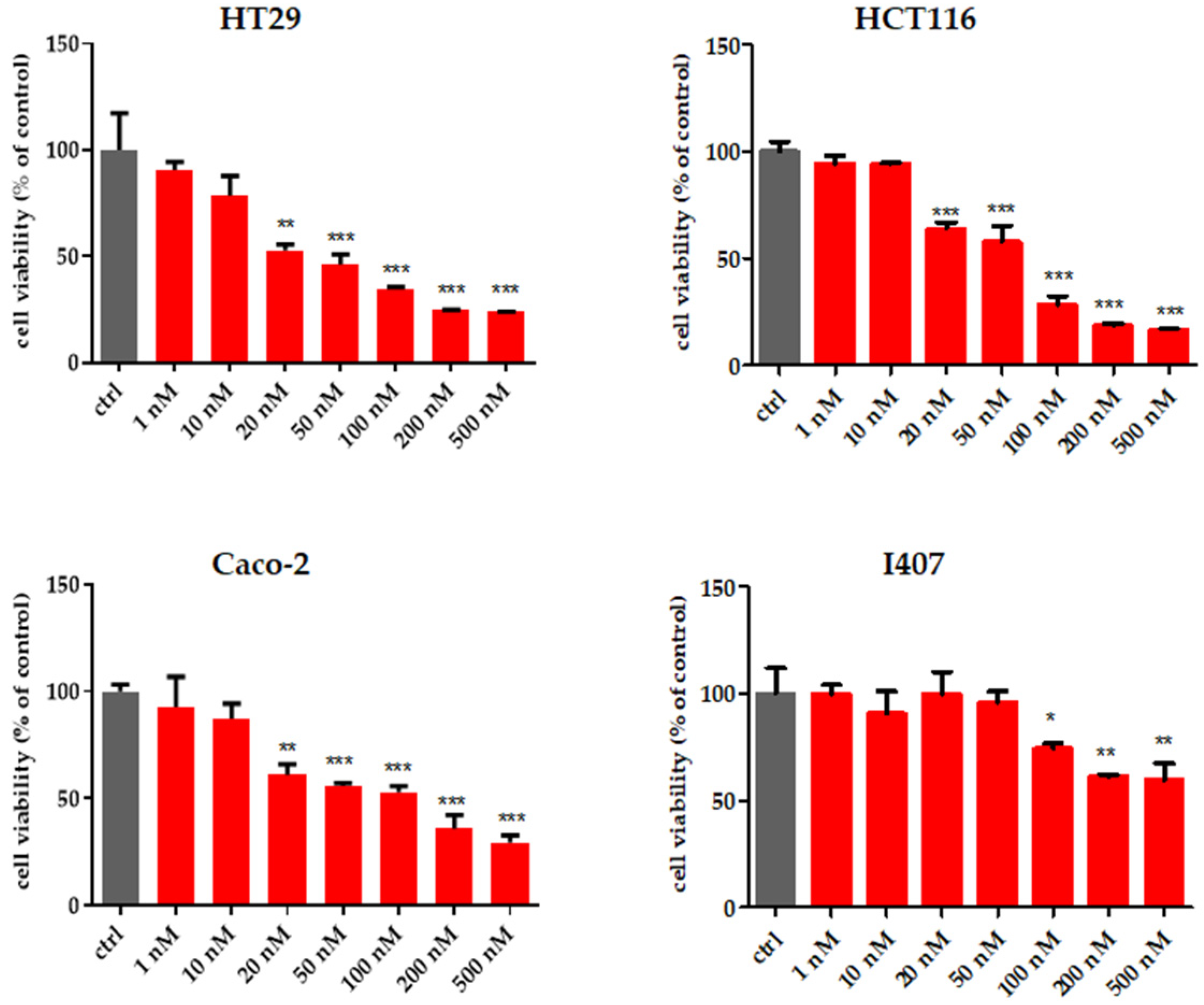
2.2.1. Cell Viability
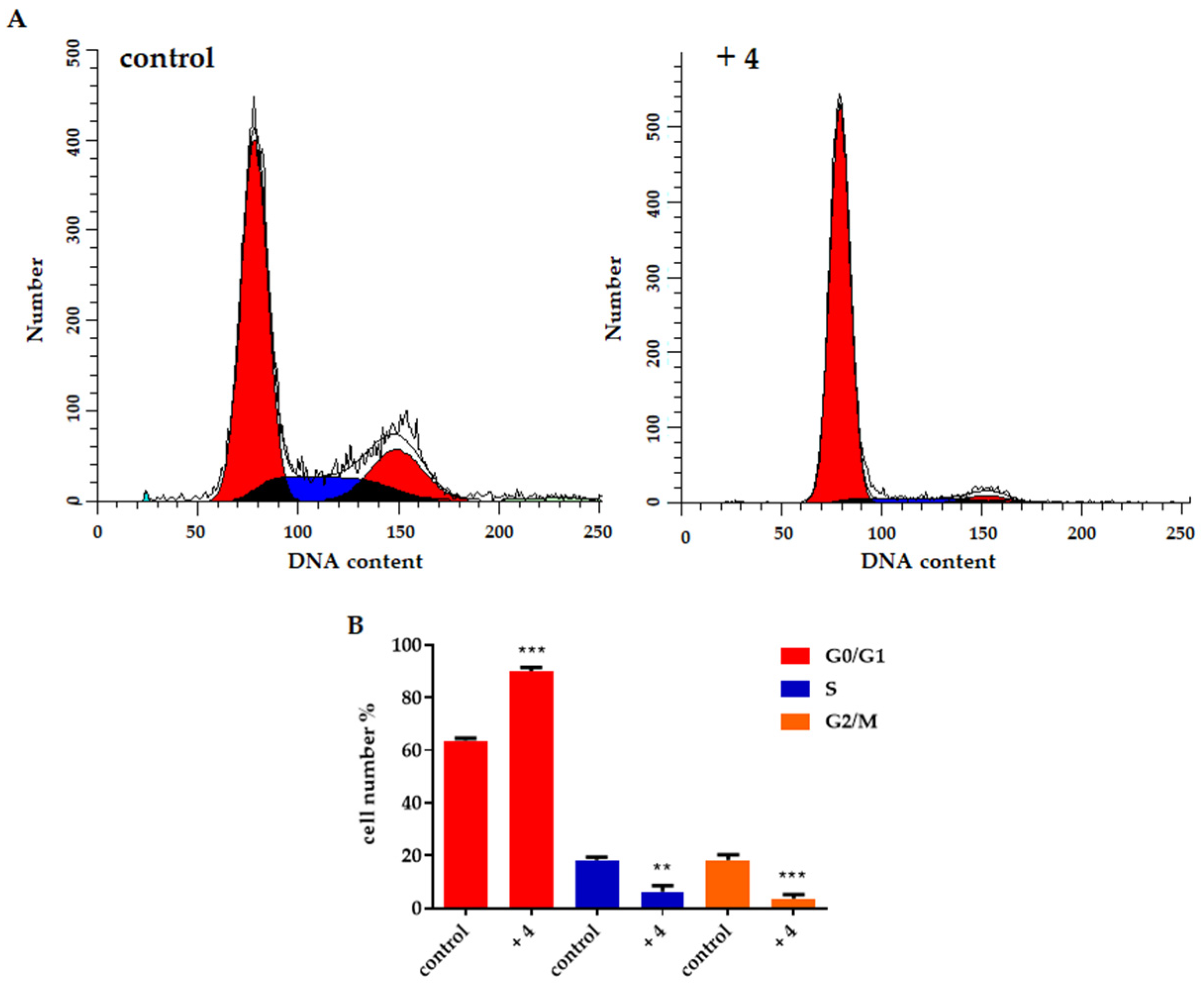
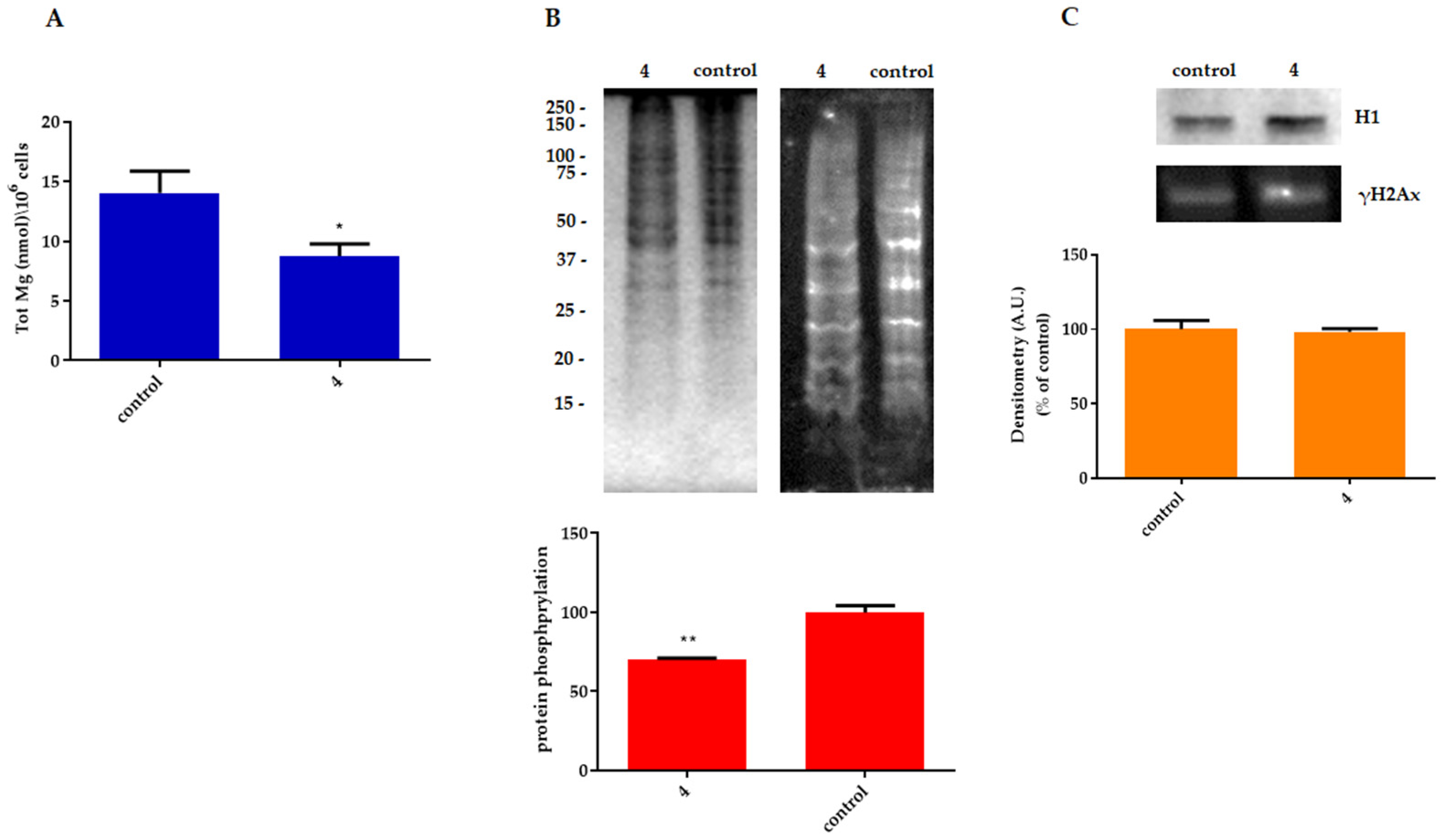
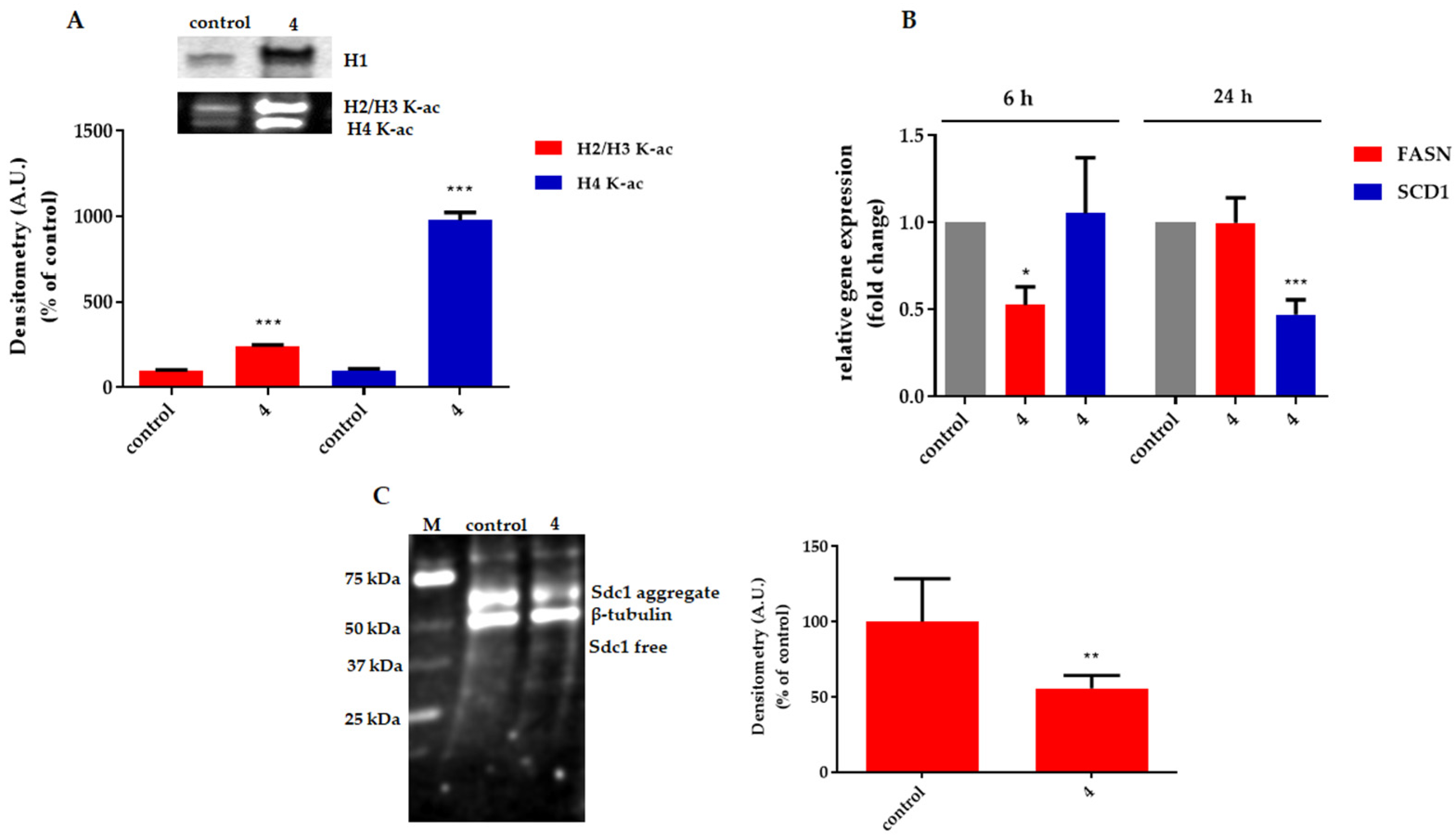
2.2.2. Effect on Cell Proliferation
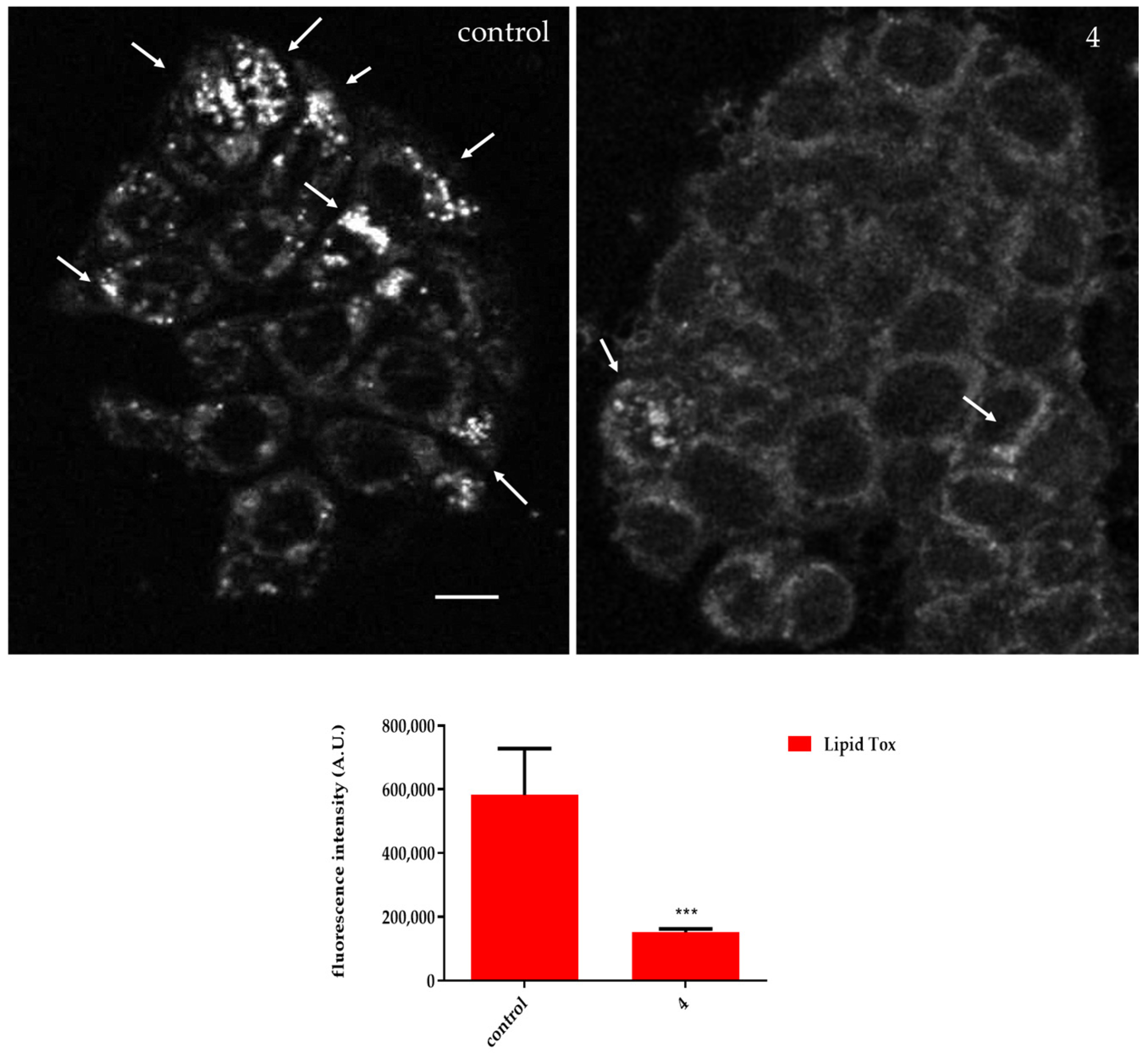
2.2.3. Compound 4 Treatment Reduced the Intracellular Lipid Accumulation
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General
3.1.2. Synthesis of Methyl (R)-9-((tert-butyldimethylsilyl)oxy)octadecanoate (3)
3.1.3. Synthesis of (R)-9-((tert-butyldimethylsilyl)oxy)octadecanoic Acid (4)
3.1.4. Synthesis of Rac-erythro-9,10-dihydroxystearic Acid (6)
3.1.5. Synthesis of Rac-erythro-methyl 9,10-dihydroxystearate (7)
3.1.6. Synthesis of Rac-erythro-methyl 9-((tert-butyldimethylsilyl)oxy)-10-hydroxyoctadecanoate and Rac-erythro-methyl 10-((tert-butyldimethylsilyl)oxy)-9-hydroxyoctadecanoate (8 and 9)
3.1.7. Synthesis of Rac-erythro-methyl 9,10-bis((tert-butyldimethylsilyl)oxy)octadecanoate (10)
3.1.8. Synthesis of Rac-erythro-9-((tert-butyldimethylsilyl)oxy)-10-hydroxyoctadecanoic acid and Rac-erythro-10-((tert-butyldimethylsilyl)oxy)-9-hydroxyoctadecanoic Acid (11 and 12)
3.1.9. Synthesis of Rac-erythro-9,10-bis((tert-butyldimethylsilyl)oxy)octadecanoic Acid (13)
3.2. Biology
3.2.1. Cell Culture and Treatments
3.2.2. MTT Assay
3.2.3. Cell Cycle Analysis
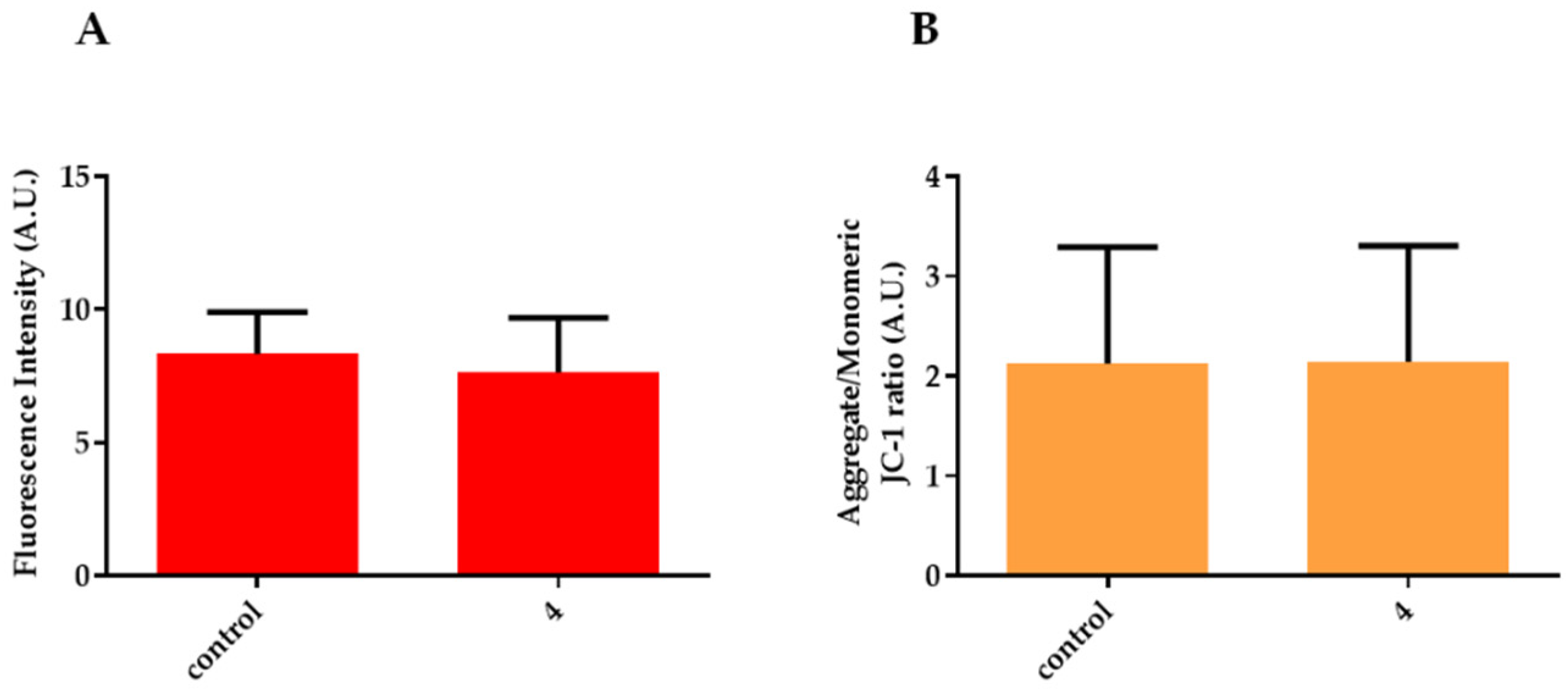
3.2.4. ROS Assay
3.2.5. Analysis of Mitochondrial Potential by JC-1 Staining
3.2.6. Cellular Content of Total Magnesium
3.2.7. Histone Post-Translational Modification
3.2.8. Protein Analysis: SDS-Page and Western Blot
3.2.9. Confocal Microscopy
3.2.10. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
3.2.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Farruggia, G.; Buontempo, F.; Boga, C.; Brusa, G.L.; Santucci, M.A.; Masotti, L. 9-Hydroxystearic Acid Upregulates p21WAF1 in HT29 Cancer Cells. Biochem. Biophys. Res. Commun. 2004, 314, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Boga, C.; Bertucci, C.; Fiori, J.; Tasco, G.; Casadio, R.; Masotti, L. Histone Deacetylase 1: A Target of 9-Hydroxystearic Acid in the Inhibition of Cell Growth in Human Colon Cancer. J. Lipid Res. 2005, 46, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Pagnotta, E.; Calonghi, N.; Boga, C.; Masotti, L. N-Methylformamide and 9-Hydroxystearic Acid: Two Anti-Proliferative and Differentiating Agents with Different Modes of Action in Colon Cancer Cells. Anti-Cancer Drugs 2006, 17, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Pagnotta, E.; Parolin, C.; Molinari, C.; Boga, C.; Dal Piaz, F.; Brusa, G.L.; Santucci, M.A.; Masotti, L. Modulation of Apoptotic Signalling by 9-Hydroxystearic Acid in Osteosarcoma Cells. Biochim. Biophys. Acta 2007, 1771, 139–146. [Google Scholar] [CrossRef]
- Parolin, C.; Calonghi, N.; Presta, E.; Boga, C.; Caruana, P.; Naldi, M.; Andrisano, V.; Masotti, L.; Sartor, G. Mechanism and Stereoselectivity of HDAC I Inhibition by (R)-9-Hydroxystearic Acid in Colon Cancer. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2012, 1821, 1334–1340. [Google Scholar] [CrossRef]
- Busi, A.; Aluigi, A.; Guerrini, A.; Boga, C.; Sartor, G.; Calonghi, N.; Sotgiu, G.; Posati, T.; Corticelli, F.; Fiori, J.; et al. Unprecedented Behavior of (9 R)-9-Hydroxystearic Acid-Loaded Keratin Nanoparticles on Cancer Cell Cycle. Mol. Pharm. 2019, 16, 931–942. [Google Scholar] [CrossRef]
- Micheletti, G.; Boga, C.; Telese, D.; Cassani, M.C.; Boanini, E.; Nitti, P.; Ballarin, B.; Ghirri, A.; Barucca, G.; Rinaldi, D. Magnetic Nanoparticles Coated with (R)-9-Acetoxystearic Acid for Biomedical Applications. ACS Omega 2020, 5, 12707–12715. [Google Scholar] [CrossRef]
- Brejchova, K.; Balas, L.; Paluchova, V.; Brezinova, M.; Durand, T.; Kuda, O. Understanding FAHFAs: From Structure to Metabolic Regulation. Prog. Lipid Res. 2020, 79, 101053. [Google Scholar] [CrossRef]
- Riecan, M.; Paluchova, V.; Lopes, M.; Brejchova, K.; Kuda, O. Branched and Linear Fatty Acid Esters of Hydroxy Fatty Acids (FAHFA) Relevant to Human Health. Pharmacol. Ther. 2022, 231, 107972. [Google Scholar] [CrossRef]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a Class of Endogenous Mammalian Lipids with Anti-Diabetic and Anti-Inflammatory Effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef]
- Nelson, A.T.; Kolar, M.J.; Chu, Q.; Syed, I.; Kahn, B.B.; Saghatelian, A.; Siegel, D. Stereochemistry of Endogenous Palmitic Acid Ester of 9-Hydroxystearic Acid and Relevance of Absolute Configuration to Regulation. J. Am. Chem. Soc. 2017, 139, 4943–4947. [Google Scholar] [CrossRef] [PubMed]
- Panayides, J.-L.; Riley, D.L.; Hasenmaile, F.; Van Otterlo, W.A.L. The Role of Silicon in Drug Discovery: A Review. RSC Med. Chem. 2024, 15, 3286–3344. [Google Scholar] [CrossRef] [PubMed]
- Showell, G.A.; Mills, J.S. Chemistry Challenges in Lead Optimization: Silicon Isosteres in Drug Discovery. Drug Discov. Today 2003, 8, 551–556. [Google Scholar] [CrossRef]
- Gabizon, A.A.; Tzemach, D.; Horowitz, A.T.; Shmeeda, H.; Yeh, J.; Zalipsky, S. Reduced Toxicity and Superior Therapeutic Activity of a Mitomycin C Lipid-Based Prodrug Incorporated in Pegylated Liposomes. Clin. Cancer Res. 2006, 12, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Ohkouchi, K.; Imoto, H.; Takakura, Y.; Hashida, M.; Sezaki, H. Disposition of Anticancer Drugs after Bolus Arterial Administration in a Tissue-Isolated Tumor Perfusion System. Cancer Res. 1990, 50, 1640–1644. [Google Scholar]
- Todd, D. Experimental Organic Chemistry; Prentice-Hall: Englewood Cliffs, NJ, USA, 1979; ISBN 978-0-13-294660-5. [Google Scholar]
- Traynard, J. Sur La Preparation de L’acide Dioxystearique. In Bulletin de la Societe Chimique de France; Scientifiques Medicales Elsevier: Paris, France, 1952; pp. 323–354. ISBN 0037-8968. [Google Scholar]
- Gensler, W.J.; Schlein, H.N. Configuration of 9,10-Dihydroxystearic Acid. J. Am. Chem. Soc. 1956, 78, 169–172. [Google Scholar] [CrossRef]
- Bender, A.D.; Berkoff, C.E.; Groves, W.G.; Sofranko, L.M.; Wellman, G.R.; Liu, J.-H.; Begosh, P.P.; Horodniak, J.W. Synthesis and Biological Properties of Some Novel Heterocyclic Homoprostanoids. J. Med. Chem. 1975, 18, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Minnikin, D.E.; Patel, D.V. Butyldimethylsilyl Ethers of Iodine-Catalysed Solvolysis Products of Long-Chain Epoxides. Chem. Phys. Lipids 1979, 23, 173–178. [Google Scholar] [CrossRef]
- Calonghi, N.; Boga, C.; Nitti, P.; Telese, D.; Bordoni, S.; Farruggia, G.; Asaro, F.; Grandi, M.; Zalambani, C.; Micheletti, G. Effects of Regioisomerism on the Antiproliferative Activity of Hydroxystearic Acids on Human Cancer Cell Lines. Molecules 2022, 27, 2396. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS Homeostasis and Metabolism: A Dangerous Liason in Cancer Cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH Oxidases and Cancer. Clin. Sci. 2015, 128, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Stanicka, J.; Russell, E.G.; Woolley, J.F.; Cotter, T.G. NADPH Oxidase-Generated Hydrogen Peroxide Induces DNA Damage in Mutant FLT3-Expressing Leukemia Cells. J. Biol. Chem. 2015, 290, 9348–9361. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Y.; Zhang, J.; Hu, C.; Jiang, J.; Li, Y.; Peng, Z. ROS-Induced Lipid Peroxidation Modulates Cell Death Outcome: Mechanisms behind Apoptosis, Autophagy, and Ferroptosis. Arch. Toxicol. 2023, 97, 1439–1451. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Li, K.; Fan, C.; Chen, J.; Xu, X.; Lu, C.; Shao, H.; Xi, Y. Role of Oxidative Stress-induced Ferroptosis in Cancer Therapy. J. Cell. Mol. Med. 2024, 28, e18399. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in Cancer Treatment: Activity, Chemoresistance and Its Overcoming. Drug Resist. Updates 2021, 54, 100742. [Google Scholar] [CrossRef]
- Azmanova, M.; Pitto-Barry, A. Oxidative Stress in Cancer Therapy: Friend or Enemy? ChemBioChem 2022, 23, e202100641. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor Tyrosine Kinases (RTKs) in Breast Cancer: Signaling, Therapeutic Implications and Challenges. Mol. Cancer 2018, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Ségaliny, A.I.; Tellez-Gabriel, M.; Heymann, M.-F.; Heymann, D. Receptor Tyrosine Kinases: Characterisation, Mechanism of Action and Therapeutic Interests for Bone Cancers. J. Bone Oncol. 2015, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, U.; Duda-Chodak, A. Magnesium: Its Role in Nutrition and Carcinogenesis. Rocz. Panstw. Zakl. Hig. 2013, 64, 165–171. [Google Scholar]
- Van Laecke, S. Hypomagnesemia and Hypermagnesemia. Acta Clin. Belg. 2019, 74, 41–47. [Google Scholar] [CrossRef]
- Wolf, F.; Trapani, V.; Simonacci, M.; Boninsegna, A.; Mazur, A.; Maier, J. Magnesium Deficiency Affects Mammary Epithelial Cell Proliferation: Involvement of Oxidative Stress. Nutr. Cancer 2009, 61, 131–136. [Google Scholar] [CrossRef]
- Færgeman, N.J.; Knudsen, J. Role of Long-Chain Fatty Acyl-CoA Esters in the Regulation of Metabolism and in Cell Signalling. Biochem. J. 1997, 323, 1–12. [Google Scholar] [CrossRef]
- Carratù, L.; Franceschelli, S.; Pardini, C.L.; Kobayashi, G.S.; Horvath, I.; Vigh, L.; Maresca, B. Membrane Lipid Perturbation Modifies the Set Point of the Temperature of Heat Shock Response in Yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 3870–3875. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of Fatty Acid Synthase (FAS) as a Predictor of Recurrence in Stage I Breast Carcinoma Patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Visca, P.; Alò, P.L.; Del Nonno, F.; Botti, C.; Trombetta, G.; Marandino, F.; Filippi, S.; Di Tondo, U.; Donnorso, R.P. Immunohistochemical Expression of Fatty Acid Synthase, Apoptotic-Regulating Genes, Proliferating Factors, and Ras Protein Product in Colorectal Adenomas, Carcinomas, and Adjacent Nonneoplastic Mucosa. Clin. Cancer Res. 1999, 5, 4111–4118. [Google Scholar]
- Epstein, J.I.; Carmichael, M.; Partin, A.W. OA-519 (Fatty Acid Synthase) as an Independent Predictor of Pathologic Stage in Adenocarcinoma of the Prostate. Urology 1995, 45, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Visca, P.; Sebastiani, V.; Pizer, E.S.; Botti, C.; De Carli, P.; Filippi, S.; Monaco, S.; Alo, P.L. Immunohistochemical Expression and Prognostic Significance of FAS and GLUT1 in Bladder Carcinoma. Anticancer Res. 2003, 23, 335–339. [Google Scholar] [PubMed]
- Gansler, T.S.; Hardman, W.; Hunt, D.A.; Schaffel, S.; Hennigar, R.A. Increased Expression of Fatty Acid Synthase (OA-519) in Ovarian Neoplasms Predicts Shorter Survival. Hum. Pathol. 1997, 28, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, T.; Nashimoto, A.; Honma, K.; Suzuki, T. Fatty Acid Synthase Is Highly Expressed in Carcinoma, Adenoma and in Regenerative Epithelium and Intestinal Metaplasia of the Stomach. Histopathology 2002, 40, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Innocenzi, D.; Alò, P.L.; Balzani, A.; Sebastiani, V.; Silipo, V.; La Torre, G.; Ricciardi, G.; Bosman, C.; Calvieri, S. Fatty Acid Synthase Expression in Melanoma. J. Cutan. Pathol. 2003, 30, 23–28. [Google Scholar] [CrossRef]
- Alo, P.L.; Amini, M.; Piro, F.; Pizzuti, L.; Sebastiani, V.; Botti, C.; Murari, R.; Zotti, G.; Di Tondo, U. Immunohistochemical Expression and Prognostic Significance of Fatty Acid Synthase in Pancreatic Carcinoma. Anticancer Res. 2007, 27, 2523–2527. [Google Scholar]
- Silva, S.D.; Agostini, M.; Nishimoto, I.N.; Coletta, R.D.; Alves, F.A.; Lopes, M.A.; Kowalski, L.P.; Graner, E. Expression of Fatty Acid Synthase, ErbB2 and Ki-67 in Head and Neck Squamous Cell Carcinoma. A Clinicopathological Study. Oral Oncol. 2004, 40, 688–696. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Miyazaki, M.; Dobrzyn, A. Regulation of stearoyl-CoA Desaturase Expression. Lipids 2004, 39, 1061–1065. [Google Scholar] [CrossRef]
- Igal, R.A. Stearoyl CoA Desaturase-1: New Insights into a Central Regulator of Cancer Metabolism. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2016, 1861, 1865–1880. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.-X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.-P.; Fargnoli, J.; Henley, B.J.; Yang, W.-P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer Cell Dependence on Unsaturated Fatty Acids Implicates Stearoyl-CoA Desaturase as a Target for Cancer Therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wan, J.; Qu, X.; Xia, K.; Wang, F.; Zhang, Z.; Yang, M.; Wu, X.; Gao, R.; Yuan, X.; et al. Nodal Promotes Colorectal Cancer Survival and Metastasis through Regulating SCD1-Mediated Ferroptosis Resistance. Cell Death Dis. 2023, 14, 229. [Google Scholar] [CrossRef] [PubMed]
- Kurdistani, S.K.; Tavazoie, S.; Grunstein, M. Mapping Global Histone Acetylation Patterns to Gene Expression. Cell 2004, 117, 721–733. [Google Scholar] [CrossRef]
- Horn, P.J.; Peterson, C.L. Chromatin Higher Order Folding—Wrapping up Transcription. Science 2002, 297, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Boanini, E.; Torricelli, P.; Boga, C.; Micheletti, G.; Cassani, M.C.; Fini, M.; Bigi, A. (9R)-9-Hydroxystearate-Functionalized Hydroxyapatite as Antiproliferative and Cytotoxic Agent toward Osteosarcoma Cells. Langmuir 2016, 32, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Julien-David, D.; Geoffroy, P.; Marchioni, E.; Raul, F.; Aoudé-Werner, D.; Miesch, M. Synthesis of Highly Pure Oxyphytosterols and (Oxy)Phytosterol Esters. Steroids 2008, 73, 1098–1109. [Google Scholar] [CrossRef]
- Dorado, V.; Herrerías, C.I.; Fraile, J.M. Catalytic Hydrolysis of Epoxyfatty Esters with Solid Sulfonic Acids. Mol. Catal. 2023, 547, 113282. [Google Scholar] [CrossRef]
- Micheletti, G.; Calonghi, N.; Farruggia, G.; Strocchi, E.; Palmacci, V.; Telese, D.; Bordoni, S.; Frisco, G.; Boga, C. Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells. Molecules 2020, 25, 404. [Google Scholar] [CrossRef]
- Sargenti, A.; Farruggia, G.; Zaccheroni, N.; Marraccini, C.; Sgarzi, M.; Cappadone, C.; Malucelli, E.; Procopio, A.; Prodi, L.; Lombardo, M.; et al. Synthesis of a Highly Mg2+-Selective Fluorescent Probe and Its Application to Quantifying and Imaging Total Intracellular Magnesium. Nat. Protoc. 2017, 12, 461–471. [Google Scholar] [CrossRef]
- Albadri, S.; Naso, F.; Thauvin, M.; Gauron, C.; Parolin, C.; Duroure, K.; Vougny, J.; Fiori, J.; Boga, C.; Vriz, S.; et al. Redox Signaling via Lipid Peroxidation Regulates Retinal Progenitor Cell Differentiation. Dev. Cell 2019, 50, 73–89.e6. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HT29 | MCF7 | HeLa | U2OS | J6 |
|---|---|---|---|---|---|
| 8 + 9 | na | na | na | na | na |
| 10 | na | na | na | na | na |
| 11 + 12 | 26.11 ± 0.43 | na | 132.5 ± 0.16 | 50.40 ± 0.30 | na |
| 13 | 25.32 ± 0.27 | na | 131.8 ± 0.21 | 41.95 ± 0.29 | na |
| 3 | 84.76 ± 0.10 | 134.0 ± 0.27 | 133.7 ± 0.32 | 29.33 ± 0.23 | 151.6 ± 0.31 |
| 4 | 20.31 ± 0.24 | 211.6 ± 0.23 | 166.1 ± 0.33 | 36.81 ± 0.18 | 214.1 ± 0.26 |
| 4 | IC50 nM | S.I. |
|---|---|---|
| HT29 | 20.88 ± 0.21 | 2.96 |
| HCT116 | 21.49 ± 0.16 | 2.88 |
| Caco-2 | 23.39 ± 0.12 | 2.65 |
| I407 | 61.91 ± 0.26 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalambani, C.; Anconelli, L.; Calonghi, N.; Telese, D.; Micheletti, G.; Boga, C.; Farruggia, G.; Pagnotta, E. New Compounds with Enhanced Biological Activity Through the Strategic Introduction of Silylated Groups into Hydroxystearic Acids. Molecules 2025, 30, 440. https://doi.org/10.3390/molecules30030440
Zalambani C, Anconelli L, Calonghi N, Telese D, Micheletti G, Boga C, Farruggia G, Pagnotta E. New Compounds with Enhanced Biological Activity Through the Strategic Introduction of Silylated Groups into Hydroxystearic Acids. Molecules. 2025; 30(3):440. https://doi.org/10.3390/molecules30030440
Chicago/Turabian StyleZalambani, Chiara, Lorenzo Anconelli, Natalia Calonghi, Dario Telese, Gabriele Micheletti, Carla Boga, Giovanna Farruggia, and Eleonora Pagnotta. 2025. "New Compounds with Enhanced Biological Activity Through the Strategic Introduction of Silylated Groups into Hydroxystearic Acids" Molecules 30, no. 3: 440. https://doi.org/10.3390/molecules30030440
APA StyleZalambani, C., Anconelli, L., Calonghi, N., Telese, D., Micheletti, G., Boga, C., Farruggia, G., & Pagnotta, E. (2025). New Compounds with Enhanced Biological Activity Through the Strategic Introduction of Silylated Groups into Hydroxystearic Acids. Molecules, 30(3), 440. https://doi.org/10.3390/molecules30030440