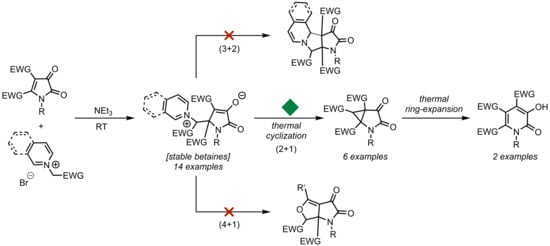
3.2. Synthesis of Betaines 3
General Procedure A: To a round-bottom flask containing a magnetic stir bar and pyrrole-2,3-dione 1 (1 equiv.), dry acetonitrile (10 mL per 1 mmol of 1) is added, followed by salt 2 (1.1 equiv.) and triethylamine (1.1 equiv.). The resulting mixture is stirred at room temperature for 1–2 h. The formed precipitate is filtered and washed with acetonitrile.
(S*)-4-Benzoyl-5-((R*)-2-ethoxy-2-oxo-1-(pyridin-1-ium-1-yl)ethyl)-5-(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3aa). Synthesized according to General Procedure A from pyrrole-2,3-dione 1a (1 mmol scale) and salt 2a. A pale-yellow/off-white powder, 376 mg (75%), m.p. 166–168 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.95–8.84 (m, 2H), 8.64–8.54 (m, 1H), 8.05 (t, J = 7.1 Hz, 2H), 7.50 (dd, J = 8.7, 6.7 Hz, 2H), 7.43–7.33 (m, 3H), 7.33–7.24 (m, 1H), 7.24–7.11 (m, 5H), 3.82 (s, 3H), 3.65 (dq, J = 10.7, 7.1 Hz, 1H), 3.05 (dq, J = 10.7, 7.1 Hz, 1H), 0.97 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 184.6, 169.4, 169.1, 167.4, 164.9, 147.0, 146.5 (2C), 140.6, 135.7, 129.1, 128.7 (2C), 127.9 (2C), 127.5, 127.3 (2C), 126.5 (4C), 106.1, 71.6, 70.8, 62.1, 52.8, 13.1. IR (mineral oil), cm−1: 1741, 1729, 1713, 1654, 1632, 1582. Anal. Calcd for C28H24N2O7: C 67.19; H 4.83; N 5.60. Found: C 67.46; H 4.78; N 5.81.
(S*)-4-Benzoyl-1-(4-chlorophenyl)-5-((R*)-2-ethoxy-2-oxo-1-(pyridin-1-ium-1-yl)ethyl)-5-(methoxycarbonyl)-2-oxo-2,5-dihydro-1H-pyrrol-3-olate (3ba). Synthesized according to General Procedure A from pyrrole-2,3-dione 1b (0.5 mmol scale) and salt 2a. An off-white powder, 192 mg (72%), m.p. 167–175 °C (dec.). 1H NMR (400 MHz, DMSO-d6), mixture of diastereomers, d.r. ∼3.8:1 (A:B): δ 9.04 (d, J = 5.7 Hz, 0.42H, B), 8.89 (d, J = 5.6 Hz, 1.58H, A), 8.59 (t, J = 7.8 Hz, 1H), 8.08–8.00 (m, 2H), 7.82 (d, J = 7.0 Hz, 0.42H, B), 7.57 (d, J = 8.8 Hz, 1.58H, A), 7.46–7.14 (m, 7.37H, A + B), 6.79 (d, J = 8.8 Hz, 0.42H, B), 6.52 (s, 0.21H, B), 4.04–3.88 (m, 0.42H, B), 3.81 (s, 2.37H, A), 3.80 (s, 0.63H, B), 3.72 (dq, J = 10.7, 7.1 Hz, 0.79H, A), 3.19 (dq, J = 10.8, 7.2 Hz, 0.79H, A), 1.10 (t, J = 7.1 Hz, 0.63H, B), 1.01 (t, J = 7.1 Hz, 2.37H, A). 13C NMR (101 MHz, DMSO-d6), A (major): δ 184.6, 169.4, 169.1, 167.0, 164.9, 147.0, 146.6 (2C), 140.5, 134.7, 132.1, 129.2, 129.1 (2C), 128.7 (2C), 127.9 (2C), 126.5 (4C), 106.0, 71.5, 70.7, 62.2, 53.0, 13.0. IR (mineral oil), cm−1:1758, 1737, 1714, 1650, 1627, 1580, 1529. Anal. Calcd for C28H23ClN2O7: C 62.87; H 4.33; N 5.24. Found: C 63.10; H 4.27; N 5.35.
(S*)-4-(4-Bromobenzoyl)-5-((R*)-2-ethoxy-2-oxo-1-(pyridin-1-ium-1-yl)ethyl)-5-(methoxycarbonyl)-2-oxo-1-(p-tolyl)-2,5-dihydro-1H-pyrrol-3-olate (3ca). Synthesized according to General Procedure A from pyrrole-2,3-dione 1c (1 mmol scale) and salt 2a. A pale-yellow powder, 408 mg (69%), m.p. 188–189 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.87 (d, J = 5.7 Hz, 2H), 8.59 (tt, J = 7.8, 1.3 Hz, 1H), 8.04 (t, J = 7.1 Hz, 2H), 7.43–7.34 (m, 2H), 7.32–7.25 (m, 2H), 7.27–7.18 (m, 2H), 7.18–7.06 (m, 3H), 3.79 (s, 3H), 3.68 (dq, J = 10.7, 7.1 Hz, 1H), 3.11 (dq, J = 10.7, 7.2 Hz, 1H), 2.35 (s, 3H), 0.98 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 182.9, 169.4, 168.9, 167.9, 164.8, 147.0, 146.4 (2C), 139.7, 137.2, 133.0, 130.0 (2C), 129.6 (2C), 129.2 (2C), 127.2 (2C), 126.5 (2C), 122.5, 105.9, 71.4, 70.7, 62.2, 52.8, 20.5, 13.0. IR (mineral oil), cm−1: 1743, 1728, 1712, 1648, 1632, 1571, 1511. Anal. Calcd for C29H25BrN2O7: C 58.70; H 4.25; N 4.72. Found: C 59.08; H 4.27; N 5.01.
(S*)-4-Cinnamoyl-5-((R*)-2-ethoxy-2-oxo-1-(pyridin-1-ium-1-yl)ethyl)-5-(ethoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3da). Synthesized according to General Procedure A from pyrrole-2,3-dione 1d (1 mmol scale) and salt 2a. A light-yellow powder, 390 mg (72%), m.p. 186–188 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.86 (d, J = 5.3 Hz, 2H), 8.57 (t, J = 7.8 Hz, 1H), 8.05 (t, J = 7.3 Hz, 2H), 8.00 (d, J = 16.0 Hz, 1H), 7.53–7.28 (m, 11H), 7.18 (d, J = 15.9 Hz, 1H), 4.33–4.20 (m, 2H), 3.64 (dq, J = 10.7, 7.1 Hz, 1H), 3.07 (dq, J = 10.8, 7.1 Hz, 1H), 1.20 (t, J = 7.1 Hz, 3H), 0.97 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 178.4, 169.3, 168.8, 168.3, 164.9, 147.0, 146.3 (2C), 135.8, 135.8, 135.7, 128.8, 128.7 (2C), 128.6 (2C), 127.5, 127.3 (2C), 127.2 (2C), 127.1, 126.4 (2C), 108.1, 71.5, 70.7, 62.1, 61.9, 13.7, 13.0. IR (mineral oil), cm−1: 1751, 1731, 1715, 1634. Anal. Calcd for C31H28N2O7: C 68.88; H 5.22; N 5.18. Found: C 69.19; H 5.34; N 5.10.
(S*)-5-((R*)-2-Ethoxy-2-oxo-1-(pyridin-1-ium-1-yl)ethyl)-4,5-bis(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3ea). Synthesized according to General Procedure A from pyrrole-2,3-dione 1e (1 mmol scale) and salt 2a. An off-white powder, 265 mg (58%), m.p. 152–154 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.88–8.82 (m, 2H), 8.65 (tt, J = 7.8, 1.3 Hz, 1H), 8.13 (t, J = 7.4 Hz, 2H), 7.49–7.43 (m, 2H), 7.40–7.32 (m, 3H), 7.11 (s, 1H), 3.79 (s, 3H), 3.60 (dq, J = 10.8, 7.1 Hz, 1H), 3.31 (s, 3H), 3.04 (dq, J = 10.8, 7.1 Hz, 1H), 0.95 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 169.2, 169.1, 166.3, 164.8, 164.2, 147.2, 146.2 (2C), 135.8, 128.6 (2C), 127.3, 127.2 (2C), 126.7 (2C), 92.4, 71.0, 70.7, 62.1, 52.9, 49.0, 13.0. IR (mineral oil), cm−1: 1743, 1712, 1655, 1629, 1607, 1589. Anal. Calcd for C23H22N2O8: C 60.79; H 4.88; N 6.16. Found: C 60.91; H 4.92; N 6.10.
(S*)-5-((R*)-2-Ethoxy-2-oxo-1-(pyridin-1-ium-1-yl)ethyl)-1-(4-methoxyphenyl)-4,5-bis(4-methylbenzoyl)-2-oxo-2,5-dihydro-1H-pyrrol-3-olate (3fa). Synthesized according to General Procedure A from pyrrole-2,3-dione 1f (1 mmol scale) and salt 2a. A light-yellow powder, 227 mg (37%), m.p. 146–148 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.94 (d, J = 5.3 Hz, 2H), 8.55 (tt, J = 7.8, 1.3 Hz, 1H), 8.05–7.97 (m, 2H), 7.61 (d, J = 8.3 Hz, 2H), 7.25–7.13 (m, 5H), 7.05–6.92 (m, 6H), 3.83–3.76 (m, 1H), 3.75 (s, 3H), 3.38 (dq, J = 10.8, 7.1 Hz, 1H), 2.29 (s, 3H), 2.24 (s, 3H), 1.03 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 192.2, 184.2, 169.0, 168.0, 165.1, 157.9, 146.7, 146.5 (2C), 146.2, 142.2, 138.7, 137.7, 133.9, 128.7 (2C), 127.9 (2C), 127.9 (2C), 127.7 (2C), 127.1 (2C), 126.3 (2C), 113.8 (2C), 105.2, 75.9, 71.2, 62.3, 55.3, 20.8 (2C), 13.1. IR (mineral oil), cm−1: 1752, 1737, 1710, 1680, 1647, 1605, 1571. Anal. Calcd for C36H32N2O7: C 71.51; H 5.33; N 4.63. Found: C 71.66; H 5.54; N 4.47.
(S*)-4,5-Dibenzoyl-5-((R*)-2-ethoxy-2-oxo-1-(pyridin-1-ium-1-yl)ethyl)-2-oxo-1-(p-tolyl)-2,5-dihydro-1H-pyrrol-3-olate (3ga). Synthesized according to General Procedure A from pyrrole-2,3-dione 1g (0.94 mmol scale) and salt 2a. An off-white powder, 247 mg (48%), m.p. 151–153 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.97 (d, J = 5.2 Hz, 2H), 8.57 (tt, J = 7.8, 1.3 Hz, 1H), 8.07–8.02 (m, 2H), 7.69–7.62 (m, 2H), 7.52–7.46 (m, 1H), 7.38–7.32 (m, 2H), 7.27–7.19 (m, 6H), 7.17–7.09 (m, 2H), 7.04–6.98 (m, 2H), 3.77 (dq, J = 10.8, 7.1 Hz, 1H), 3.35 (dq, J = 10.8, 7.1 Hz, 1H), 2.30 (s, 3H), 1.02 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 192.8, 184.4, 168.8, 168.1, 165.0, 146.8, 146.5 (2C), 140.6, 136.7, 136.6, 132.7, 131.9, 129.1 (2C), 129.0, 128.2 (2C), 127.7 (2C), 127.6 (2C), 126.5 (2C), 126.4 (2C), 126.0 (2C), 104.9, 75.6, 71.1, 62.3, 20.4, 13.1. IR (mineral oil), cm−1: 1747, 1719, 1681, 1647, 1597, 1581. Anal. Calcd for C34H28N2O6: C 72.85; H 5.03; N 5.00. Found: C 73.12; H 5.09; N 4.96.
(S*)-4-Benzoyl-5-(methoxycarbonyl)-2-oxo-1-phenyl-5-((R*)-(3-phenyl-1,2,4-oxadiazol-5-yl)(pyridin-1-ium-1-yl)methyl)-2,5-dihydro-1H-pyrrol-3-olate (3ab). Synthesized according to General Procedure A from pyrrole-2,3-dione 1a (1 mmol scale) and salt 2b. An off-white powder, 444 mg (78%), m.p. 173–175 °C (dec.). 1H NMR (400 MHz, DMSO-d6), mixture of diastereomers, d.r. ∼33:1 (A:B): δ 9.26 (d, J = 6.5 Hz, 0.06H, B), 9.17 (d, J = 5.8 Hz, 1.94H, A), 8.65 (t, J = 7.8 Hz, 0.97H, A), 8.61 (t, J = 7.7 Hz, 0.03H, B), 8.12 (t, J = 7.0 Hz, 1.94H, A), 8.02 (t, J = 7.2 Hz, 0.06H, B), 7.97 (s, 1H), 7.74 (d, J = 6.8 Hz, 2H), 7.64–7.50 (m, 3H), 7.39–7.30 (m, 3H), 7.28–7.18 (m, 4H), 7.13–7.02 (m, 2.94H), 6.88 (d, J = 7.8 Hz, 0.06H, B), 3.86 (s, 2.91H, A), 3.83 (s, 0.09H, B). 13C NMR (101 MHz, DMSO-d6), A (major): δ 184.8, 172.2, 170.2, 168.7, 167.4, 167.0, 147.7, 146.4 (2C), 140.1, 135.3, 131.6, 129.4, 129.0 (2C), 128.7 (2C), 128.1 (2C), 127.1 (2C), 127.0 (2C), 126.8, 126.7 (2C), 125.5 (2C), 125.1, 105.5, 72.2, 65.8, 53.1.IR (mineral oil), cm−1: 1743, 1732, 1651, 1629, 1582. Anal. Calcd for C33H24N4O6: C 69.22; H 4.23; N 9.79. Found: C 69.51; H 4.45; N 9.71.
(S*)-4-Benzoyl-5-((R*)-cyano(pyridin-1-ium-1-yl)methyl)-5-(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3ac). Synthesized according to General Procedure A from pyrrole-2,3-dione 1a (1 mmol scale) and salt 2c. A pale-beige powder, 370 mg (82%), m.p. 180–182 °C (dec.). 1H NMR (400 MHz, DMSO-d6), mixture of diastereomers, d.r. ∼50:1, A (major): δ 8.91 (d, J = 5.5 Hz, 2H), 8.67 (tt, J = 7.9, 1.3 Hz, 1H), 8.12 (dd, J = 7.8, 6.7 Hz, 2H), 7.61–7.54 (m, 4H), 7.51–7.45 (m, 1H), 7.42–7.28 (m, 5H), 7.27 (s, 1H), 3.77 (s, 3H). 13C NMR (101 MHz, DMSO-d6), A (major): δ 184.9, 170.0, 168.3, 166.9, 148.3, 145.1 (2C), 139.5, 134.8, 129.8, 129.5 (2C), 128.5 (2C), 128.3, 127.7 (2C), 127.2 (2C), 126.8 (2C), 113.9, 103.6, 71.2, 63.2, 53.1. IR (mineral oil), cm−1:1734, 1714, 1653, 1629, 1595, 1581 (CN-peak was not observed even in KBr-tablet). Anal. Calcd for C26H19N3O5: C 68.87; H 4.22; N 9.27. Found: C 69.16; H 4.13; N 9.26.
(5-(Cyano(pyridin-1-ium-1-yl)methyl)-4,5-bis(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3ec). Synthesized according to General Procedure A from pyrrol-2,3-dione 1e (0.4 mmol scale) and salt 2c. A pale-beige powder, 110 mg (68%), m.p. 176–178 °C (dec.). 1H NMR (400 MHz, DMSO-d6), mixture of diastereomers, d.r. ∼9:1 (A:B): δ 9.08 (d, J = 5.5 Hz, 1.8H, A), 8.87 (d, J = 5.5 Hz, 0.2H, B), 8.73 (tt, J = 7.9, 1.3 Hz, 0.1H, B), 8.64 (tt, J = 7.8, 1.3 Hz, 0.9H, A), 8.20 (dd, J = 7.8, 6.7 Hz, 0.2H, B), 8.15–8.06 (m, 1.8H, A), 7.58–7.53 (m, 0.2H, B), 7.52–7.39 (m, 2.8H, A + B), 7.28–7.25 (m, 0.2H, B), 7.24–7.17 (m, 1.8H, A), 7.08 (s, 0.1H, B), 6.64 (s, 0.9H, A), 3.78 (s, 2.7H, A), 3.74 (s, 0.3H, B), 3.33 (s, 0.3H, B), 3.14 (s, 2.7H, A). 13C spectral signals are listed together for both diastereomers due to rapid epimerization in solution (see NMR 1H spectrum recorded 30 min after dissolution in the SI, d.r. ∼1:1.5 (A:B)). 13C NMR (101 MHz, DMSO-d6), A + B: δ 170.2, 170.1, 168.0, 167.9, 167.0, 166.5, 164.9, 164.6, 148.3, 147.6, 145.8, 145.3, 135.4, 134.8, 129.5, 129.4, 128.4, 128.1, 127.6, 127.6, 127.1, 126.9, 113.9, 113.1, 91.3, 89.9, 71.1, 70.9, 65.0, 64.0, 53.2, 53.0, 49.5, 49.1. IR (mineral oil), cm−1: 1748, 1723, 1694, 1679, 1634 (CN-peak was not observed even in KBr-tablet). Anal. Calcd for C21H17N3O6: C 61.92; H 4.21; N 10.31. Found: C 61.70; H 4.05; N 9.98.
(S*)-4-benzoyl-5-((R*)-1-(3-carbamoylpyridin-1-ium-1-yl)-2-ethoxy-2-oxoethyl)-5-(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3ad). Synthesized according to General Procedure A from pyrrole-2,3-dione 1a (0.5 mmol scale) and salt 2d. An off-white powder, 197 mg (72%), m.p. 148–150 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 9.34 (s, 1H), 9.03 (d, J = 6.3 Hz, 1H), 8.97 (d, J = 8.2 Hz, 1H), 8.43 (s, 1H), 8.18 (dd, J = 8.1, 6.3 Hz, 1H), 8.04 (s, 1H), 7.54–7.47 (m, 2H), 7.42–7.34 (m, 3H), 7.31–7.26 (m, 1H), 7.24–7.15 (m, 5H), 3.79 (s, 3H), 3.71 (dq, J = 10.8, 7.1 Hz, 1H), 3.26–3.18 (m, 1H), 1.01 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6), A (major): δ 184.6, 169.4, 168.8, 167.5, 164.6, 162.1, 144.4, 140.6, 135.6, 131.7, 129.1, 128.8 (2C), 127.9 (2C), 127.5, 126.8 (2C), 126.6 (2C), 126.2, 105.6, 71.4, 71.3, 62.3, 52.9, 13.1. IR (mineral oil), cm−1: 3404, 3302, 1754, 1746, 1710, 1688, 1645, 1634, 1581. Anal. Calcd for C29H25N3O8: C 64.08; H 4.64; N 7.73. Found: C 64.29; H 4.75; N 7.63.
(S*)-4-benzoyl-5-((R*)-1-(4-benzoylpyridin-1-ium-1-yl)-2-ethoxy-2-oxoethyl)-5-(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3ae). Synthesized according to General Procedure A from pyrrole-2,3-dione 1a (0.5 mmol scale) and salt 2e. A beige powder, 253 mg (84%), m.p. 145–148 °C (dec.). 1H NMR (400 MHz, DMSO-d6), mixture of diastereomers, d.r. ∼30:1, A (major): δ 9.18 (d, J = 7.1 Hz, 2H), 8.21 (d, J = 6.9 Hz, 2H), 7.76 (tt, J = 7.2, 1.4 Hz, 1H), 7.61–7.47 (m, 6H), 7.42–7.31 (m, 6H), 7.28–7.20 (m, 3H), 3.80 (s, 3H), 3.74 (dq, J = 10.7, 7.1 Hz, 1H), 3.25–3.17 (m, 1H), 0.99 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6), A (major): δ 191.5, 184.6, 169.7, 169.0, 167.7, 164.6, 152.8, 147.6 (2C), 140.2, 135.7, 134.6, 133.8, 129.9 (2C), 129.4, 128.9 (2C), 128.8 (2C), 128.2 (2C), 127.4, 126.9 (2C), 126.7 (2C), 125.0 (2C), 106.0, 71.3, 71.1, 62.3, 52.9, 13.1. IR (mineral oil), cm−1: 1747, 1734, 1701, 1675, 1641, 1595, 1582, 1513. Anal. Calcd for C35H28N2O8: C 69.53; H 4.67; N 4.63. Found: C 69.20; H 4.79; N 4.61.
(S*)-4-benzoyl-5-((R*)-2-ethoxy-1-(3-methylpyridin-1-ium-1-yl)-2-oxoethyl)-5-(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3af). Synthesized according to General Procedure A from pyrrole-2,3-dione 1a (0.5 mmol scale) and salt 2f. An off-white powder, 98 mg (38%), m.p. 172–174 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.78 (d, J = 6.3 Hz, 1H), 8.70 (s, 1H), 8.44–8.41 (m, 1H), 8.02–7.97 (m, 1H), 7.55–7.45 (m, 2H), 7.42–7.18 (m, 8H), 7.01 (s, 1H), 3.79 (s, 3H), 3.72 (dq, J = 10.7, 7.1 Hz, 1H), 3.26–3.18 (m, 1H), 2.24 (s, 3H), 0.99 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6), A (major): δ 184.5, 169.6, 169.1, 167.3, 164.8, 147.2, 145.9, 143.3, 140.4, 136.7, 135.7, 129.3, 128.8 (2C), 128.0 (2C), 127.4, 126.9 (2C), 126.6 (2C), 126.1, 105.9, 71.4, 70.8, 62.2, 52.7, 17.5, 13.1. IR (mineral oil), cm−1: 1740, 1732, 1725, 1631, 1584, 1534, 1518. Anal. Calcd for C29H26N2O7: C 67.70; H 5.09; N 5.44. Found: C 67.82; H 4.95; N 5.49.
(S*)-4-benzoyl-5-((R*)-2-ethoxy-1-(isoquinolin-2-ium-2-yl)-2-oxoethyl)-5-(methoxycarbonyl)-2-oxo-1-phenyl-2,5-dihydro-1H-pyrrol-3-olate (3ag). Synthesized according to General Procedure A from pyrrole-2,3-dione 1a (0.5 mmol scale) and salt 2g. A pale-yellow powder, 198 mg (72%), m.p. 175–177 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 9.86 (s, 1H), 8.66 (d, J = 7.6 Hz, 1H), 8.46 (d, J = 7.0 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 8.30 (d, J = 8.3 Hz, 1H), 8.22 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H), 7.96–7.91 (m, 1H), 7.56–7.49 (m, 2H), 7.44–7.35 (m, 3H), 7.24–7.11 (m, 2H), 7.10–6.97 (m, 4H), 3.80 (s, 3H), 3.78–3.67 (m, 1H), 0.99 (t, J = 7.1 Hz, 3H). The signal of one diastereotopic atom of the CH2 group is in the water region in 1H NMR. 13C NMR (101 MHz, DMSO-d6): δ 169.8, 169.1, 137.7, 135.8, 130.8, 129.1, 128.8, 127.8, 127.4, 127.0, 126.3, 125.8, 123.8, 71.5, 70.9, 62.2, 52.8, 13.1. Some of the carbon atom signals are not visible due to the very low solubility of the compound. IR (mineral oil), cm−1: 1744, 1732, 1714, 1646. Anal. Calcd for C32H26N2O7: C 69.81; H 4.76; N 5.09. Found: C 70.17; H 4.81; N 4.86.
3.3. Synthesis of Cyclopropanes 4
General Procedure B: To a V-vial containing a magnetic stir bar and betaine 3 (1 equiv.), dry chlorobenzene (5 mL per 1 mmol of 3) is added. The vial is connected to a Vigreux column (serving as a condenser) and placed in a preheated-to-140 °C metal heating block. The resulting mixture is stirred at 140 °C until complete dissolution of the starting betaine 3 (5–50 min depending on the betaine used). The solution is cooled, and cyclopropanes 4 are isolated by flash-column chromatography (loaded directly on a column in PhCl) or crystallization.
6-Ethyl 1-methyl (1
R*,5
S*,6
R*)-5-benzoyl-3,4-dioxo-2-phenyl-2-azabicyclo[3.1.0]hexane-1,6-dicarboxylate (
4aa). Synthesized according to General Procedure B from betaine
3aa (0.3 mmol scale). Isolated by FCC, Hexanes/EtOAc 2/1, R
f 0.33. An off-white powder, 102 mg (81%), m.p. 158–160 °C (lit. [
18] 159–161 °C).
1H NMR (400 MHz, CDCl
3): δ 7.87–7.80 (m, 2H), 7.66 (t,
J = 7.5 Hz, 1H), 7.60–7.49 (m, 4H), 7.48–7.41 (m, 2H), 7.30 (t,
J = 7.4 Hz, 1H), 4.18 (q,
J = 7.1 Hz, 2H), 4.15 (s, 1H), 3.58 (s, 3H), 1.16 (t,
J = 7.1 Hz, 3H).
13C NMR (101 MHz, CDCl
3): δ 186.6, 185.1, 165.4, 164.2, 156.7, 136.7, 135.0, 134.2, 129.7 (2C), 129.6 (2C), 129.3 (2C), 127.1, 120.1 (2C), 63.9, 54.8, 54.0, 46.8, 45.7, 13.9. IR (mineral oil), cm
−1: 1785, 1756, 1742, 1724, 1671, 1595. The spectral data was consistent with the data reported in the literature [
18].
6-Ethyl 1-methyl (1
R*,5
S*,6
R*)-5-benzoyl-2-(4-chlorophenyl)-3,4-dioxo-2-azabicyclo[3.1.0]hexane-1,6-dicarboxylate (
4ba). Synthesized according to General Procedure B from betaine
3ba (0.37 mmol scale). Isolated by crystallization from EtOH. An off-white powder, 128 mg (75%), m.p. 166–169 °C (lit. [
18] 167–169 °C).
1H NMR (400 MHz, CDCl
3): δ 7.87–7.78 (m, 2H), 7.72–7.62 (m, 1H), 7.57–7.49 (m, 4H), 7.47–7.37 (m, 2H), 4.18 (q,
J = 7.1 Hz, 2H), 4.13 (s, 1H), 3.60 (s, 3H), 1.17 (t,
J = 7.1 Hz, 3H).
13C NMR (101 MHz, CDCl
3): δ 186.3, 184.6, 165.3, 164.0, 156.6, 135.2, 135.1, 134.1, 132.7, 129.7 (2C), 129.7 (2C), 129.4 (2C), 121.3 (2C), 64.0, 54.6, 54.1, 46.8, 45.5, 14.0. IR (mineral oil), cm
−1: 1782, 1758, 1749, 1722, 1674, 1596. The spectral data was consistent with the data reported in the literature [
18].
6-Ethyl 1,5-dimethyl (1
R*,5
R*,6
R*)-3,4-dioxo-2-phenyl-2-azabicyclo[3.1.0]hexane-1,5,6-tricarboxylate (
4ea). Synthesized according to General Procedure B from betaine
3ea (0.3 mmol scale). Isolated by FCC, Hexanes/EtOAc 3/2, R
f 0.32. An off-white powder, 102 mg (91%), m.p. 129–131 °C.
1H NMR (400 MHz, CDCl
3): δ 7.50–7.46 (m, 2H), 7.45–7.39 (m, 2H), 7.30–7.25 (m, 1H), 4.16 (s, 1H), 4.12 (qd,
J = 7.2, 1.0 Hz, 2H), 3.84 (s, 3H), 3.69 (s, 3H), 1.13 (t,
J = 7.1 Hz, 3H).
13C NMR (101 MHz, CDCl
3): δ 183.6, 164.9, 163.7, 162.7, 156.6, 136.5, 129.6 (2C), 127.2, 120.2 (2C), 63.8, 55.1, 54.2 (2C), 45.4, 41.9, 13.9. IR (mineral oil), cm
−1: 1777, 1747, 1724, 1597. The spectral data was consistent with the data reported in the literature [
18].
Ethyl (1
R*,5
S*,6
R*)-2-(4-methoxyphenyl)-1,5-bis(4-methylbenzoyl)-3,4-dioxo-2-azabicyclo[3.1.0]hexane-6-carboxylate (
4fa). Synthesized according to General Procedure
B from betaine
3fa (0.3 mmol scale). Isolated by FCC, Hexanes/EtOAc 2/1, R
f 0.5. A yellow powder, 90 mg (57%), m.p. 177–179 °C (lit. [
18] 178–180 °C).
1H NMR (400 MHz, CDCl
3): δ 7.84 (d,
J = 8.0 Hz, 2H), 7.64 (d,
J = 8.0 Hz, 2H), 7.32 (d,
J = 8.0 Hz, 2H), 7.26 (d,
J = 9.0 Hz, 2H), 7.12 (d,
J = 8.0 Hz, 2H), 6.75 (d,
J = 9.0 Hz, 2H), 4.25 (s, 1H), 4.25–4.18 (m, 2H), 3.71 (s, 3H), 2.45 (s, 3H), 2.33 (s, 3H), 1.19 (t,
J = 7.1 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 190.4, 186.3, 185.3, 165.9, 158.2, 156.7, 146.5, 144.9, 132.9, 132.3, 130.6 (2C), 129.9 (2C), 129.3 (2C), 129.2, 128.9 (2C), 122.0 (2C), 114.6 (2C), 63.8, 60.0, 55.5, 49.8, 45.6, 22.0, 21.8, 14.0. IR (mineral oil), cm
−1: 1778, 1769, 1737, 1678, 1602. The spectral data was consistent with the data reported in the literature [
18].
Ethyl (1R*,5S*,6R*)-1,5-dibenzoyl-3,4-dioxo-2-(p-tolyl)-2-azabicyclo[3.1.0]hexane-6-carboxylate (4ga). Synthesized according to General Procedure B from betaine 3ga (0.3 mmol scale). Isolated by FCC, Hexanes/EtOAc 3/1, Rf 0.42. A pale-yellow/off-white powder, 57 mg (40%), m.p. 177–180 °C (dec.). 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 7.2 Hz, 2H), 7.72–7.65 (m, 3H), 7.56–7.50 (m, 2H), 7.49–7.42 (m, 1H), 7.34–7.28 (m, 2H), 7.20 (d, J = 8.5 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 4.29 (s, 1H), 4.27–4.17 (m, 2H), 2.22 (s, 3H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 191.3, 186.9, 185.1, 165.8, 156.6, 137.0, 135.5, 135.2, 134.6, 133.7, 133.6, 130.4 (2C), 130.0 (2C), 129.2 (2C), 128.7 (2C), 128.6 (2C), 120.4 (2C), 63.9, 59.9, 50.0, 45.5, 21.0, 14.0. IR (mineral oil), cm−1: 1772, 1764, 1742, 1715, 1685, 1678, 1599, 1577. Anal. Calcd for C29H23NO6: C 72.34; H 4.81; N 2.91. Found: C 72.62; H 4.96; N 2.68.
Methyl (1
R*,5
S*,6
R*)-5-benzoyl-3,4-dioxo-2-phenyl-6-(3-phenyl-1,2,4-oxadiazol-5-yl)-2-azabicyclo[3.1.0]hexane-1-carboxylate (
4ab)
. Synthesized according to General Procedure B from betaine
3ab (0.3 mmol scale). Isolated by FCC, Hexanes/EtOAc 4/1, R
f 0.31. An off-white powder, 104 mg (70%), m.p. 194–196 °C (lit. [
18] 195–197 °C).
1H NMR (400 MHz, CDCl
3): δ 7.92–7.79 (m, 4H), 7.68 (t,
J = 7.4 Hz, 1H), 7.53 (t,
J = 7.7 Hz, 2H), 7.50–7.33 (m, 7H), 7.25–7.18 (m, 1H), 4.76 (s, 1H), 3.64 (s, 3H).
13C NMR (101 MHz, CDCl
3): δ 186.2, 184.2, 170.7, 168.9, 163.9, 156.7, 136.3, 135.3, 134.1, 132.0, 129.8 (2C), 129.7 (2C), 129.5 (2C), 129.1 (2C), 127.9 (2C), 127.4, 125.3, 120.0 (2C), 55.7, 54.2, 47.1, 38.1. IR (mineral oil), cm
−1: 1774, 1755, 1739, 1682, 1597, 1579. The spectral data was consistent with the data reported in the literature [
18].



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
