

Design, Synthesis, Molecular Docking, and Anticancer Activity of Chalcone Derivatives as VEGFR-2 Inhibitors


Abstract
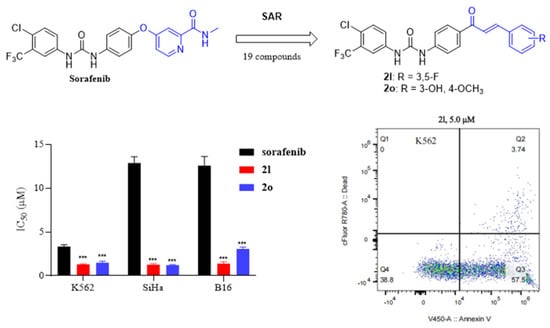
1. Introduction
2. Results and Discussion
2.1. Synthesis of Compounds 2a–2s
2.2. Biological Evaluation
2.2.1. In Vitro Cytotoxic Activities Study
2.2.2. VEGFR-2 Enzyme Inhibition Assay Results
2.2.3. Compound 2l Induced K562 Apoptosis
2.2.4. Compound 2l Induced K562 Cycle Analysis
2.3. Molecular Docking Results
3. Materials and Methods
3.1. Chemistry
3.1.1. Procedure for Synthesis of Compound 1a
3.1.2. General Procedure for Preparation of 2a–2n
3.1.3. General Procedure for Preparation of 2o–2s
3.1.4. Structural Characterization of Synthesized Compounds
3.2. In Vitro Cytotoxicity
3.3. VEGFR-2 Enzyme Inhibition Assay
3.4. Cell Apoptosis Assay
3.5. Cell Cycle Analysis
3.6. Molecular Docking
3.7. Statistical Analysis
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VEGFR-1 | Vascular endothelial growth factor receptor-1 |
| VEGFR-2 | Vascular endothelial growth factor receptor-2 |
| VEGFR-3 | Vascular endothelial growth factor receptor-3 |
| FDA | Food and Drug Administration |
| DFG | Dynamic form generator |
| Et3N | Triethylamine |
| DCM | Dichloromethane |
| CCK-8 | Cell Counting Kit 8 |
| MOE | Molecular Operating Environment |
| Glu | Glutamic acid |
| Asp | Aspartic acid |
| Cys | Cysteine |
| Phe | Phenylalanine |
| TLC | Thin-layer chromatography |
References
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef]
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021, 28, 100422. [Google Scholar] [CrossRef]
- Liu, Z.L.; Chen, H.H.; Zheng, L.L.; Sun, L.P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Tigu, A.B.; Munteanu, R.; Moldovan, C.S.; Kegyes, D.; Onaciu, A.; Gulei, D.; Ghiaur, G.; Einsele, H.; Croce, C.M. Therapeutic advances of targeting receptor tyrosine kinases in cancer. Signal Transduct. Target. Ther. 2024, 9, 201. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Wang, Y.; Lin, C.; Zhang, D.; Chen, J.; Ouyang, L.; Wu, F.; Zhang, J.; Chen, L. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J. Hematol. Oncol. 2022, 15, 89. [Google Scholar] [CrossRef]
- Kaufman, N.E.M.; Dhingra, S.; Jois, S.D.; Vicente, M.D.G.H. Molecular Targeting of Epidermal Growth Factor Receptor (EGFR) and Vascular Endothelial Growth Factor Receptor (VEGFR). Molecules 2021, 26, 1076. [Google Scholar] [CrossRef]
- Abdulkadir, S.; Li, C.; Jiang, W.; Zhao, X.; Sang, P.; Wei, L.; Hu, Y.; Li, Q.; Cai, J. Modulating Angiogenesis by Proteomimetics of Vascular Endothelial Growth Factor. J. Am. Chem. Soc. 2022, 144, 270–281. [Google Scholar] [CrossRef]
- Cheng, K.; Liu, C.F.; Rao, G.W. Anti-angiogenic Agents: A Review on Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) Inhibitors. Curr. Med. Chem. 2021, 28, 2540–2564. [Google Scholar] [CrossRef]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.H.; Nam, Y.S.; Bang, J.Y.; Hwang, I.S.; Kim, D.H.; Ki, M.; Lee, H.W. Targeting vascular endothelial growth receptor-2 (VEGFR-2): Structural biology, functional insights, and therapeutic resistance. Arch. Pharm. Res. 2025, 48, 404–425. [Google Scholar] [CrossRef]
- Zha, H.; Li, F.; Cai, L.; Liu, W.; Zhang, M.; Gu, S.; Feng, H.; Xia, Z.; Guo, C.; Wu, X.; et al. Design, synthesis and biological evaluation of indazole derivatives as VEGFR-2 kinase inhibitors with anti-angiogenic properties. Eur. J. Med. Chem. 2024, 279, 116889. [Google Scholar] [CrossRef]
- Shah, A.A.; Kamal, M.A.; Akhtar, S. Tumor Angiogenesis and VEGFR-2: Mechanism, Pathways and Current Biological Therapeutic Interventions. Curr. Drug Metab. 2021, 22, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Ryu, M.H.; Di Bartolomeo, M.; Chau, I.; Yoon, H.; Kim, J.G.; Lee, K.W.; Oh, S.C.; Takashima, A.; Kryzhanivska, A.; et al. Rivoceranib, a VEGFR-2 inhibitor, monotherapy in previously treated patients with advanced or metastatic gastric or gastroesophageal junction cancer (ANGEL study): An international, randomized, placebo-controlled, phase 3 trial. Gastric Cancer 2024, 27, 375–386. [Google Scholar] [CrossRef]
- Marques, C.S.; Brandão, P.; Burke, A.J. Targeting Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2): Latest Insights on Synthetic Strategies. Molecules 2024, 29, 5341. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Zhao, H.C.; Hou, S.J.; Zhang, H.J.; Cheng, L.; Yuan, S.; Zhang, L.R.; Song, J.; Zhang, S.Y.; Chen, S.W. Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg. Chem. 2023, 133, 106425. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, P.J.; Nemade, A.R.; Shirkhedkar, A.A. Recent updates on potential of VEGFR-2 small-molecule inhibitors as anticancer agents. RSC Adv. 2024, 14, 33384–33417. [Google Scholar] [CrossRef]
- Farghaly, T.A.; Al-Hasani, W.A.; Abdulwahab, H.G. An updated patent review of VEGFR-2 inhibitors (2017-present). Expert Opin. Ther. Patents 2021, 31, 989–1007. [Google Scholar] [CrossRef]
- Modi, S.J.; Kulkarni, V.M. Exploration of structural requirements for the inhibition of VEGFR-2 tyrosine kinase: Binding site analysis of type II, ‘DFG-out’ inhibitors. J. Biomol. Struct. Dyn. 2022, 40, 5712–5727. [Google Scholar] [CrossRef]
- Sroor, F.M.; Othman, A.M.; Tantawy, M.A.; Mahrous, K.F.; El-Naggar, M.E. Synthesis, antimicrobial, anti-cancer and in silico studies of new urea derivatives. Bioorg. Chem. 2021, 112, 104953. [Google Scholar] [CrossRef]
- Listro, R.; Rossino, G.; Piaggi, F.; Sonekan, F.F.; Rossi, D.; Linciano, P.; Collina, S. Urea-based anticancer agents. Exploring 100-years of research with an eye to the future. Front. Chem. 2022, 10, 995351. [Google Scholar] [CrossRef]
- Martín-Beltrán, C.; Gil-Edo, R.; Hernández-Ribelles, G.; Agut, R.; Marí-Mezquita, P.; Carda, M.; Falomir, E. Aryl Urea based scaffolds for multitarget drug discovery in anticancer immunotherapies. Pharmaceuticals 2021, 14, 337. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khan, J.; Dukhyil, A.A.B.; Alarousy, R.M.I.I.; Attah, E.I.; Sharma, T.; Khairnar, S.J.; Bendale, A.R. Chalcone Scaffolds, Bioprecursors of Flavonoids: Chemistry, Bioactivities, and Pharmacokinetics. Molecules 2021, 26, 7177. [Google Scholar] [CrossRef]
- Zhou, K.; Yang, S.; Li, S.M. Naturally occurring prenylated chalcones from plants: Structural diversity, distribution, activities and biosynthesis. Nat. Prod. Rep. 2021, 38, 2236–2260. [Google Scholar] [CrossRef]
- Guazelli, C.F.S.; Fattori, V.; Ferraz, C.R.; Borghi, S.M.; Casagrande, R.; Baracat, M.M.; Verri, W.A., Jr. Antioxidant and anti-inflammatory effects of hesperidin methyl chalcone in experimental ulcerative colitis. Chem. Biol. Interact. 2021, 333, 109315. [Google Scholar] [CrossRef]
- Pan, Q.; Yang, H.; Du, Z.; Ni, Z.; Zhu, Q.; Tu, S.; Zhao, Y.; Ye, F. Synthesis, characterization, and anticancer activity of syringaldehyde-derived chalcones against female cancers. Med. Chem. Res. 2024, 33, 532–547. [Google Scholar] [CrossRef]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A.O.; et al. Pharmacological properties of chalcones: A review of preclinical including molecular mechanisms and clinical evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef]
- Elkanzi, N.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of chalcones derivatives and their biological activities: A review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef]
- Alswah, M.; Bayoumi, A.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, synthesis and cytotoxic evaluation of novel chalcone derivatives bearing triazolo[4,3-a]-quinoxaline moieties as potent anticancer agents with dual EGFR kinase and tubulin polymerization inhibitory effects. Molecules 2017, 23, 48. [Google Scholar] [CrossRef]
- Emam, S.H.; Sonousi, A.; Osman, E.O.; Hwang, D.; Kim, G.D.; Hassan, R.A. Design and synthesis of methoxyphenyl- and coumarin-based chalcone derivatives as anti-inflammatory agents by inhibition of NO production and down-regulation of NF-κB in LPS-induced RAW264.7 macrophage cells. Bioorg. Chem. 2021, 107, 104630. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, S.; Wu, C.; Liu, X.; Tao, H.; Huang, Y.; Liu, Y.; Zheng, P.; Zhu, W. Design, synthesis and activity of novel sorafenib analogues bearing chalcone unit. Bioorg. Med. Chem. Lett. 2016, 26, 5450–5454. [Google Scholar] [CrossRef]
- Ahmed, M.F.; Santali, E.Y.; El-Haggar, R. Novel piperazine–chalcone hybrids and related pyrazoline analogues targeting VEGFR-2 kinase; design, synthesis, molecular docking studies, and anticancer evaluation. J. Enzym. Inhib. Med. Chem. 2020, 36, 308–319. [Google Scholar] [CrossRef]
- Shalaby, M.A.; Rizk, S.A.; Fahim, A.M. Synthesis, reactions and application of chalcones: A systematic review. Org. Biomol. Chem. 2023, 21, 5317–5346. [Google Scholar] [CrossRef]
- Dutta, U.; Das, J.; Goswami, M.J.; Bhardwaj, S.; Verma, A.K.; Kakati, D. Design and synthesis of O-glycoside derivatives with promising antidiabetic and anticancer potential. Med. Chem. Res. 2025, 34, 1025–1039. [Google Scholar] [CrossRef]
- Othman, E.M.; Fayed, E.A.; Husseiny, E.M.; Abulkhair, H.S. The effect of novel synthetic semicarbazone-and thiosemicarbazone-linked 1, 2, 3-triazoles on the apoptotic markers, VEGFR-2, and cell cycle of myeloid leukemia. Bioorg. Chem. 2022, 127, 105968. [Google Scholar] [CrossRef]
- Shirogane, Y.; Usami, Y.; Okumura, M.; Hirose, K.; Naniwa, K.; Ikebe, K.; Toyosawa, S. Anti-VEGFR2 neutralising antibody slows the progression of multistep oral carcinogenesis. J. Pathol. 2024, 4, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Padró, T.; Bieker, R.; Ruiz, S.; Steins, M.; Retzlaff, S.; Bürger, H.; Büchner, T.; Kessler, T.; Herrera, F.; Kienast, J.; et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 2002, 7, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Molhoek, K.R.; Erdag, G.; Rasamny, J.K.; Murphy, C.; Deacon, D.; Patterson, J.W.; Slingluff, C.L., Jr.; Brautigan, D.L. VEGFR-2 expression in human melanoma: Revised assessment. Int. J. Cancer 2011, 12, 2807–2815. [Google Scholar] [CrossRef]
- Sano, D.; Fooshee, D.R.; Zhao, M.; Andrews, G.A.; Frederick, M.J.; Galer, C.; Milas, Z.L.; Morrow, P.K.; Myers, J.N. Targeted molecular therapy of head and neck squamous cell carcinoma with the tyrosine kinase inhibitor vandetanib in a mouse model. Head Neck 2011, 3, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.L.; Li, G.B.; Ma, S.; Zou, C.; Zhou, S.; Sun, Q.Z.; Chen, C.; Wang, L.J.; Feng, S.; Li, L.L.; et al. Structure-activity relationship studies of pyrazolo[3,4-d]pyrimidine derivatives leading to the discovery of a novel multikinase inhibitor that potently inhibits FLT3 and VEGFR2 and evaluation of its activity against acute myeloid leukemia in vitro and in vivo. J. Med. Chem. 2013, 4, 1641–1655. [Google Scholar] [CrossRef]
- Sabt, A.; Khedr, M.A.; Eldehna, W.M.; Elshamy, A.I.; Abdelhameed, M.F.; Allam, R.M.; Batran, R.Z. New pyrazolylindolin-2-one based coumarin derivatives as anti-melanoma agents: Design, synthesis, dual BRAFV600E/VEGFR-2 inhibition, and computational studies. RSC Adv. 2024, 9, 5907–5925. [Google Scholar] [CrossRef]
- Moradi, M.; Mousavi, A.; Emamgholipour, Z.; Giovannini, J.; Moghimi, S.; Peytam, F.; Honarmand, A.; Bach, S.; Foroumadi, A. Quinazoline-based VEGFR-2 inhibitors as potential anti-angiogenic agents: A contemporary perspective of SAR and molecular docking studies. Eur. J. Med. Chem. 2023, 259, 115626. [Google Scholar] [CrossRef]
- Chaudhry, G.E.; Md Akim, A.; Sung, Y.Y.; Sifzizul, T.M.T. Cancer and apoptosis: The apoptotic activity of plant and marine natural products and their potential as targeted cancer therapeutics. Front. Pharmacol. 2022, 13, 842376. [Google Scholar] [CrossRef]
- Ma, C.; Gurkan-Cavusoglu, E. A comprehensive review of computational cell cycle models in guiding cancer treatment strategies. NPJ Syst. Biol. Appl. 2024, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Lim, J.H.; Jang, J.H.; Lee, J.Y. The use of machine learning modeling, virtual screening, molecular docking, and molecular dynamics simulations to identify potential VEGFR2 kinase inhibitors. Sci. Rep. 2022, 12, 18825. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Huang, T.; Wang, Y.; Wang, L.; Feng, S.; Cheng, W.; Yang, L.; Duan, Y. Angiogenesis and anti-leukaemia activity of novel indole derivatives as potent colchicine binding site inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Adel, M.; Abouzid, K.A. New fluorinated diarylureas linked to pyrrolo[2,3-d] pyrimidine scaffold as VEGFR-2 inhibitors: Molecular docking and biological evaluation. Bioorg. Chem. 2022, 127, 106006. [Google Scholar] [CrossRef]
- Brestoff, J.R. Full spectrum flow cytometry in the clinical laboratory. Int. J. Lab. Hematol. 2023, 45, 44–49. [Google Scholar] [CrossRef]
- Fouad, M.A.; Osman, A.A.; Abdelhamid, N.M.; Rashad, M.W.; Nabawy, A.Y.; El Kerdawy, A.M. Discovery of dual kinase inhibitors targeting VEGFR2 and FAK: Structure-based pharmacophore modeling, virtual screening, and molecular docking studies. BMC Chem. 2024, 18, 29. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | IC50, μM | |||
|---|---|---|---|---|
| SiHa | K562 | B16 | HK-2 | |
| 2a | 4.43 ± 0.16 | 12.65 ± 0.45 | 10.69 ± 0.78 | nd |
| 2b | 5.55 ± 0.26 | 5.08 ± 0.31 | 8.32 ± 0.61 | nd |
| 2c | 1.03 ± 0.02 | 2.05 ± 0.13 | 5.02 ± 0.18 | 15.68 ± 1.22 |
| 2d | 4.78 ± 0.14 | 8.41 ± 0.57 | 12.77 ± 0.84 | nd |
| 2e | 4.71 ± 0.29 | 2.91 ± 0.17 | 11.28 ± 0.64 | nd |
| 2f | 4.89 ± 0.21 | 1.68 ± 0.09 | 1.65 ± 0.29 | 5.85 ± 0.36 |
| 2g | 6.03 ± 0.38 | 4.19 ± 0.26 | 7.15 ± 0.49 | nd |
| 2h | 2.99 ± 0.27 | 2.02 ± 0.12 | 1.62 ± 0.14 | 9.87± 0.89 |
| 2i | 7.96 ± 0.26 | 4.45 ± 0.53 | 8.66 ± 0.58 | nd |
| 2j | 6.62 ± 0.28 | 2.40 ± 0.22 | 9.73 ± 0.98 | nd |
| 2k | 8.35 ± 0.09 | 1.73 ± 0.23 | 6.42 ± 0.54 | nd |
| 2l | 1.25 ± 0.13 | 1.30 ± 0.07 | 1.39 ± 0.21 | 25.36 ± 1.47 |
| 2m | 6.53 ± 0.24 | 8.95 ± 0.76 | nd | nd |
| 2n | >20 | 15.2 ± 1.2 | nd | nd |
| 2o | 1.22 ± 0.07 | 1.51 ± 0.16 | 3.06 ± 0.22 | 19.14± 1.05 |
| 2p | 4.25 ± 0.14 | 3.42 ± 0.18 | 4.37 ± 0.28 | 13.89± 0.58 |
| 2q | 2.53 ± 0.09 | 1.12 ± 0.10 | 4.49 ± 0.24 | 24.32± 1.71 |
| 2r | 2.22 ± 0.10 | 0.97 ± 0.06 | 2.93 ± 0.29 | 11.27± 0.66 |
| 2s | 8.96 ± 0.03 | 6.47 ± 0.36 | 4.29 ± 0.52 | nd |
| sorafenib | 12.82 ± 0.71 | 3.34 ± 0.22 | 12.57 ± 1.03 | 10.87 ± 0.69 |
| Comp. | IC50, μM |
|---|---|
| 2c | 0.68 ± 0.03 |
| 2f | 0.39 ± 0.02 |
| 2l | 0.42 ± 0.03 |
| 2n | 2.2 ± 0.1 |
| 2o | 0.31 ± 0.02 |
| 2q | 0.55 ± 0.04 |
| sorafenib | 0.12 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, M.; Zhang, X.; Zhu, H.; Zhang, X. Design, Synthesis, Molecular Docking, and Anticancer Activity of Chalcone Derivatives as VEGFR-2 Inhibitors. Molecules 2025, 30, 4526. https://doi.org/10.3390/molecules30234526
Yu M, Zhang X, Zhu H, Zhang X. Design, Synthesis, Molecular Docking, and Anticancer Activity of Chalcone Derivatives as VEGFR-2 Inhibitors. Molecules. 2025; 30(23):4526. https://doi.org/10.3390/molecules30234526
Chicago/Turabian StyleYu, Mingjun, Xin Zhang, Hui Zhu, and Xiaoqian Zhang. 2025. "Design, Synthesis, Molecular Docking, and Anticancer Activity of Chalcone Derivatives as VEGFR-2 Inhibitors" Molecules 30, no. 23: 4526. https://doi.org/10.3390/molecules30234526
APA StyleYu, M., Zhang, X., Zhu, H., & Zhang, X. (2025). Design, Synthesis, Molecular Docking, and Anticancer Activity of Chalcone Derivatives as VEGFR-2 Inhibitors. Molecules, 30(23), 4526. https://doi.org/10.3390/molecules30234526