
Lithium-Decorated C26 Fullerene in DFT Investigation: Tuning Electronic Structures for Enhanced Hydrogen Storage


Abstract

1. Introduction
2. Results and Discussion
2.1. Hydrogen Adsorption on C26 Fullerene
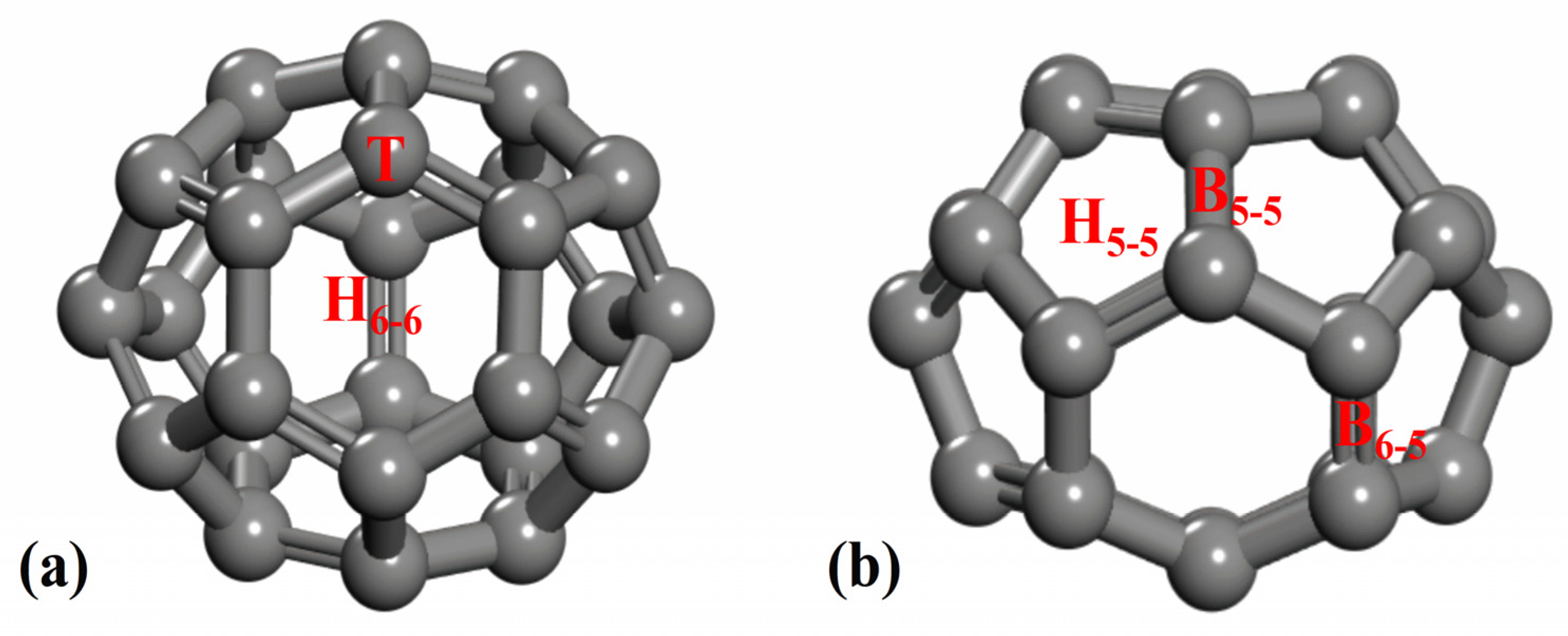
2.2. Li-Decorated C26 Fullerene
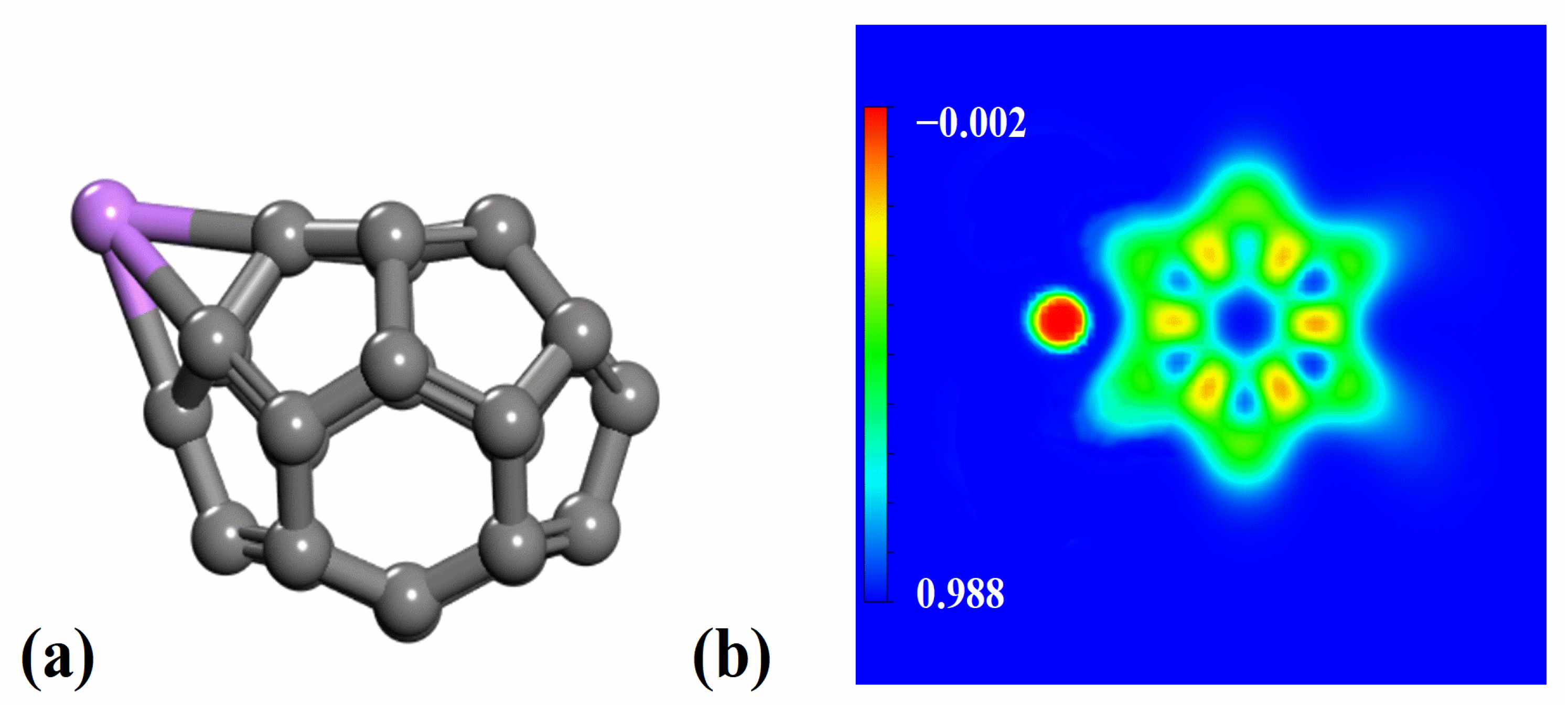
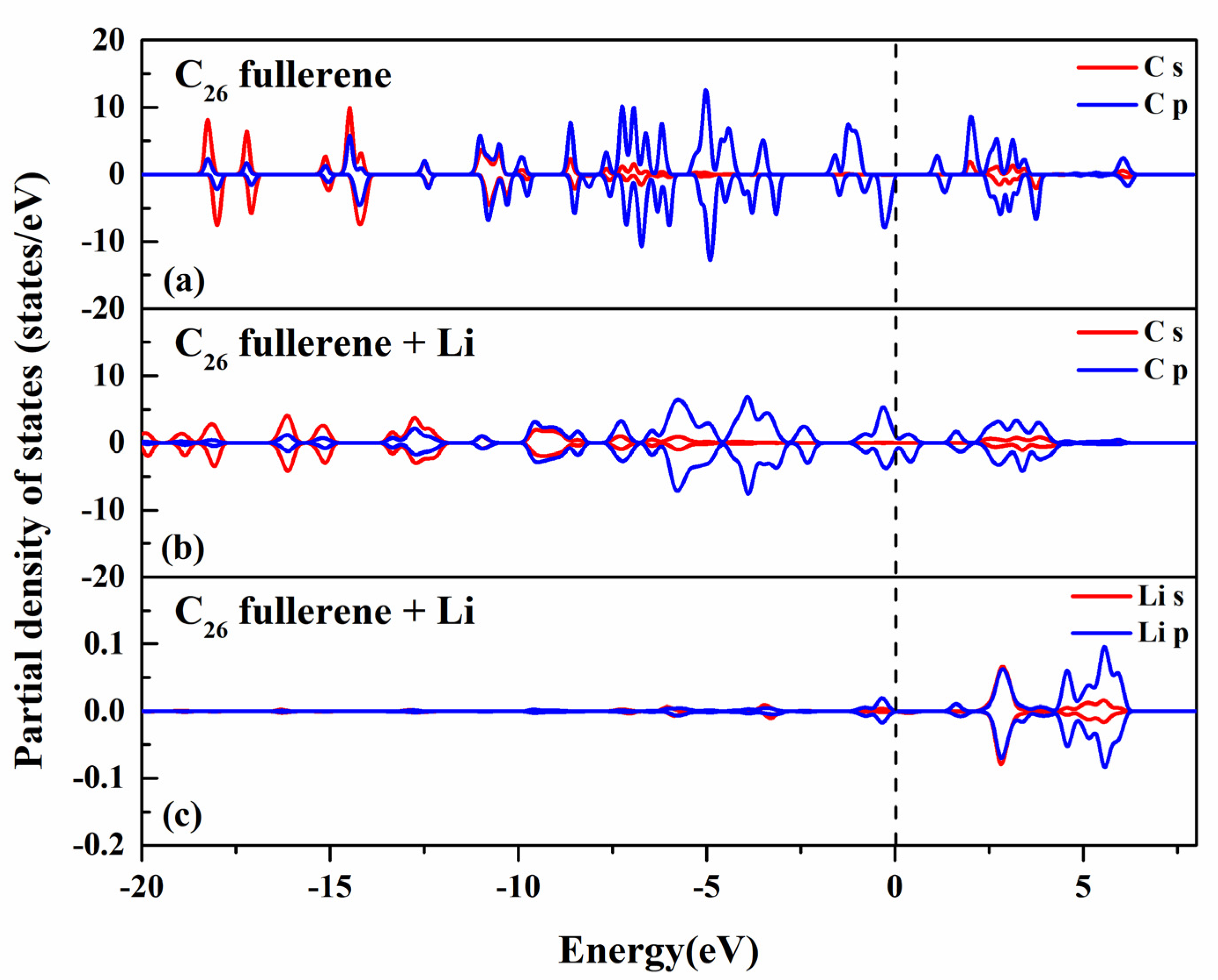
2.3. Interaction Between Li and C26 Fullerene
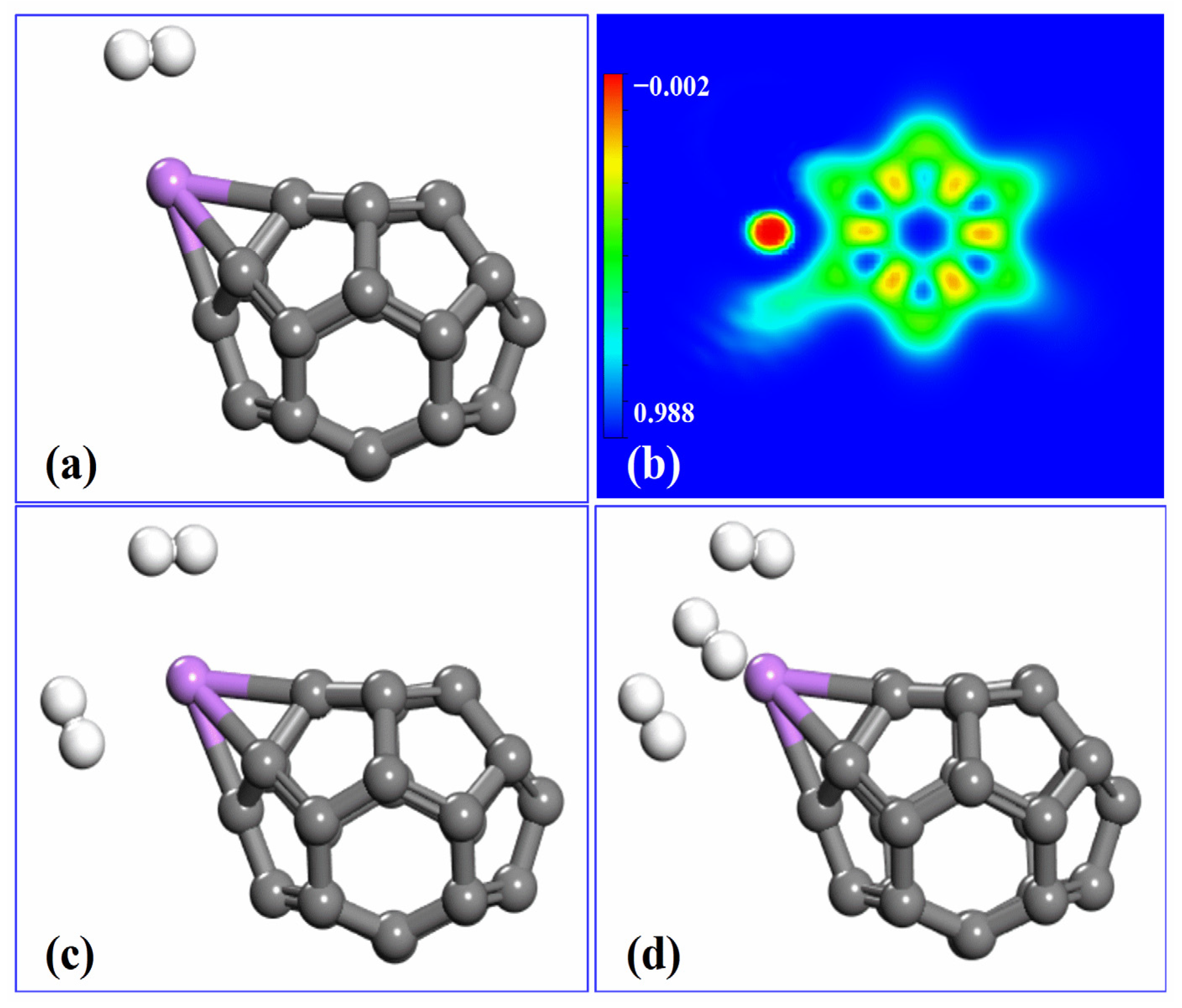
2.4. Hydrogen Adsorption on Li-Decorated C26 Fullerene
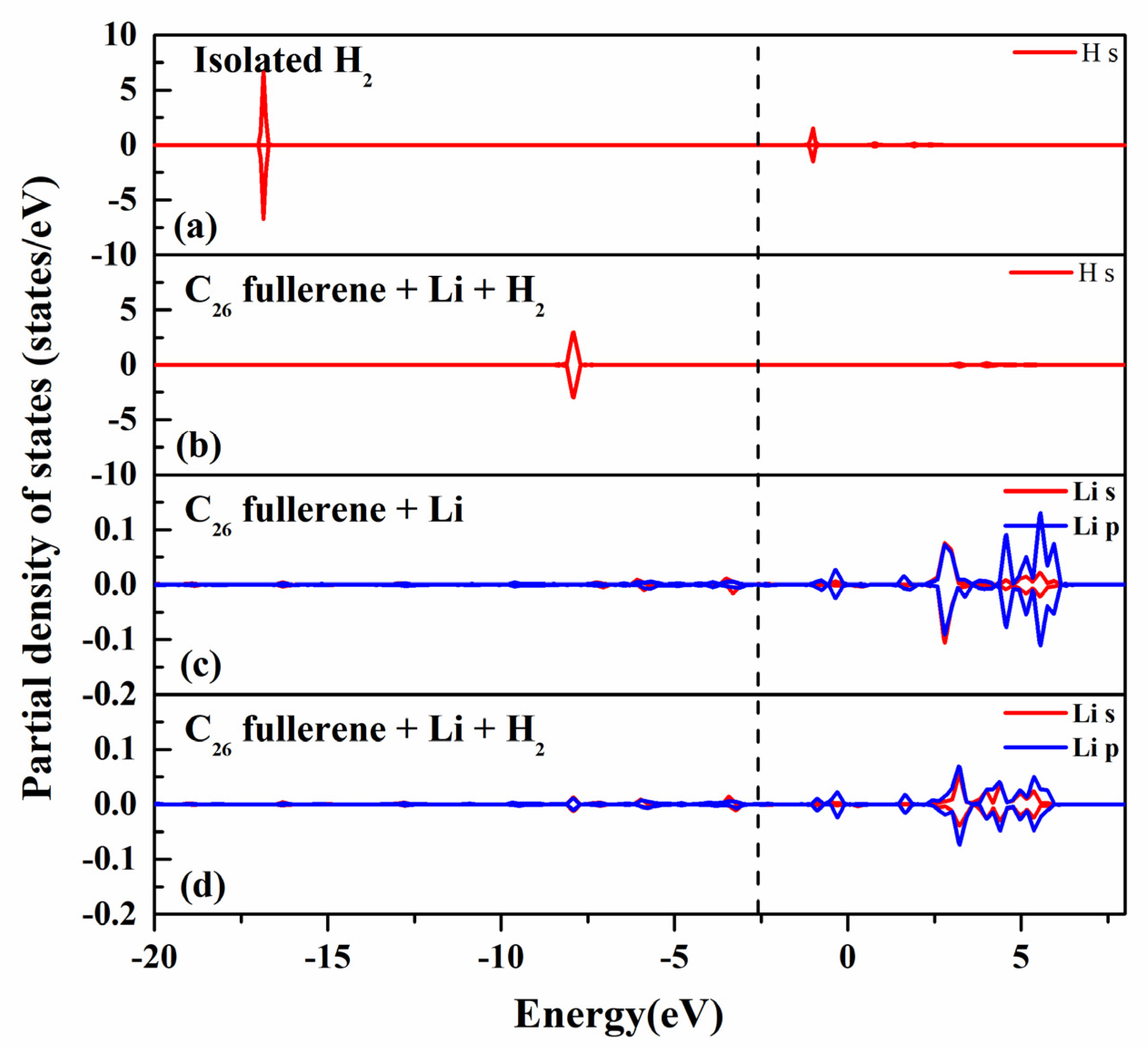
2.5. Interaction of Hydrogen with Li-Decorated C26 Fullerene
2.6. Kubas-Type Interaction
3. Computational Details
Computational Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, J.; Ji, W.; Cao, X.; He, W.; Fan, J.; Yuan, Y. A current perspective on the renewable energy hydrogen production process. J. Therm. Sci. 2023, 32, 542–596. [Google Scholar] [CrossRef]
- Yang, J.; Lam, T.Y.; Luo, Z.; Cheng, Q.; Wang, G.; Yao, H. Renewable energy driven electrolysis of water for hydrogen production, storage, and transportation. Renew. Sustain. Energy Rev. 2025, 218, 115804. [Google Scholar] [CrossRef]
- Dai, S.; Shen, P.; Deng, W.; Yu, Q. Hydrogen energy in electrical power systems: A review and future outlook. Electronics 2024, 13, 3370. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, B.; Huang, W.; Li, Y.; Hou, L.; Xiao, J.; Mao, Z.; Li, X. A review of hydrogen generation, storage, and applications in power system. J. Energy Storage 2024, 75, 109307. [Google Scholar] [CrossRef]
- Wang, Z.; Lao, G. Hydrogen production from renewable sources: Bridging the gap to sustainable energy and economic viability. Int. J. Hydrogen Energy 2025, 117, 121–134. [Google Scholar] [CrossRef]
- Buckley, C.E.; Chen, P.; van Hassel, B.A.; Hirscher, M. Hydrogen-based Energy Storage (IEA-HIA Task 32). Appl. Phys. A-Mater. Sci. Process. 2016, 122, 141. [Google Scholar] [CrossRef]
- Li, C.; Li, H.; Yue, H.; Lv, J.; Zhang, J. Flexibility Value of Multimodal Hydrogen Energy Utilization in Electric-Hydrogen-Thermal Systems. Sustainability 2024, 16, 4939. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, H.; Cheng, X.; Tang, Y. Ti-decorated B38 fullerene: A high capacity hydrogen storage material. Int. J. Hydrogen Energy 2016, 41, 19123–19128. [Google Scholar] [CrossRef]
- Li, C.; Li, J.; Wu, F.; Li, S.-S.; Xia, J.-B.; Wang, L.-W. High capacity hydrogen storage in Ca decorated graphyne: A first-principles study. J. Phys. Chem. C 2011, 115, 23221–23225. [Google Scholar] [CrossRef]
- Srinivasu, K.; Ghosh, S.K. Transition metal decorated porphyrin-like porous fullerene: Promising materials for molecular hydrogen adsorption. J. Phys. Chem. C 2012, 116, 25184–25189. [Google Scholar] [CrossRef]
- Salem, M.A.; Katin, K.P.; Kaya, S.; Kochaev, A.I.; Maslov, M.M. Interaction of dopants and functional groups adsorbed on the carbon fullerenes: Computational study. Phys. E Low-Dimens. Syst. Nanostructures 2020, 124, 114319. [Google Scholar] [CrossRef]
- Dai, W.; Xiao, M.; Chen, M.-Q.; Xu, J.-J.; Tang, Y.-J. A simulation study on the hydrogen storage properties of fullerene family molecules Cx (x = 56, 60, 70) and their hydrides. Mod. Phys. Lett. B 2016, 30, 1650303. [Google Scholar] [CrossRef]
- Wang, Y.; Alcami, M.; Martin, F. Understanding the Supramolecular Self-Assembly of the Fullerene Derivative PCBM on Gold Surfaces. Chemphyschem 2008, 9, 1030–1035. [Google Scholar] [CrossRef]
- Chandrakumar, K.R.S.; Ghosh, S.K. Alkali-metal-induced enhancement of hydrogen adsorption in C60 fullerene: An ab initio study. Nano Lett. 2008, 8, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Rai, P.K.; Kumar, P. How an electric field makes endohedral fullerene an improved catalyst for hydrogen evolution reaction. Comput. Theor. Chem. 2023, 1221, 114026. [Google Scholar] [CrossRef]
- Chen, Z.; Jiao, H.; Bühl, M.; Hirsch, A.; Thiel, W. Theoretical Investigation into Structures and Magnetic Properties of Smaller Fullerenes and their Heteroanalogues. Theor. Chem. Acc. Theory Comput. Model. (Theor. Chim. Acta) 2001, 106, 352–363. [Google Scholar] [CrossRef]
- Dunk, P.W.; Kaiser, N.K.; Mulet-Gas, M.; Rodriguez-Fortea, A.; Poblet, J.M.; Shinohara, H.; Hendrickson, C.L.; Marshall, A.G.; Kroto, H.W. The smallest stable fullerene, M@C28 (M = Ti, Zr, U): Stabilization and growth from carbon vapor. J. Am. Chem. Soc. 2012, 134, 9380–9389. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.; Kalafat, M.Y.; Kizilduman, B.K.; Bicil, Z.; Turhan, Y.; Yanmaz, E.; Duman, B. Hydrogen storage analysis of fullerene and defective fullerenes: The first experimental study. Fuel 2025, 390, 134705. [Google Scholar] [CrossRef]
- Lee, H.; Park, D.G.; Park, J.; Kim, Y.-H.; Kang, J.K. Amorphized Defective Fullerene with a Single-Atom Platinum for Room-Temperature Hydrogen Storage. Adv. Energy Mater. 2023, 13, 2300041. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Yar, M.; Lakhani, A.; Sheikh, N.S.; Bayach, I.; Ayub, K. Probing the catalytic activity of first-row transition metal doped C20 fullerene as remarkable HER electrocatalysts: A DFT study. Mater. Sci. Semicond. Process. 2023, 167, 107785. [Google Scholar] [CrossRef]
- Chai, Z.; Liu, L.; Liang, C.; Liu, Y.; Wang, Q. Insight into the Reversible Hydrogen Storage of Titanium-Decorated Boron-Doped C20 Fullerene: A Theoretical Prediction. Mol. Catal. 2024, 29, 4728. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Mane, P.; Sarkar, U.; Chakraborty, B. Yttrium decorated fullerene C30 as potential hydrogen storage material: Perspectives from DFT simulations. Theor. Chem. Acc. 2023, 142, 94. [Google Scholar] [CrossRef]
- Mahamiya, V.; Shukla, A.; Chakraborty, B. Applied Surface Science Scandium decorated C24 fullerene as high capacity reversible hydrogen storage material: Insights from density functional theory simulations. Appl. Surf. Sci. 2022, 573, 151389. [Google Scholar] [CrossRef]
- Pandey, A.K.; Singh, V.; Dubey, D.D.; Pandey, K.K.; Avaish, M.; Dwivedi, A. Alkali Metal-Doped Fullerenes as Hydrogen Storage-A Quantum Chemical Investigation. In Proceedings of the 4th National Conference on Material and Device (NCMD), Moradabad, India, 28–29 December 2023. [Google Scholar]
- Sahoo, R.K.; Chakraborty, B.; Sahu, S. Reversible hydrogen storage on alkali metal (Li and Na) decorated C20 fullerene: A density functional study. Int. J. Hydrogen Energy 2021, 46, 40251–40261. [Google Scholar] [CrossRef]
- Deniz, C.U.; Mert, H.; Baykasoglu, C. Li-doped fullerene pillared graphene nanocomposites for enhancing hydrogen storage: A computational study. Comp. Mater. Sci. 2021, 186, 110023. [Google Scholar] [CrossRef]
- Dong, S.; Lv, E.; Wang, J.; Li, C.; Ma, K.; Gao, Z.; Yang, W.; Ding, Z.; Wu, C.; Gates, I.D. Construction of transition metal-decorated boron doped twin-graphene for hydrogen storage: A theoretical prediction. Fuel 2021, 304, 121351. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Bader, R. Atoms in molecules: A quantum theory. Int. Ser. Monogr. Chem. 1990, 22, 9–15. [Google Scholar]
- Mahamiya, V.; Shukla, A.; Chakraborty, B. Exploring yttrium doped C24 fullerene as a high-capacity reversible hydrogen storage material: DFT investigations. J. Alloys Compd. 2022, 897, 162797. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Wang, Y.; Burke, K.; Perdew, J. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Eb (eV) | dC-Li (Å) |
|---|---|---|
| H5-5 | −2.68 | 2.22 |
| H6-6 | −2.59 | 2.28 |
| B5-5 | −2.42 | 2.11 |
| B6-5 | −2.38 | 2.12 |
| T | unstable and move to H5-5 site | |
| nH2 | Eads (eV) | Ec (eV) | dLi-H2 (Å) |
|---|---|---|---|
| 1H2 | −0.15 | −0.15 | 2.09 |
| 2H2 | −0.13 | −0.11 | 2.13 |
| 3H2 | −0.11 | −0.07 | 2.25 |
| 4H2 | −0.09 | −0.02 | 2.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Liu, L.; Li, Q.; Xu, Z.; Shi, Y.; Lei, C. Lithium-Decorated C26 Fullerene in DFT Investigation: Tuning Electronic Structures for Enhanced Hydrogen Storage. Molecules 2025, 30, 3223. https://doi.org/10.3390/molecules30153223
Yu J, Liu L, Li Q, Xu Z, Shi Y, Lei C. Lithium-Decorated C26 Fullerene in DFT Investigation: Tuning Electronic Structures for Enhanced Hydrogen Storage. Molecules. 2025; 30(15):3223. https://doi.org/10.3390/molecules30153223
Chicago/Turabian StyleYu, Jiangang, Lili Liu, Quansheng Li, Zhidong Xu, Yujia Shi, and Cheng Lei. 2025. "Lithium-Decorated C26 Fullerene in DFT Investigation: Tuning Electronic Structures for Enhanced Hydrogen Storage" Molecules 30, no. 15: 3223. https://doi.org/10.3390/molecules30153223
APA StyleYu, J., Liu, L., Li, Q., Xu, Z., Shi, Y., & Lei, C. (2025). Lithium-Decorated C26 Fullerene in DFT Investigation: Tuning Electronic Structures for Enhanced Hydrogen Storage. Molecules, 30(15), 3223. https://doi.org/10.3390/molecules30153223