
A Comparative Study on ZrO2- and MgO-Based Sulfonic Acid Materials for the Reactive Adsorption of o-Xylene



Abstract

1. Introduction
2. Results and Discussion
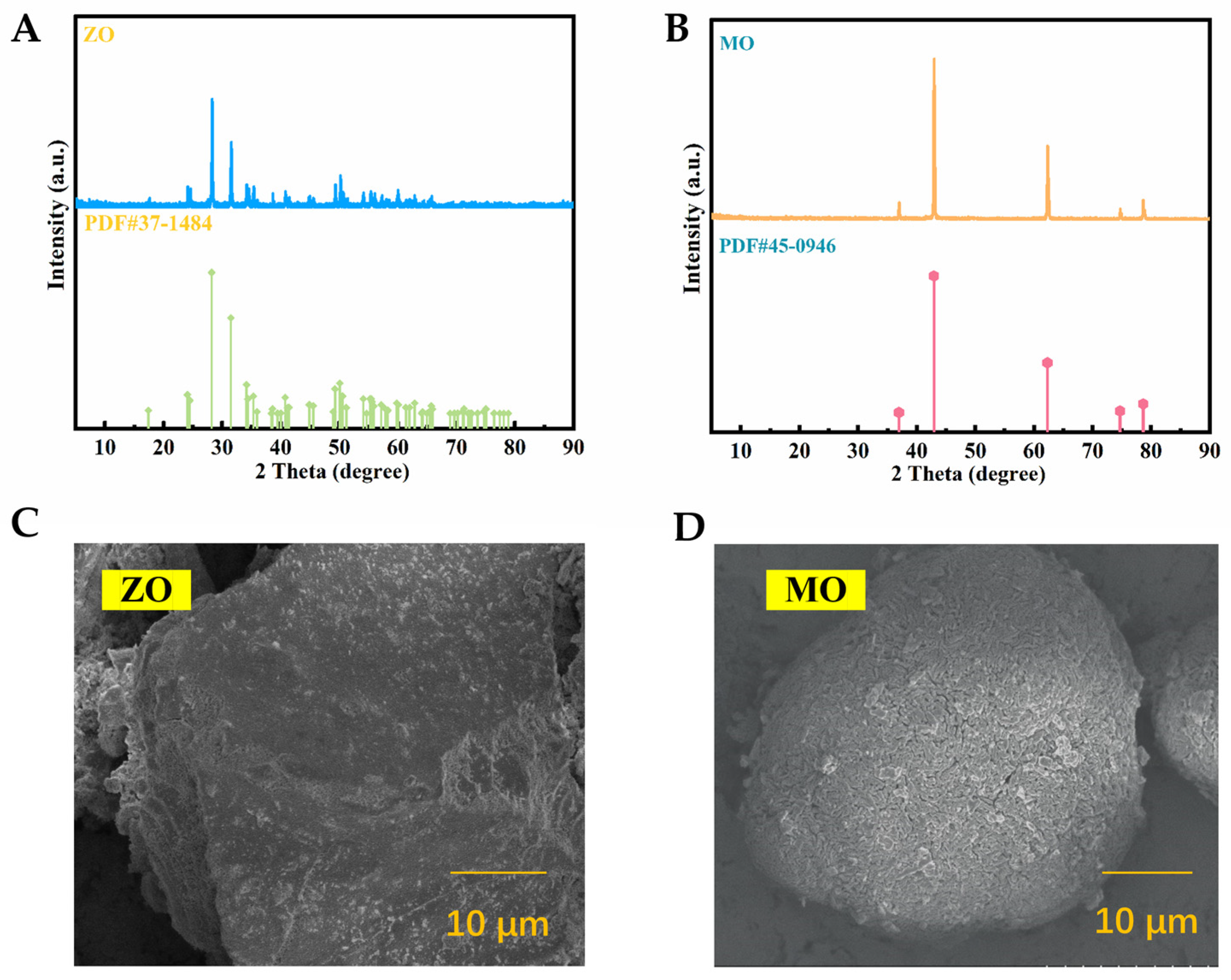
2.1. Analysis of the Basic Properties of ZO and MO Carrier Materials
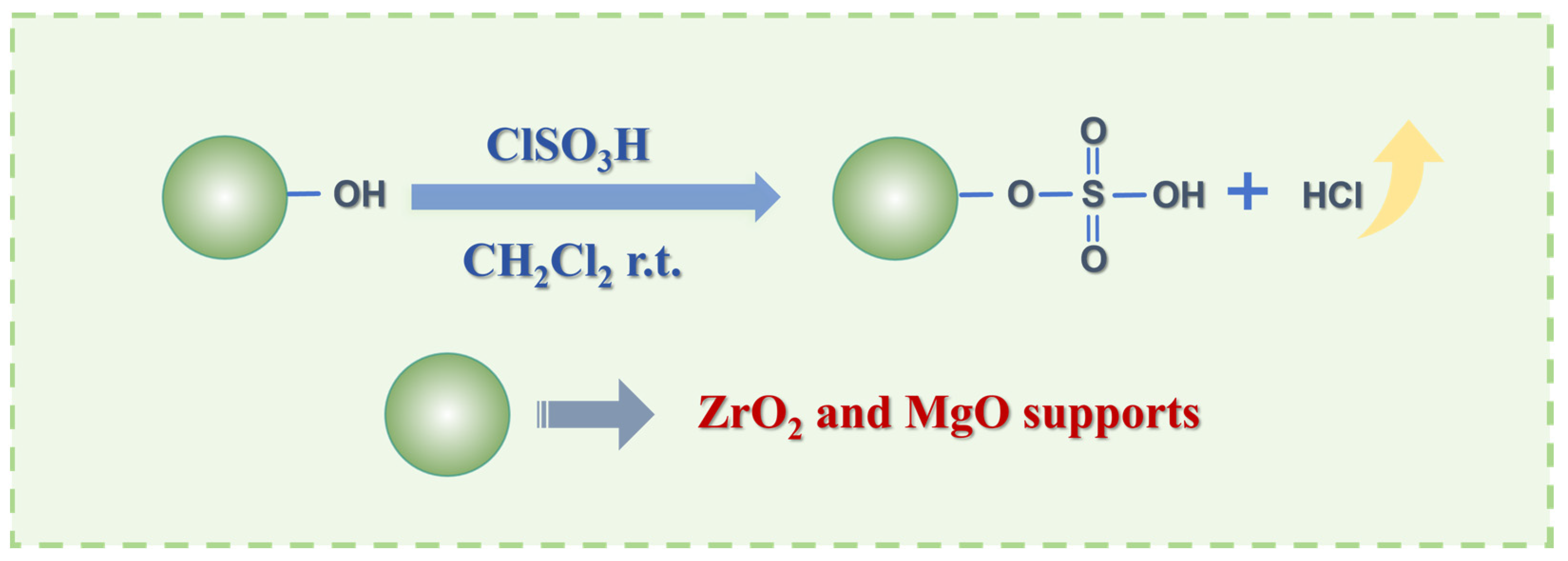
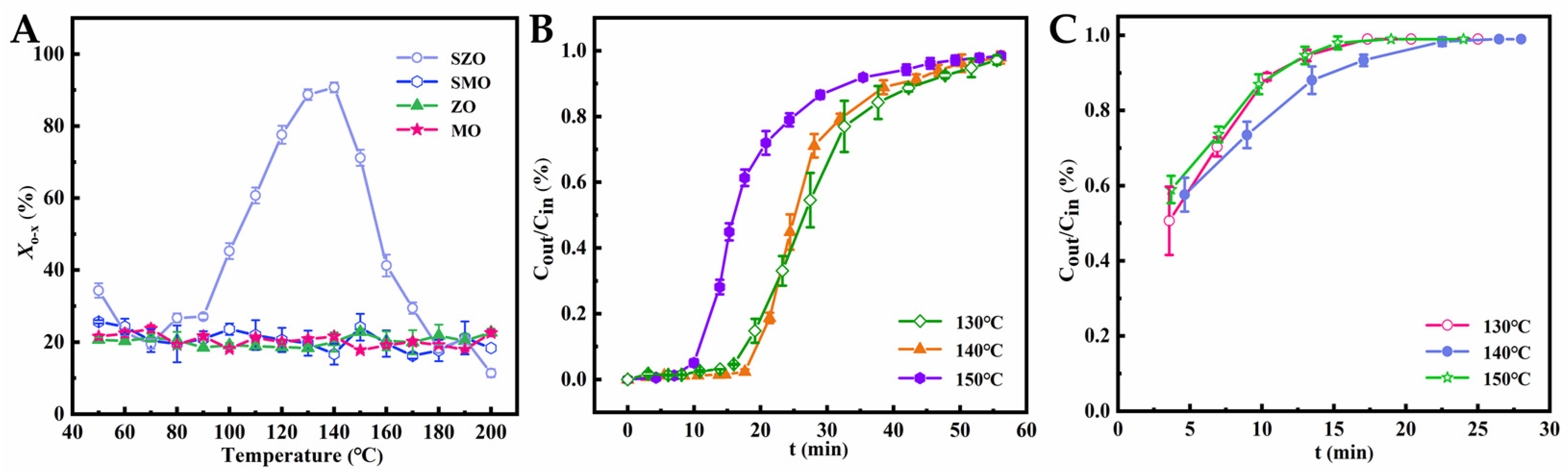
2.2. Comparison of Sulfonic Acid Loading in SZO and SMO Materials
2.3. Comparative Analysis of o-Xylene Removal Reactivity by SZO and SMO
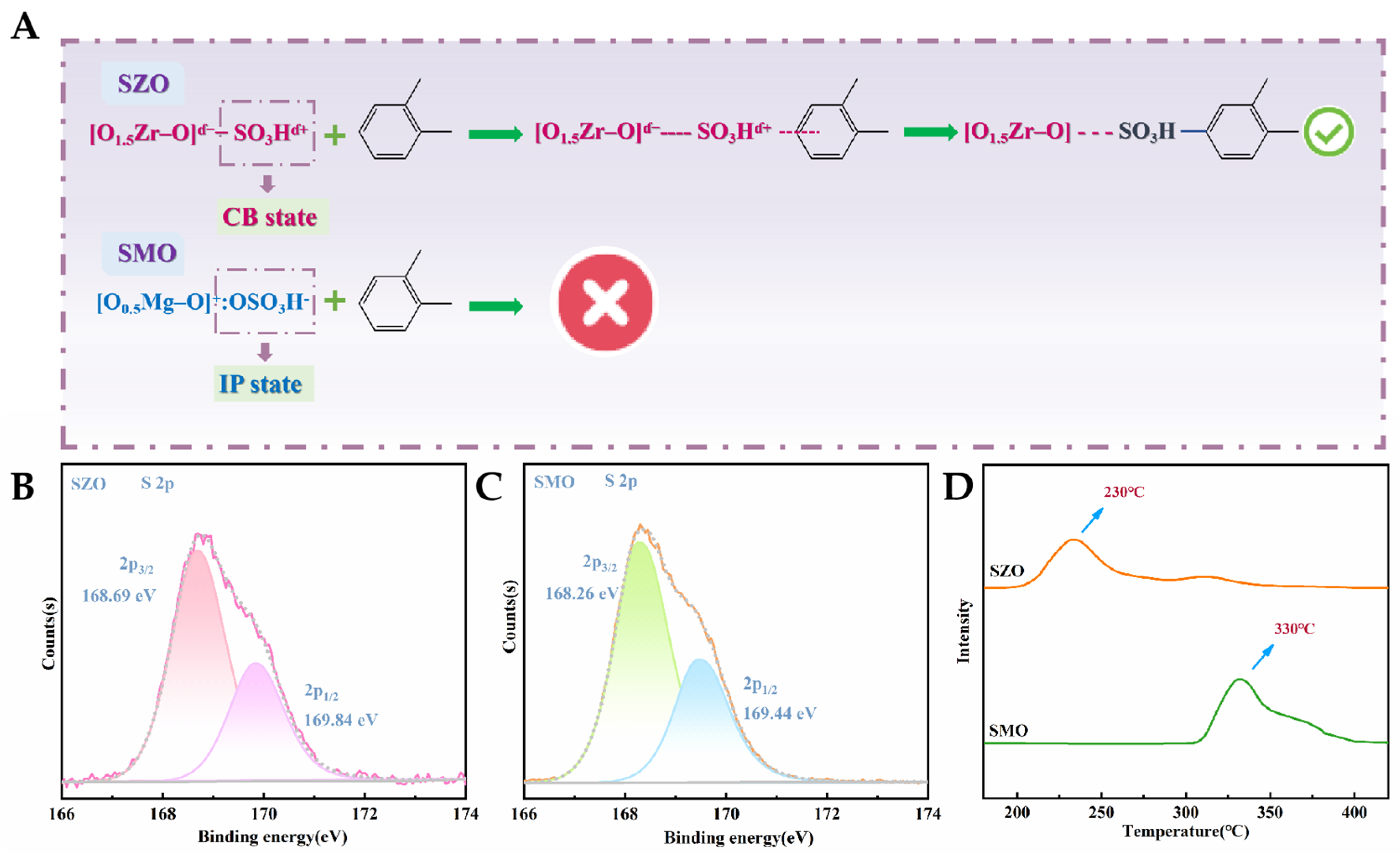
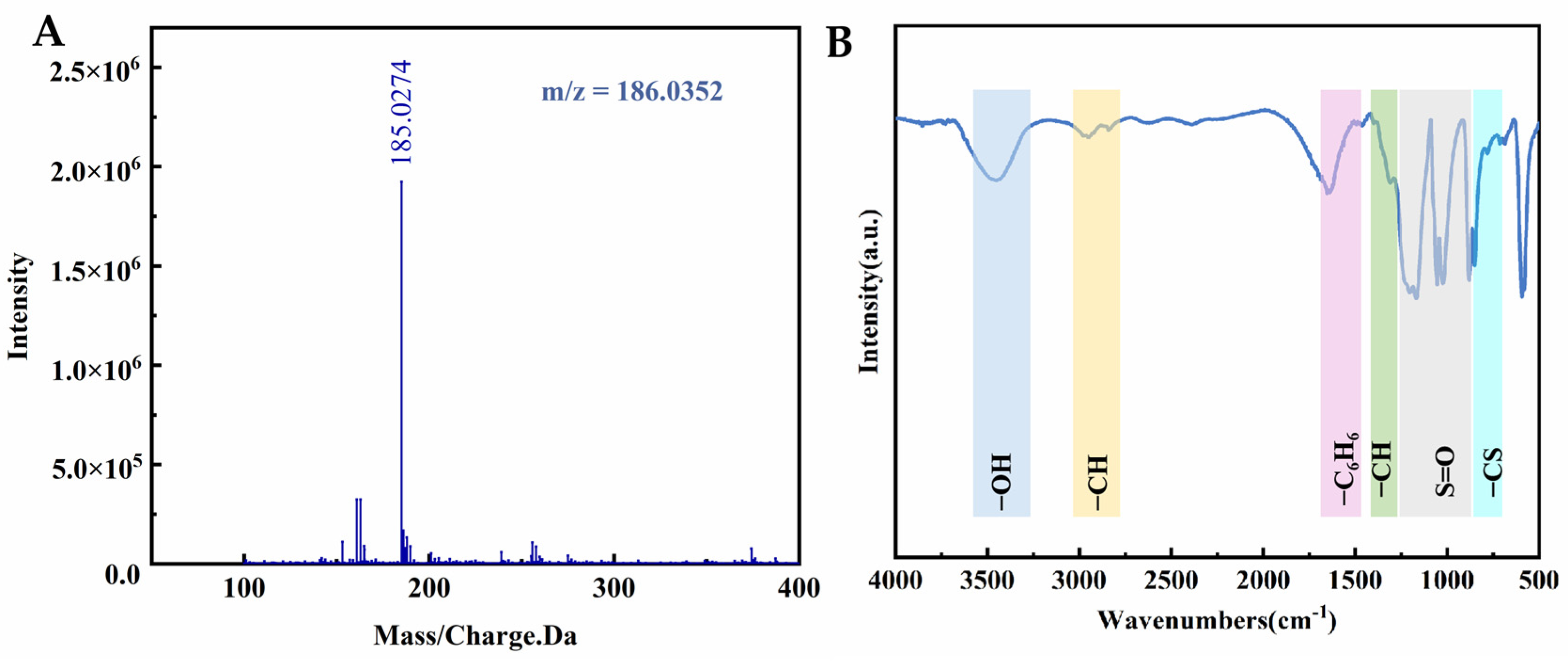
2.4. Investigation into the Fixation State of Sulfonic Acid Groups and Their Operational Mechanism in o-Xylene Elimination Procedures
3. Materials and Methods
3.1. Reagents and Materials
3.2. Synthesis of SMO and SZO
3.3. Determination of the Sulfonic Acid Group Loading Quantity
3.4. Evaluation of the Dynamic Adsorption Capacity for o-Xylene
3.5. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, C.S.; Li, H.Y.; Kim, B.Y.; Jo, Y.M.; Byun, H.G.; Hwang, I.S.; Abdel-Hady, F.; Wazzan, A.A.; Lee, J.H. Discriminative detection of indoor volatile organic compounds using a sensor array based on pure and Fe-doped In2O3 nanofibers. Sens. Actuators B 2019, 285, 193–200. [Google Scholar] [CrossRef]
- Li, N.; Jiang, Q.; Wang, F.; Xie, J.; Li, Y.; Li, J.; Wu, S. Emission behavior, environmental impact and priority-controlled pollutants assessment of volatile organic compounds (VOCs) during asphalt pavement construction based on laboratory experiment. J. Hazard. Mater. 2020, 398, 122904. [Google Scholar] [CrossRef]
- Siu, B.; Chowdhury, A.R.; Yan, Z.; Humphrey, S.M.; Hutter, T. Selective adsorption of volatile organic compounds in metal-organic frameworks (MOFs). Coord. Chem. Rev. 2023, 485, 215119. [Google Scholar] [CrossRef]
- Chang, C.; Chen, B. Toxicity assessment of volatile organic compounds and polycyclic aromatic hydrocarbons in motorcycle exhaust. J. Hazard. Mater. 2008, 153, 1262–1269. [Google Scholar] [CrossRef]
- Partha, D.B.; Cassidy-Bushrow, A.E.; Huang, Y. Global preterm births attributable to BTEX (benzene, toluene, ethylbenzene, and xylene) exposure. Sci. Total Environ. 2022, 838, 156390. [Google Scholar] [CrossRef]
- Moufawad, T.; Costa Gomes, M.; Fourmentin, S. Deep eutectic solvents as absorbents for VOC and VOC mixtures in static and dynamic processes. Chem. Eng. J. 2022, 448, 137619. [Google Scholar] [CrossRef]
- Ha Chi, N.N.; Kim Oanh, N.T. Photochemical smog modeling of PM2.5 for assessment of associated health impacts in crowded urban area of Southeast Asia. Environ. Technol. Innov. 2021, 21, 101214. [Google Scholar] [CrossRef]
- Unnithan, A.; Bekele, D.N.; Chadalavada, S.; Naidu, R. Insights into vapour intrusion phenomena: Current outlook and preferential pathway scenario. Sci. Total Environ. 2021, 796, 148885. [Google Scholar] [CrossRef]
- Khadra, W.M.; Elias, A.R.; Majdalani, M.A. A systematic approach to derive natural background levels in groundwater: Application to an aquifer in North Lebanon perturbed by various pollution sources. Sci. Total Environ. 2022, 847, 157586. [Google Scholar] [CrossRef] [PubMed]
- Passi, A.; Nagendra, S.M.S.; Maiya, M.P. Characteristics of indoor air quality in underground metro stations: A critical review. Build. Environ. 2021, 198, 107907. [Google Scholar] [CrossRef]
- Wang, L.; Huang, C.; Gao, Z.; Cui, B.; Zhao, M.; Xiao, M.; Yu, X. Recent Research on the Anti-Poisoning Catalysts in the Catalytic Oxidation of VOCs: A Review. Catalysts 2025, 15, 234. [Google Scholar] [CrossRef]
- Zhu, L.; Shen, D.; Luo, K.H. A critical review on VOCs adsorption by different porous materials: Species, mechanisms and modification methods. J. Hazard. Mater. 2020, 389, 122102. [Google Scholar] [CrossRef]
- Rodríguez, M.L.; Cadús, L.E.; Borio, D.O. Monolithic reactor for VOCs abatement: Influence of non-uniformity in the coating. J. Environ. Chem. Eng. 2017, 5, 292–302. [Google Scholar] [CrossRef]
- Xie, H.; Gao, W.; Zhao, W.; Han, Y.; Gao, Y.; Liu, B.; Han, Y. Source profile study of VOCs unorganized emissions from typical aromatic devices in petrochemical industry. Sci. Total Environ. 2023, 889, 164098. [Google Scholar] [CrossRef]
- Liu, J.; Wang, T.; Zhong, L.; Serageldin, M.A.; Pan, W.-P. Review of organic pollutants in coal combustion processes and control technologies. Prog. Energy Combust. Sci. 2025, 109, 101213. [Google Scholar] [CrossRef]
- Shen, B.; Zhao, S.; Yang, X.; Carta, M.; Zhou, H.; Jin, W. Relation between permeate pressure and operational parameters in VOC/nitrogen separation by a PDMS composite membrane. Sep. Purif. Technol. 2022, 280, 119974. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Zhou, X.; Xu, X.; Pan, M. Synergistic mechanisms of carbon-based materials for VOCs photocatalytic degradation: A critical review. J. Environ. Manag. 2024, 367, 122087. [Google Scholar] [CrossRef]
- Wu, P.; Jin, X.; Qiu, Y.; Ye, D. Recent Progress of Thermocatalytic and photo/thermocatalytic oxidation for VOCs purification over manganese-based oxide catalysts. Environ. Sci. Technol. 2021, 55, 4268–4286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; You, J.; Kennes, C.; Cheng, Z.; Ye, J.; Chen, D.; Chen, J.; Wang, L. Current advances of VOCs degradation by bioelectrochemical systems: A review. Chem. Eng. J. 2018, 334, 2625–2637. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, Y.; Qin, J.; Yang, Z.; Fu, M. TiO2-UiO-66-NH2 nanocomposites as efficient photocatalysts for the oxidation of VOCs. Chem. Eng. J. 2020, 385, 123814. [Google Scholar] [CrossRef]
- Liu, L.; Shao, G.; Ma, C.; Nikiforov, A.; De Geyter, N.; Morent, R. Plasma-catalysis for VOCs decomposition: A review on micro- and macroscopic modeling. J. Hazard. Mater. 2023, 451, 131100. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhao, Y.; Sun, J.; Shen, Y.; Zhao, X.; Wang, W.; Song, Z.; Mao, Y. A Promising Monolithic Catalyst for Advanced VOCs Oxidation by Graphene-Doped α-MnO2 Loaded on Cordierite Honeycomb. Catalysts 2025, 15, 321. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, L.; Xu, J.; Shen, B. A Mini-Review of Recent Progress in Zeolite-Based Catalysts for Photocatalytic or Photothermal Environmental Pollutant Treatment. Catalysts 2025, 15, 158. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Guo, Y.; Zhu, T.; Xu, W. Adsorption and desorption characteristics of hydrophobic hierarchical zeolites for the removal of volatile organic compounds. Chem. Eng. J. 2021, 411, 215119. [Google Scholar] [CrossRef]
- Xu, M.; Wu, H.C.; Lin, Y.S.; Deng, S. Simulation and optimization of pressure swing adsorption process for high-temperature air separation by perovskite sorbents. Chem. Eng. J. 2018, 354, 62–74. [Google Scholar] [CrossRef]
- Zhu, X.; Li, S.; Shi, Y.; Cai, N. Recent advances in elevated-temperature pressure swing adsorption for carbon capture and hydrogen production. Prog. Energy Combust. Sci. 2019, 75, 200784. [Google Scholar] [CrossRef]
- Zhan, G.; Bai, L.; Wu, B.; Cao, F.; Duan, Y.; Chang, F.; Shang, D.; Bai, Y.; Li, Z.; Zhang, X.; et al. Dynamic process simulation and optimization of CO2 removal from confined space with pressure and temperature swing adsorption. Chem. Eng. J. 2021, 416, 129104. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, W.; Alston, S.; Xiao, Y.; Ajayan, P.; Bu, X.; Feng, P. Multi-stage optimization of pore size and shape in pore-space-partitioned metal–organic frameworks for highly selective and sensitive benzene capture. Angew. Chem. Int. Ed. 2024, 64, e202415576. [Google Scholar] [CrossRef]
- Hu, L.; Wu, W.; Hu, M.; Jiang, L.; Lin, D.; Wu, J.; Yang, K. Double-walled Al-based MOF with large microporous specific surface area for trace benzene adsorption. Nat. Commun. 2024, 15, 3204. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, L.; Fu, Y.; Sun, Z.; Wang, T.; Shi, Y.; Yang, G.; Ma, R.; Liu, Z.; Wu, L.; et al. Fluorine doping modulating pore structure and adsorption capability of carbon matrix boosting potassium storage performance of red phosphorus anode. Adv. Funct. Mater. 2024, 34, 2409090. [Google Scholar] [CrossRef]
- Kim, N.S.; Numan, M.; Nam, S.C.; Park, S.-E.; Jo, C. Dynamic adsorption/desorption of p-xylene on nanomorphic MFI zeolites: Effect of zeolite crystal thickness and mesopore architecture. J. Hazard. Mater. 2021, 403, 123659. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, Y.; Zhang, H.; Cheng, F.; Jiao, Z. Coke powder improving the performance of desulfurized activated carbon from the cyclic thermal regeneration. Chem. Eng. J. 2022, 448, 137459. [Google Scholar] [CrossRef]
- Gao, K.; Ma, M.; Liu, Y.; Ma, Z. A comparative study of the removal of o-xylene from gas streams using mesoporous silicas and their silica supported sulfuric acids. J. Hazard. Mater. 2021, 409, 124965. [Google Scholar] [CrossRef]
- Ma, M.; Gao, K.; Ma, Z.; Ding, J. Influence of preparation method on the adsorptive performance of silica sulfuric acid for the removal of gaseous o-xylene. Sep. Purif. Technol. 2021, 265, 118484. [Google Scholar] [CrossRef]
- Zhao, D.; Ma, M.; Qian, J.; Wang, Y.; Ma, Z.; Ma, X. Influence of impregnation medium on the adsorptive performance of silica sulfuric acid for the removal of gaseous o-xylene: Comparison on ethyl acetate and water. Catalysts 2022, 12, 737. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, X.; Wang, H.; Zhao, D.; Liu, Y.; Ma, Z. Enhancement of gaseous o-xylene elimination by chlorosulfonic acid-modified H-zeolite socony mobil-5. Molecules 2024, 29, 3507. [Google Scholar] [CrossRef]
- Basahel, S.N.; Ali, T.T.; Mokhtar, M.; Narasimharao, K. Influence of crystal structure of nanosized ZrO2 on photocatalytic degradation of methyl orange. Nanoscale Res. Lett. 2015, 10, 73. [Google Scholar] [CrossRef]
- Meng, G.; Lan, W.; Zhang, L.; Wang, S.; Zhang, T.; Zhang, S.; Xu, M.; Wang, Y.; Zhang, J.; Yue, F.; et al. Synergy of single atoms and lewis acid sites for efficient and selective lignin disassembly into monolignol derivatives. J. Am. Chem. Soc. 2023, 145, 12884–12893. [Google Scholar] [CrossRef]
- Kostyukov, A.I.; Nashivochnikov, A.A.; Rakhmanova, M.I.; Panchenko, V.N.; Pochtar, A.A.; Cherepanova, S.V.; Snytnikov, V.N. Photoluminescence of tetragonal ZrO2 nanoparticles: Contribution from OH-groups, oxygen vacancies, and impurities. Opt. Mater. 2025, 159, 116648. [Google Scholar] [CrossRef]
- Brown, M.A.; Fujimori, Y.; Ringleb, F.; Shao, X.; Stavale, F.; Nilius, N.; Sterrer, M.; Freund, H.-J. Oxidation of Au by surface OH: Nucleation and electronic structure of gold on hydroxylated MgO(001). J. Am. Chem. Soc. 2011, 133, 10668–10676. [Google Scholar] [CrossRef]
- Li, D.; Yao, J.; Liu, B.; Sun, H.; van Agtmaal, S.; Feng, C. Preparation and characterization of surface grafting polymer of ZrO2 membrane and ZrO2 powder. Appl. Surf. Sci. 2019, 471, 394–402. [Google Scholar] [CrossRef]
- Dabhane, H.; Ghotekar, S.; Zate, M.; Kute, S.; Jadhav, G.; Medhane, V. Green synthesis of MgO nanoparticles using aqueous leaf extract of Ajwain (Trachyspermum ammi) and evaluation of their catalytic and biological activities. Inorg. Chem. Commun. 2022, 138, 109270. [Google Scholar] [CrossRef]
- Dacquin, J.P.; Cross, H.E.; Brown, D.R.; Düren, T.; Williams, J.J.; Lee, A.F.; Wilson, K. Interdependent lateral interactions, hydrophobicity and acid strength and their influence on the catalytic activity of nanoporous sulfonic acid silicas. Green Chem. 2010, 12, 1383–1391. [Google Scholar] [CrossRef]
- Lv, S.; Ma, X.; Wang, Y.; Zheng, Y.; Ma, Z.; Liu, T. Cyclic siloxane removal by ring-opening polymerization on silica gel-supported sulfuric acid. Chem. Eng. J. 2025, 504, 158842. [Google Scholar] [CrossRef]
- Lv, S.; Zhang, R.; He, Y.; Ma, Z.; Ma, X. Efficient reactive adsorption of hexamethyldisiloxane on MCM-41 supported sulfuric acid. Renew. Energ. 2024, 224, 120174. [Google Scholar] [CrossRef]
- Allred, A.L. Electronegativity values from thermochemical data. J. Inorg. Nucl. Chem. 1961, 17, 215–221. [Google Scholar] [CrossRef]
- Parreño, R.P. The correlation of sulfonation reaction kinetics with the degree of sulfonation (DS) and its effects on microstructure and morphology of electrospun fibers for the membrane of fuel cells. RSC Adv. 2023, 13, 2523–2529. [Google Scholar] [CrossRef]
- Wang, P.C.; Chen, J.; Lu, M. Electrophilic aromatic nitration: Substituent effects of monosubstituted benzenes. J. Chin. Chem. Soc. 2013, 57, 967–971. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, D.; Tan, M.; Jiang, B.; Zheng, J.; Tsubaki, N.; Wu, M. Monodispersed hollow SO3H-functionalized carbon/silica as efficient solid acid catalyst for esterification of oleic acid. ACS Appl. Mater. Interfaces 2015, 7, 26767–26775. [Google Scholar] [CrossRef]
- Wang, Y.; Chai, J.; Li, Y.; Ma, Z. Influence of aluminum incorporation on the adsorptive performance of silica-based supported sulfonic acid for the chemical recovery of gaseous o-xylene. Molecules 2025, 30, 1073. [Google Scholar] [CrossRef]
- Karthick, N.K.; Arivazhagan, G.; Kannan, P.P.; Kumbharkhane, A.C.; Joshi, Y.S. Homo/hetero interactions in the binary solutions of toluene with acetonitrile: FTIR spectroscopic, theoretical and dielectric studies. J. Mol. Struct. 2019, 1192, 208–216. [Google Scholar] [CrossRef]
- Yin, Z.; Liu, B.; Fan, S.; Wang, P.; Wang, X.; Long, D.; Zhang, L.; Yang, X.; Li, X. In situ FTIR spectra investigation of the photocatalytic degradation of gaseous toluene over a novel hedgehog-like CaFe2O4 hollow-structured materials. Catal. Commun. 2019, 130, 105754. [Google Scholar] [CrossRef]
- Sáez del Bosque, I.F.; Martínez-Ramírez, S.; Blanco-Varela, M.T. FTIR study of the effect of temperature and nanosilica on the nano structure of C–S–H gel formed by hydrating tricalcium silicate. Constr. Build. Mater. 2014, 52, 314–323. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Bai, N.; Zhang, X.; Han, X.; da Silva, I.; Morris, C.G.; Xu, S.; Wilary, D.M.; Sun, Y.; et al. Refinement of pore size at sub-angstrom precision in robust metal–organic frameworks for separation of xylenes. Nat. Commun. 2020, 11, 4280. [Google Scholar] [CrossRef]
- Wang, C.; Chang, K.; Chung, T. Adsorption equilibria of aromatic compounds on activated carbon. silica gel, and 13X zeolite. J. Chem. Eng. Data 2004, 49, 527–531. [Google Scholar] [CrossRef]
- Gatica, J.M.; Rodríguez-Izquierdo, J.M.; Sánchez, D.; Chafik, T.; Harti, S.; Zaitan, H.; Vidal, H. Originally prepared carbon-based honeycomb monoliths with potential application as VOCs adsorbents. C. R. Chim. 2006, 9, 1215–1220. [Google Scholar] [CrossRef]
- Zaitan, H.; Bianchi, D.; Achak, O.; Chafik, T. A comparative study of the adsorption and desorption of o-xylene onto bentonite clay and alumina. J. Hazard. Mater. 2008, 153, 852–859. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T/(°C) | tB/(min) | QB/(mg/g) |
|---|---|---|---|
| SZO | 130 | 17.72 | 69.64 ± 0.22 |
| 140 | 20.52 | 81.18 ± 0.26 | |
| 150 | 9.65 | 38.17 ± 0.18 | |
| SMO | 130 | 0 | 0 |
| 140 | 0 | 0 | |
| 150 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zhang, X.; Shen, Z.; Ma, Z. A Comparative Study on ZrO2- and MgO-Based Sulfonic Acid Materials for the Reactive Adsorption of o-Xylene. Molecules 2025, 30, 3171. https://doi.org/10.3390/molecules30153171
Wang H, Zhang X, Shen Z, Ma Z. A Comparative Study on ZrO2- and MgO-Based Sulfonic Acid Materials for the Reactive Adsorption of o-Xylene. Molecules. 2025; 30(15):3171. https://doi.org/10.3390/molecules30153171
Chicago/Turabian StyleWang, Hongmei, Xiaoxu Zhang, Ziqi Shen, and Zichuan Ma. 2025. "A Comparative Study on ZrO2- and MgO-Based Sulfonic Acid Materials for the Reactive Adsorption of o-Xylene" Molecules 30, no. 15: 3171. https://doi.org/10.3390/molecules30153171
APA StyleWang, H., Zhang, X., Shen, Z., & Ma, Z. (2025). A Comparative Study on ZrO2- and MgO-Based Sulfonic Acid Materials for the Reactive Adsorption of o-Xylene. Molecules, 30(15), 3171. https://doi.org/10.3390/molecules30153171