
Synthesis of Monothiacalix[4]arene Using the Fragment Condensation Approach

 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
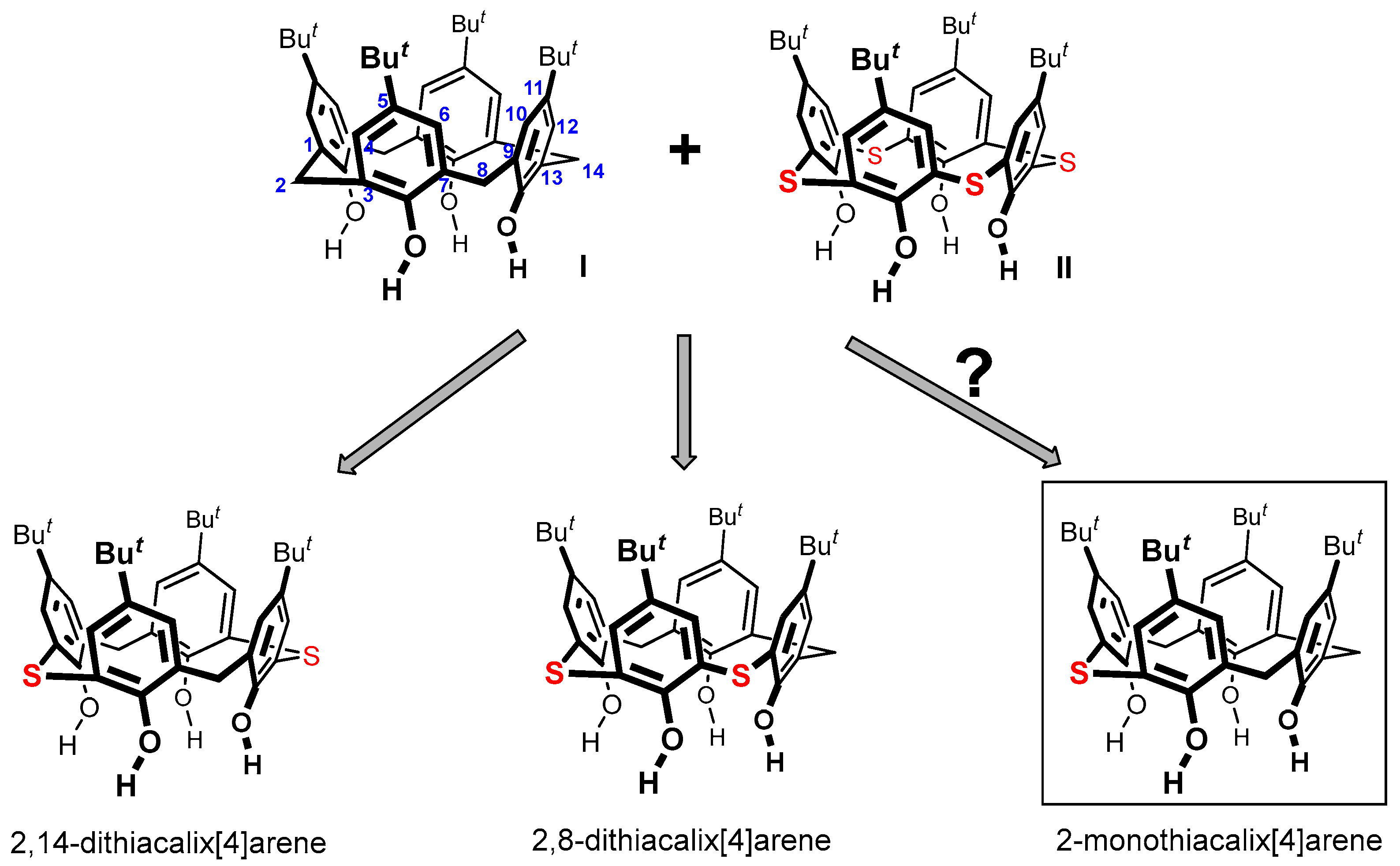
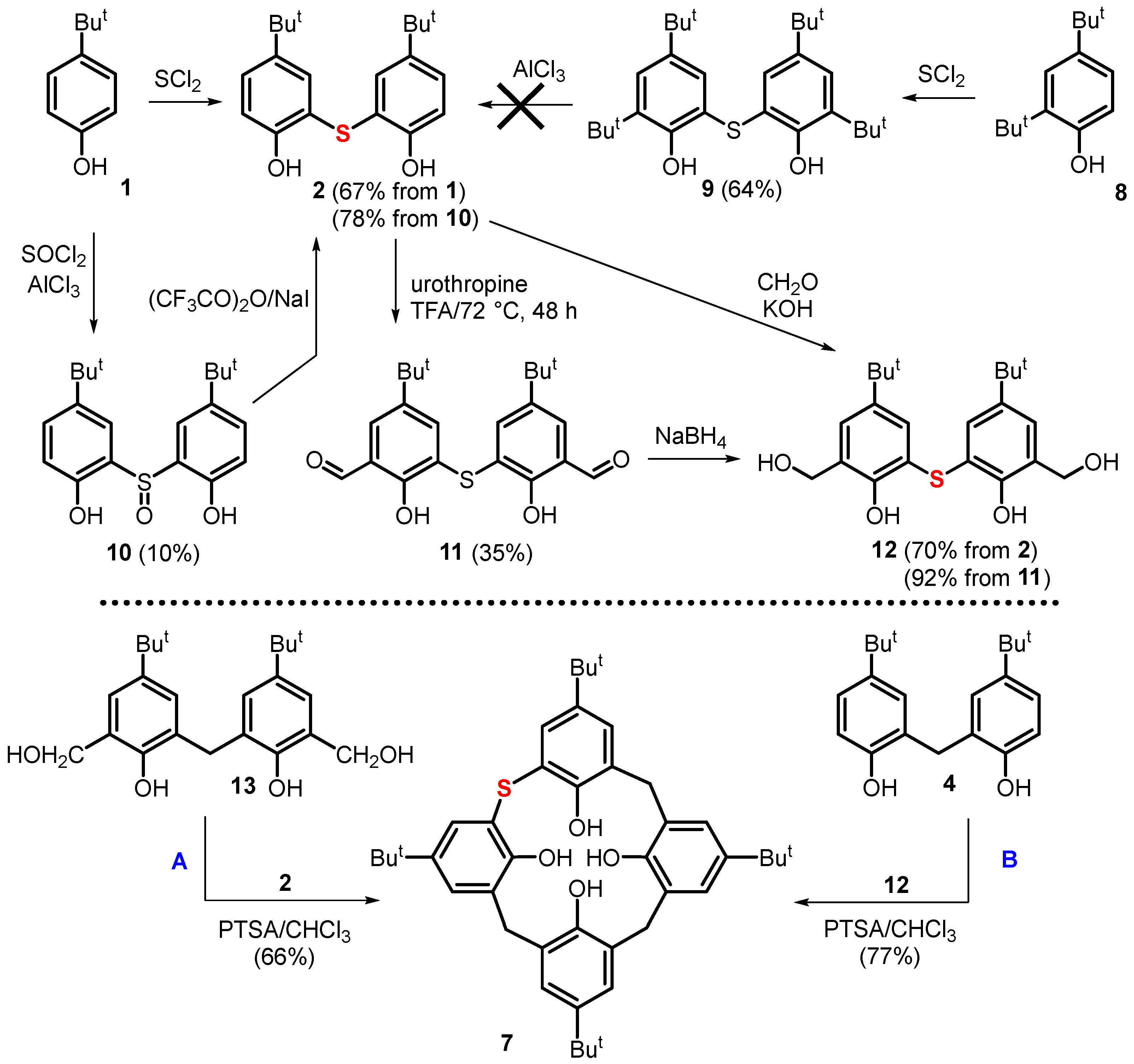
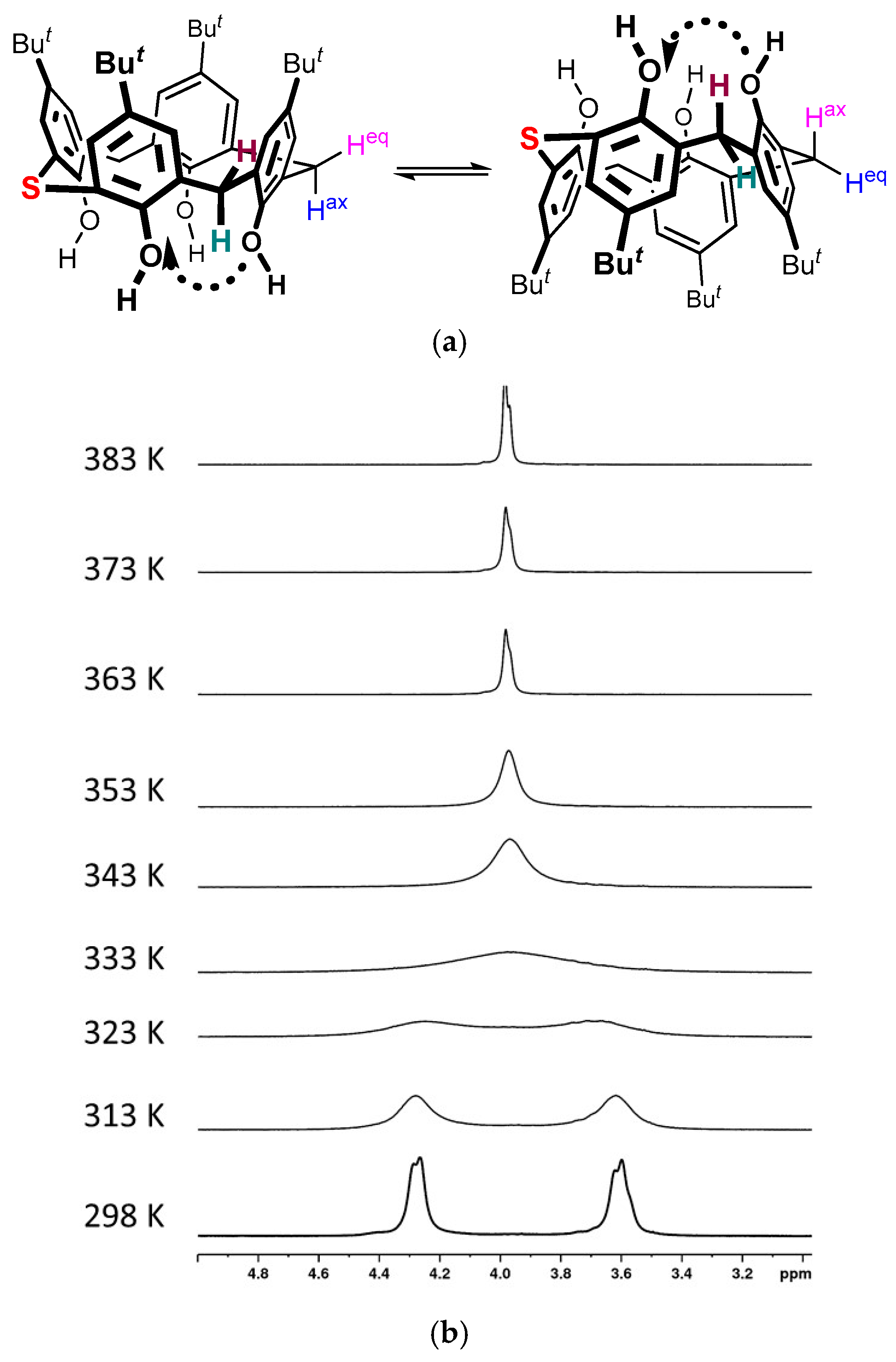
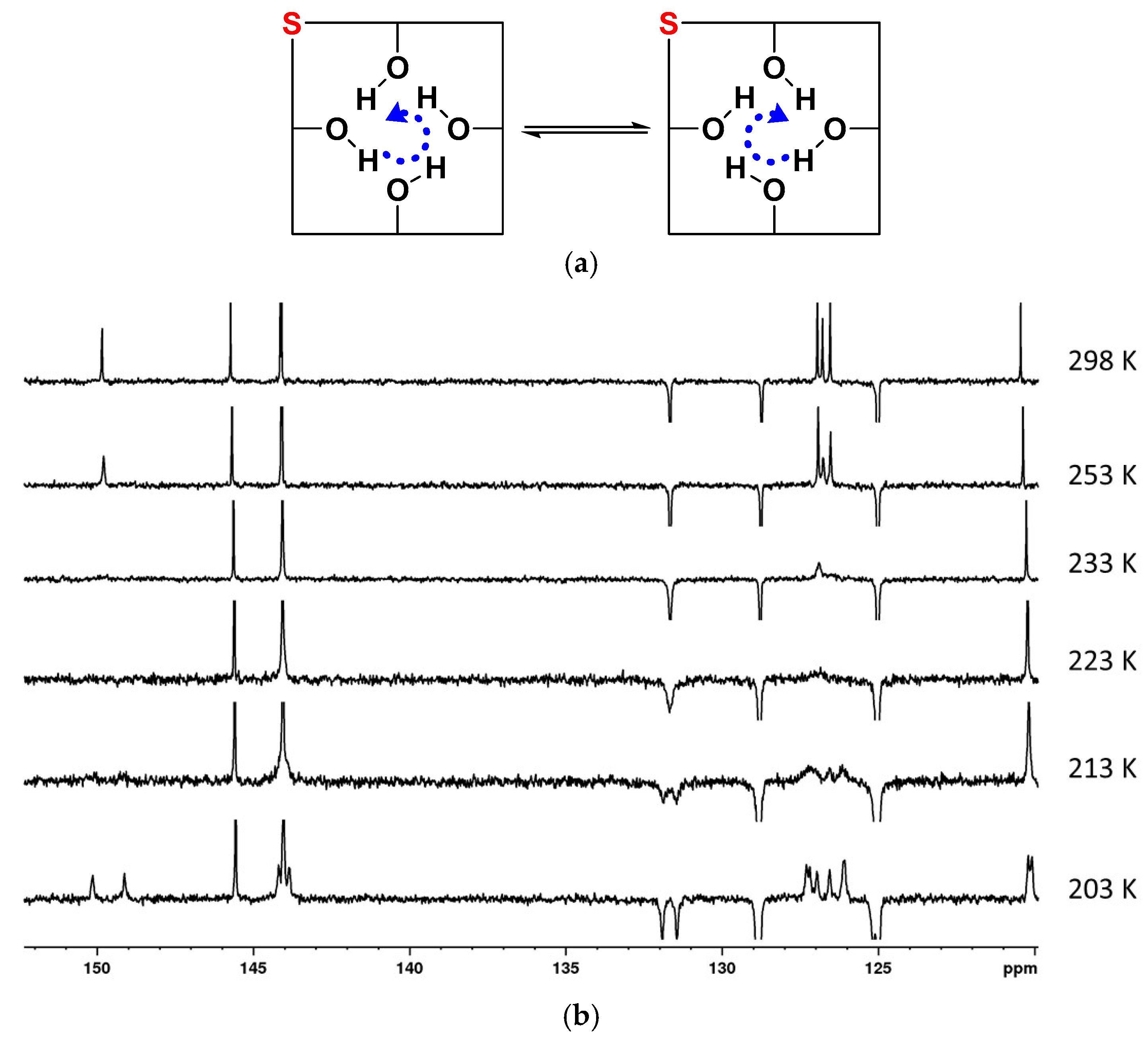
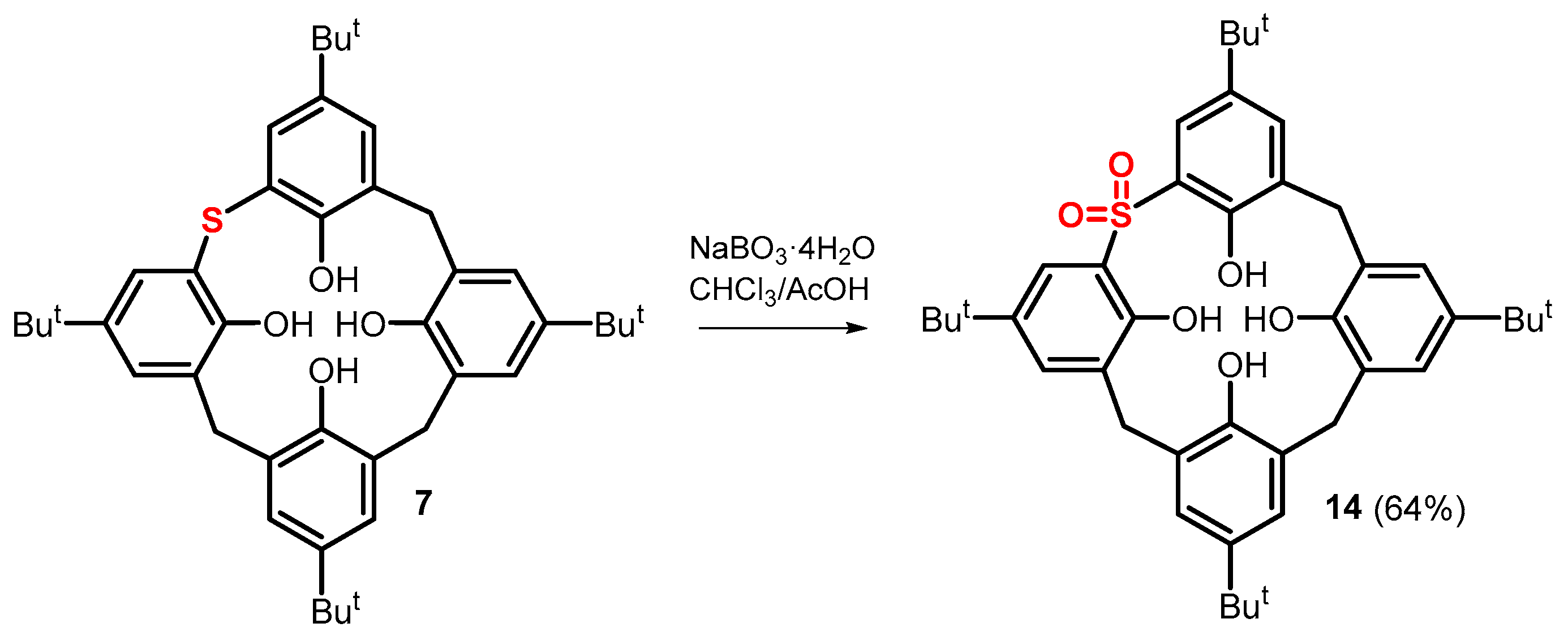
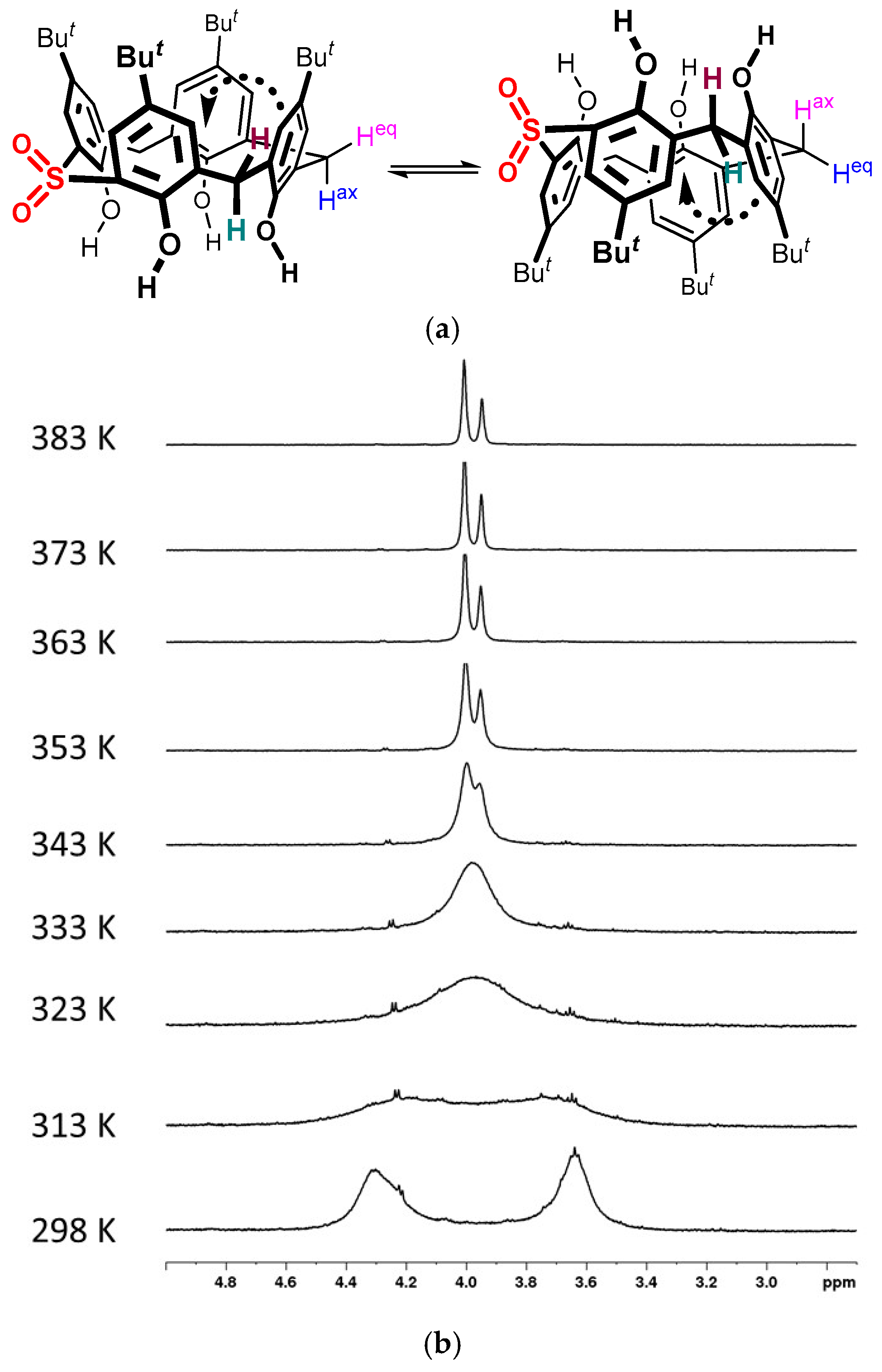
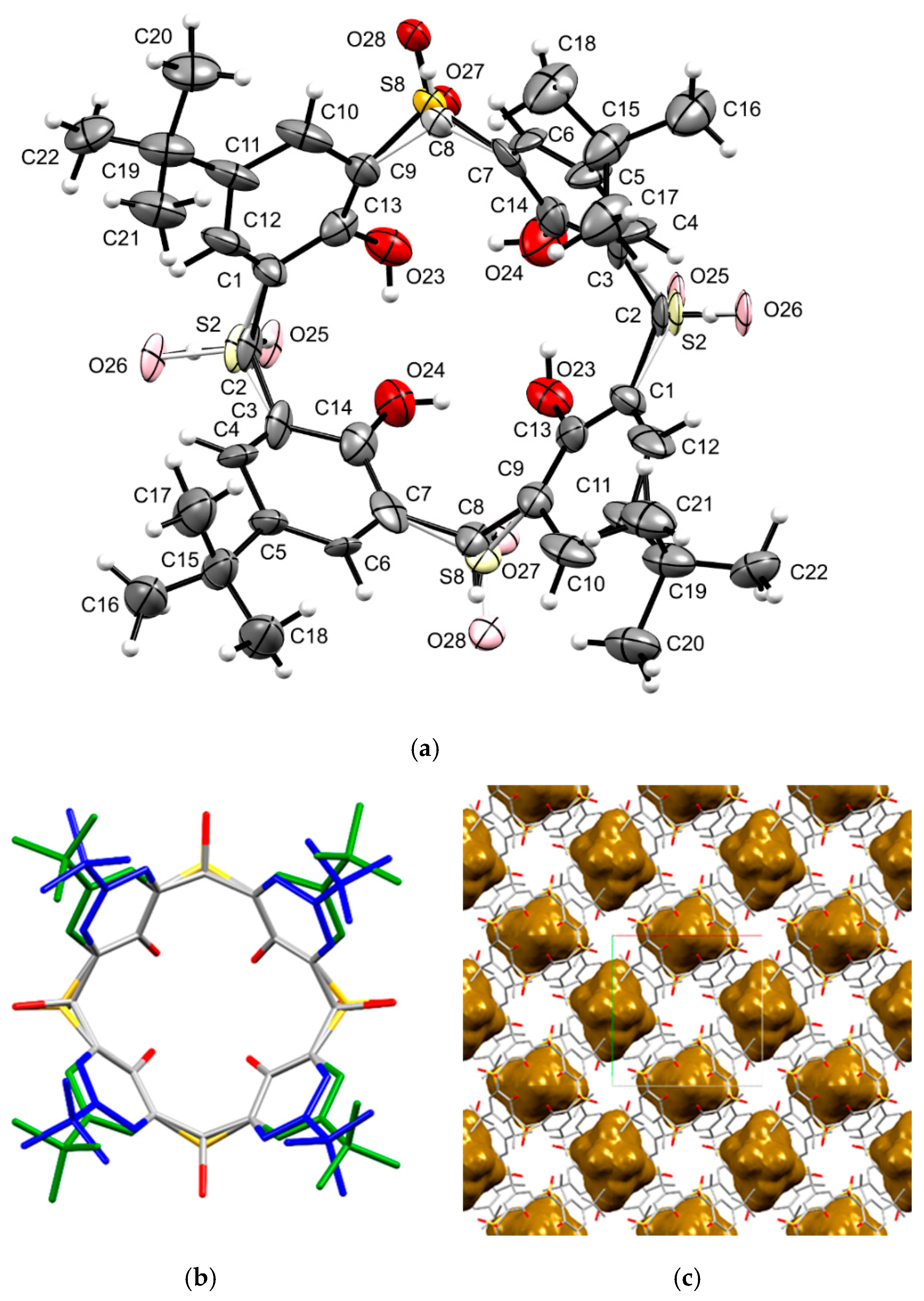
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Synthetic Procedures
3.2.1. Preparation of Compound 2
From Compound 1
From Compound 10
3.2.2. Preparation of Compound 7
Route A (from Compounds 2 and 13)
Route B (from Compounds 4 and 12)
3.2.3. Preparation of Compound 9
3.2.4. Preparation of Compound 10
3.2.5. Preparation of Compound 11
3.2.6. Preparation of Compound 12
From Compound 2
From Compound 11
3.2.7. Preparation of Compound 14
3.3. X-Ray Measurements
Crystallographic Data for 14
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gutsche, C.D. Calixarenes: An Introduction; Monographs in Supramolecular Chemistry; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Neri, P.; Sessler, J.L.; Wang, M.X. Calixarenes and Beyond; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Mandolini, L.; Ungaro, R. Calixarenes in Action; World Scientific Publishing Company: Singapore, 2000. [Google Scholar]
- Asfari, Z.; Böhmer, V.; Harrowfield, J.; Vicens, J. Calixarenes 2001; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Kumagai, H.; Hasegawa, M.; Miyanari, S.; Sugawa, Y.; Sato, Y.; Hori, T.; Ueda, S.; Kamiyama, H.; Miyano, S. Facile synthesis of p-tert-butylthiacalix[4]arene by the reaction of p-tert-butylphenol with elemental sulfur in the presence of a base. Tetrahedron Lett. 1997, 38, 3971–3972. [Google Scholar] [CrossRef]
- Lhoták, P. Chemistry of Thiacalixarenes. Eur. J. Org. Chem. 2004, 2004, 1675–1692. [Google Scholar] [CrossRef]
- Morohashi, N.; Narumi, F.; Iki, N.; Hattori, T.; Miyano, S. Thiacalixarenes. Chem. Rev. 2006, 106, 5291–5316. [Google Scholar] [CrossRef]
- Kumar, R.; Lee, Y.O.; Bhalla, V.; Kumar, M.; Kim, J.S. Recent developments of thiacalixarene based molecular motifs. Chem. Soc. Rev. 2014, 43, 4824–4870. [Google Scholar] [CrossRef]
- Hucko, M.; Dvorakova, H.; Eigner, V.; Lhotak, P. 2,14-Dithiacalix[4]arene and its homooxa analogues: Synthesis and dynamic NMR study of conformational behaviour. Chem. Commun. 2015, 51, 7051–7053. [Google Scholar] [CrossRef]
- Kortus, D.; Krizova, K.; Dvorakova, H.; Eigner, V.; Lhotak, P. Synthesis of 2,8-dithiacalix[4]arene based on fragment condensation. Tetrahedron Lett. 2021, 69, 152924. [Google Scholar] [CrossRef]
- Kortus, D.; Miksatko, J.; Kundrat, O.; Babor, M.; Eigner, V.; Dvorakova, H.; Lhotak, P. Chemistry of 2,14-Dithiacalix[4]arene: Alkylation and Conformational Behavior of Peralkylated Products. J. Org. Chem. 2019, 84, 11572–11580. [Google Scholar] [CrossRef]
- Kortus, D.; Kundrat, O.; Cejka, J.; Dvorakova, H.; Lhotak, P. Chemistry of 2,14-Dithiacalix[4]arene: Searching for the Missing Fifth Conformer. J. Org. Chem. 2021, 86, 9788–9801. [Google Scholar] [CrossRef]
- Kortus, D.; Kundrat, O.; Tlusty, M.; Cejka, J.; Dvorakova, H.; Lhotak, P. Inherent chirality through a simple dialkylation of 2,14-dithiacalix[4]arene. New J. Chem. 2020, 44, 14496–14504. [Google Scholar] [CrossRef]
- Sone, T.; Ohba, Y.; Moriya, K.; Kumada, H.; Ito, K. Synthesis and properties of sulfur-bridged analogs of p-tert-Butylcalix[4] arene. Tetrahedron 1997, 53, 10689–10698. [Google Scholar] [CrossRef]
- Ohba, Y.; Moriya, K.; Sone, T. Synthesis and Inclusion Properties of Sulfur-Bridged Analogs of Acyclic Phenol-Formaldehyde Oligomers. Bull. Chem. Soc. Jpn. 1991, 64, 576–582. [Google Scholar] [CrossRef]
- Morohashi, N.; Kojima, M.; Suzuki, A.; Ohba, Y. Conversion of Mono- and Tetra-Thiacalix[4]arenes to Sulfilimine Derivatives and Unexpected Formation of Monospirodienone Derivatives. Heterocycl. Commun. 2005, 11, 249–254. [Google Scholar] [CrossRef]
- Wasserman, E.P.; Annis, I.; Chopin, L.J.; Price, P.C.; Petersen, J.L.; Abboud, K.A. Ethylene Oxide Polymerization Catalyzed by Aluminum Complexes of Sulfur-Bridged Polyphenols. Macromolecules 2005, 38, 322–333. [Google Scholar] [CrossRef]
- Ballmann, J.; Fuchs, M.G.G.; Dechert, S.; John, M.; Meyer, F. Synthesis and Coordination Properties of Chelating Dithiophenolate Ligands. Inorg. Chem. 2009, 48, 90–99. [Google Scholar] [CrossRef]
- Sartori, G.; Bigi, F.; Maggi, R.; Porta, C. Metal-template ortho-regioselective mono- and bis-de-tert-butylation of poly-tert-butylated phenols. Tetrahedron Lett. 1994, 35, 7073–7076. [Google Scholar] [CrossRef]
- Bikas, R.; Hosseini-Monfared, H.; Sanchiz, J.; Siczek, M.; Lis, T. Synthesis, crystal structure and magnetic properties of a trinuclear phenolate bridged manganese complex containing Mn(ii)–Mn(iii) ions. RSC Adv. 2014, 4, 36175–36182. [Google Scholar] [CrossRef]
- Drabowicz, J.; Oae, S. Mild Reductions of Sulfoxides with Trifluoroacetic Anhydride/Sodium Iodide System. Synthesis 1977, 1977, 404–405. [Google Scholar] [CrossRef]
- Ingenfeld, B.; Straub, S.; Frömbgen, C.; Lützen, A. Synthesis of Monofunctionalized Calix[5]arenes. Synthesis 2018, 50, 676–684. [Google Scholar]
- Fischer, S.; Grootenhuis, P.D.J.; Groenen, L.C.; van Hoorn, W.P.; vn van Veggel, F.C.J.M.; Reinhoudt, D.N.; Karplus, M. Pathways for Conformational Interconversion of Calix[4]arenes. J. Am. Chem. Soc. 1995, 117, 1611–1620. [Google Scholar] [CrossRef]
- Kusano, T.; Tabatabai, M.; Okamoto, Y.; Böhmer, V. The Cone-to-Cone Interconversion of Partially O-Methylated Calix[4]arenes: First Experimental Values for the Energy Barriers. J. Am. Chem. Soc. 1999, 121, 3789–3790. [Google Scholar] [CrossRef]
- Israeli, G.; Shalev, O.; Biali, S.E. Enantiomerization Barrier of all-cis C-Me-tetrahydroxy p-tert-butylcalix[4]arene Atropisomeric Equilibrium of its Tetraacetoxy Derivatives. Eur. J. Org. Chem. 2020, 2020, 1968–1975. [Google Scholar] [CrossRef]
- Lang, J.; Deckerova, V.; Czernek, J.; Lhotak, P. Dynamics of circular hydrogen bond array in calix[4]arene in a nonpolar solvent: A nuclear magnetic resonance study. J. Chem. Phys. 2005, 122, 044506. [Google Scholar] [CrossRef]
- Watanabe, D.; Ito, T.; Ito, K.; Ohba, Y. Synthesis and conformational behavior of sulfinyl- or sulfonyl-bridge containing p-tert-butylcalix[4]arenes. Heterocycl. Commun. 2002, 8, 13–18. [Google Scholar] [CrossRef]
- Pastor, S.D.; Spivack, J.D.; Steinhuebel, L.P. Eight-membered organosulfur heterocycles Synthesis of dibenzo [d,g][1,3,6,2] dioxathiaphosphocin and dibenzo [d,g][1,3,6,2] dioxathiasilocin ring systems. J. Heterocycl. Chem. 1984, 21, 1285–1287. [Google Scholar] [CrossRef]
- Bruker. APEX4, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2021. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIRPOW.92–a program for automatic solution of crystal structures by direct methods optimized for powder data. J. Appl. Crystallogr. 1994, 27, 435–436. [Google Scholar] [CrossRef]
- Betteridge, P.; Carruthers, J.; Cooper, R.; Prout, K.; Watkin, D. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Rohlicek, J.; Husak, M. MCE2005–a new version of a program for fast interactive visualization of electron and similar density maps optimized for small molecules. J. Appl. Crystallogr. 2007, 40, 600–601. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kortus, D.; Moravec, O.; Varga, H.; Churý, M.; Mamleev, K.; Čejka, J.; Dvořáková, H.; Lhoták, P. Synthesis of Monothiacalix[4]arene Using the Fragment Condensation Approach. Molecules 2025, 30, 3145. https://doi.org/10.3390/molecules30153145
Kortus D, Moravec O, Varga H, Churý M, Mamleev K, Čejka J, Dvořáková H, Lhoták P. Synthesis of Monothiacalix[4]arene Using the Fragment Condensation Approach. Molecules. 2025; 30(15):3145. https://doi.org/10.3390/molecules30153145
Chicago/Turabian StyleKortus, Daniel, Oliver Moravec, Hynek Varga, Michal Churý, Kamil Mamleev, Jan Čejka, Hana Dvořáková, and Pavel Lhoták. 2025. "Synthesis of Monothiacalix[4]arene Using the Fragment Condensation Approach" Molecules 30, no. 15: 3145. https://doi.org/10.3390/molecules30153145
APA StyleKortus, D., Moravec, O., Varga, H., Churý, M., Mamleev, K., Čejka, J., Dvořáková, H., & Lhoták, P. (2025). Synthesis of Monothiacalix[4]arene Using the Fragment Condensation Approach. Molecules, 30(15), 3145. https://doi.org/10.3390/molecules30153145