
Anti-Trypanosoma cruzi Potential of New Pyrazole-Imidazoline Derivatives

 , , ,
, , , 
 and
and
Abstract

1. Introduction
2. Results and Discussion
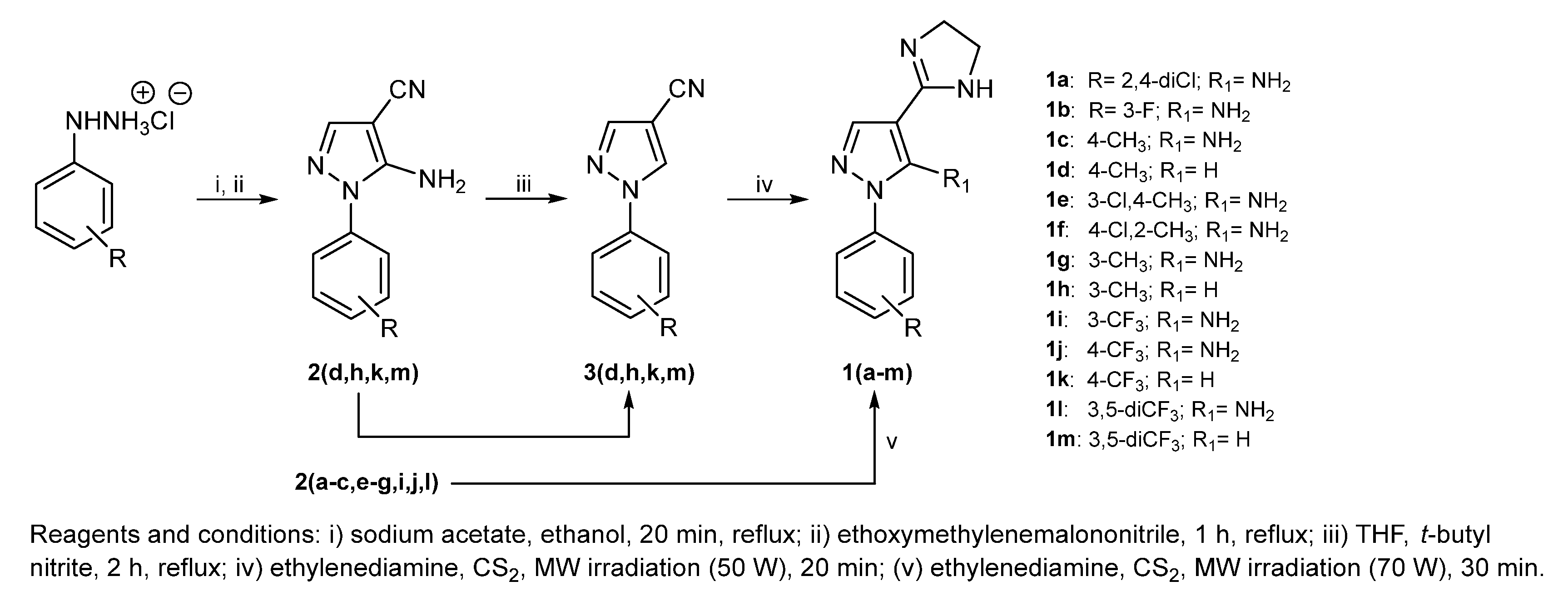
2.1. Synthesis and Chemical Analysis
2.2. Physicochemical Properties of Pyrazole-Imidazoline Derivatives
2.3. Antiparasitic Effect of New Pyrazole-Imidazoline Derivatives
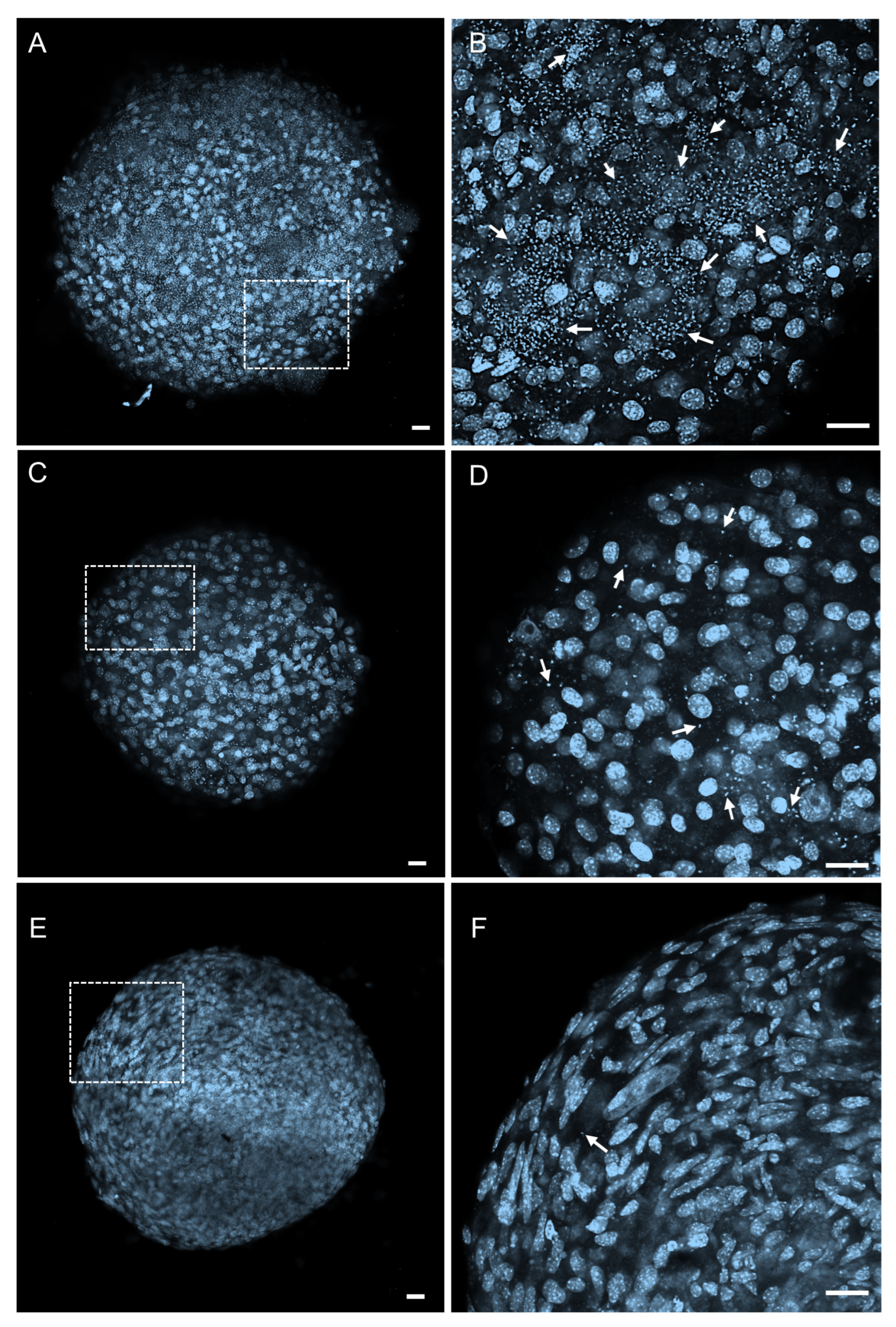
2.4. Permeability and Effectiveness of 1k in T. cruzi-Infected 3D Cardiac Microtissues
2.5. Potential of 1k in Inhibiting the Resurgence of the Parasites
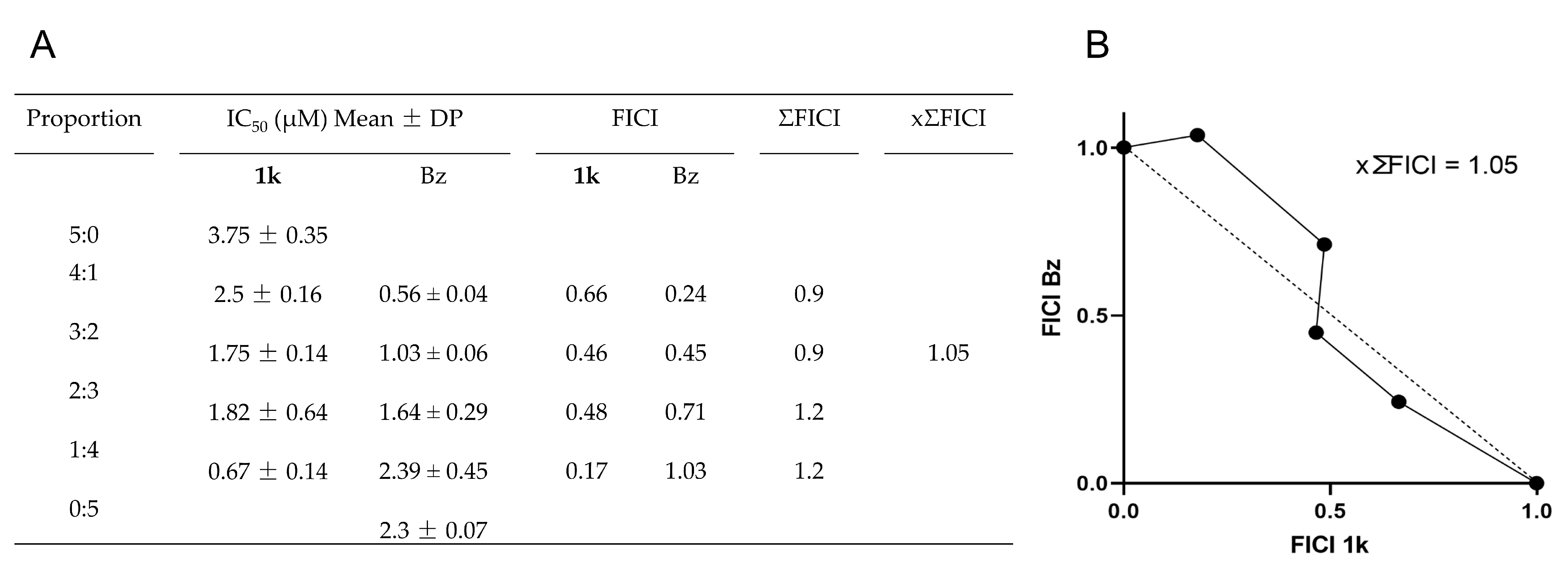
2.6. Drug Combination Effect
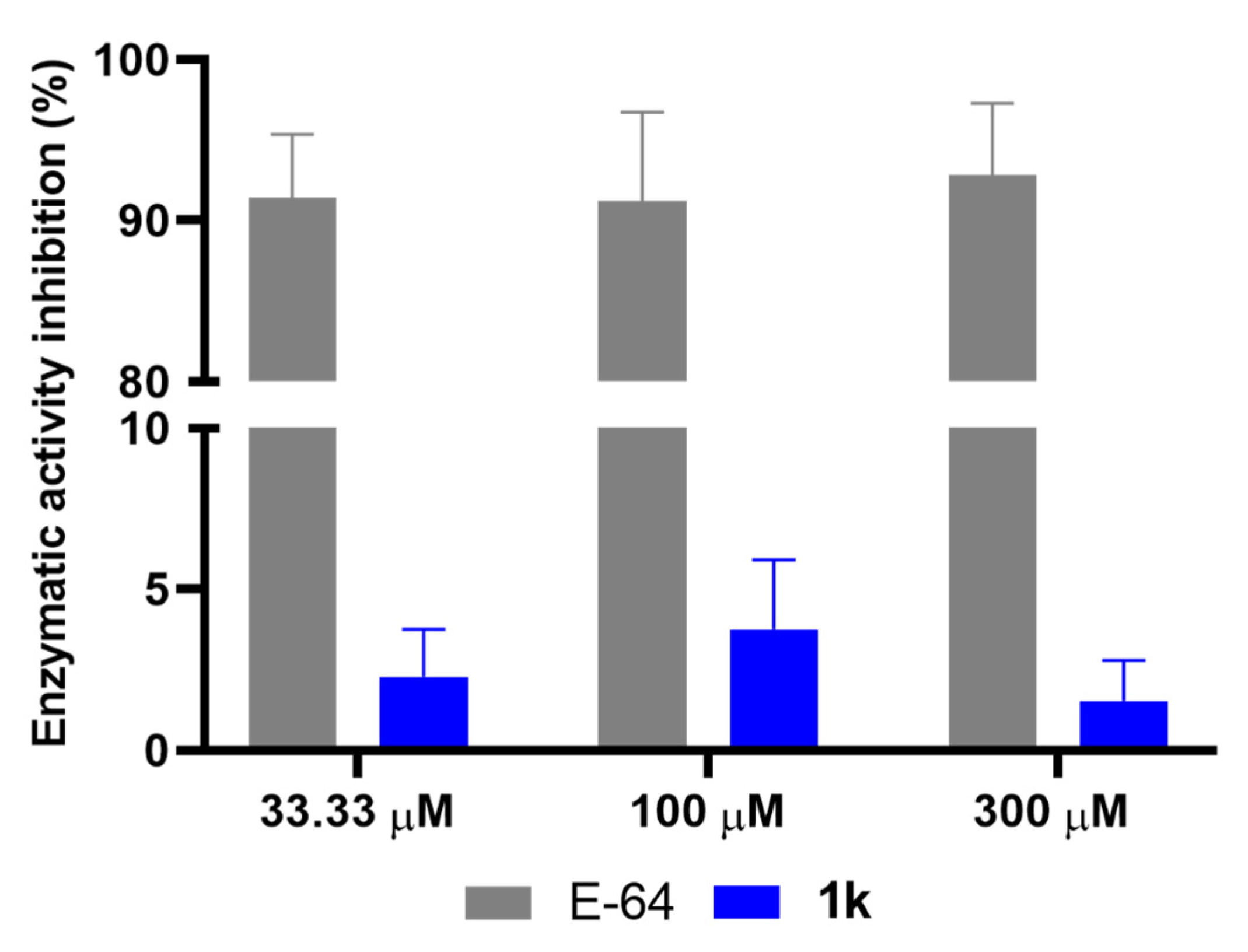
2.7. Effect of 1k on Cysteine Protease Activity
3. Materials and Methods
3.1. Chemistry
3.2. In Silico Analysis
3.3. Cell Culture
3.4. Trypanosoma cruzi
3.5. Cytotoxicity Assay
3.6. Anti-T. cruzi In Vitro Assay
3.7. Reversibility Assay
3.8. Drug Combination
3.9. Enzymatic Activity
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Chagas Disease (Also Known as American Trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 27 March 2025).
- Antinori, S.; Galimberti, L.; Bianco, R.; Grande, R.; Gali, M.; Corbellino, M. Chagas disease in Europe: A review for the internist in the globalized world. Eur. J. Intern. Med. 2017, 43, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Coura, J.R.; Viñas, P.A. Chagas disease: A new worldwide challenge. Nature 2010, 465, S6–S7. [Google Scholar] [CrossRef] [PubMed]
- Guhl, F.; Ramírez, J.D. Poverty, Migration, and Chagas Disease. Curr. Trop. Med. Rep. 2021, 8, 52–58. [Google Scholar] [CrossRef]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.V.; Romano, M.M.D.; Schmidt, A.; Martins, K.S.M.; Marin-Neto, J.A. Chagas Disease Cardiomyopathy. Int. J. Cardiovasc. Sci. 2018, 31, 173–189. [Google Scholar] [CrossRef]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Keegan, R.; Yeung, C.; Baranchuk, A. Sudden Cardiac Death Risk Stratification and Prevention in Chagas Disease: A Non-systematic Review of the Literature. Arrhythmia Electrophysiol. Rev. 2020, 9, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ochoa, S.A.; Rojas, L.Z.; Echeverría, L.E.; Muka, T.; Franco, O.H. Global, Regional, and National Trends of Chagas Disease from 1990 to 2019: Comprehensive Analysis of the Global Burden of Disease Study. Glob. Heart 2022, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Kratz, J.M.; Garcia, B.F.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Molina, J.A.; Crespillo-Andújar, C.; Bosch-Nicolau, P.; Molina, I. Trypanocidal treatment of Chagas disease. Enfermedades Infecc. Microbiol. Clin. 2021, 39, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Kann, S.; Concha, G.; Frickmann, H.; Hagen, R.M.; Warnke, P.; Molitor, E.; Hoerauf, A.; Backhaus, J. Chagas Disease: Comparison of Therapy with Nifurtimox and Benznidazole in Indigenous Communities in Colombia. J. Clin. Med. 2024, 13, 2565. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A., Jr.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. BENEFIT Investigators. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; Gómez, P.J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Waskin, H.; Sosa-Estani, S.; Del Carmen, B.M.; Cuneo, C.; Milesi, R.; Mallagray, M.; Apt, W.; Beloscar, J.; Gascon, J.; et al. STOP-CHAGAS Investigators. Benznidazole and Posaconazole in Eliminating Parasites in Asymptomatic T. cruzi Carriers: The STOP-CHAGAS Trial. J. Am. Coll. Cardiol. 2017, 69, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Torrico, F.; Gascon, J.; Ortiz, L.; Alonso-Veja, C.; Pinazo, M.J.; Schijman, A.; Almeida, I.C.; Alves, F.; Strub-Wourgaft, N.; Ribeiro, I.; et al. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: A proof-of-concept, randomised, placebo-controlled trial. Lancet Infect. Dis. 2018, 18, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Pinazo, M.J.; Forsyth, C.; Losada, I.; Esteban, E.T.; García-Rodríguez, M.; Villegas, M.L.; Molina, I.; Crespillo-Andújar, C.; Gállego, M.; Ballart, C.; et al. FEXI-12 Study Team. Efficacy and safety of fexinidazole for treatment of chronic indeterminate Chagas disease (FEXI-12): A multicentre, randomised, double-blind, phase 2 trial. Lancet Infect. Dis. 2024, 24, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Ravindar, L.; Hasbullah, S.A.; Rakesh, K.P.; Hassan, N.I. Pyrazole and pyrazoline derivatives as antimalarial agents: A key review. Eur. J. Pharm. Sci. 2023, 183, 106365. [Google Scholar] [CrossRef] [PubMed]
- Alnufaie, R.; Raj, K.C.H.; Alsup, N.; Whitt, J.; Chambers, S.A.; Gilmore, D.; Alam, M.A. Synthesis and antimicrobial studies of Coumarin-substituted pyrazole derivatives as potent anti-Staphylococcus aureus agents. Molecules 2020, 25, 2758. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Sun, Y.; Meng, Y.; Liu, L.; Dai, J.; Yan, G.; Pan, X.; Guan, X.; Song, L.; Lin, R. Design, Synthesis and Antifungal Activities of Novel Pyrazole Analogues Containing the Aryl Trifluoromethoxy Group. Molecules 2023, 28, 6279. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.A.; Nasralla, S.N.; El-Agroudy, E.J.; Hamouda, N.; El-Fattah, A.A.; Bekhit, S.A.; Amagase, K.; Ibrahim, T.M. Investigation of the anti-inflammatory and analgesic activities of promising pyrazole derivative. Eur. J. Pharm. Sci. 2022, 168, 106080. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, P.; Gan, X. Novel Pyrazole-Hydrazone Derivatives Containing an Isoxazole Moiety: Design, Synthesis, and Antiviral Activity. Molecules 2018, 23, 1798. [Google Scholar] [CrossRef] [PubMed]
- Ashourpour, M.; Mostafavi, H.F.; Amini, M.; Saeedian, M.E.; Kazerouni, F.; Arman, S.Y.; Shahsavari, Z. Pyrazole Derivatives Induce Apoptosis via ROS Generation in the Triple Negative Breast Cancer Cells, MDA-MB. Asian Pac. J. Cancer Prev. 2021, 22, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Cheng, Y.; Han, C.; Song, C.; Huang, N.; Du, Y. Pyrazole-containing pharmaceuticals: Target, pharmacological activity, and their SAR studies. RSC Med. Chem. 2022, 13, 1300–1321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, R.; Sharma, D.K. Pyrazole; A Privileged Scaffold of Medicinal Chemistry: A Comprehensive Review. Curr. Top. Med. Chem. 2023, 23, 2097–2115. [Google Scholar] [CrossRef] [PubMed]
- Alshabani, L.A.; Kumar, A.; Willcocks, S.J.; Srithiran, G.; Bhakta, S.; Estrada, D.F.; Simons, C. Synthesis, biological evaluation and computational studies of pyrazole derivatives as Mycobacterium tuberculosis CYP121A1 inhibitors. RSC Med. Chem. 2022, 13, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Martín-Montes, Á.; Martínez-Camarena, Á.; Lopera, A.; Bonastre-Sabater, I.; Clares, M.P.; Verdejo, B.; García-España, E.; Marín, C. The Bioactivity of Xylene, Pyridine, and Pyrazole Aza Macrocycles against Three Representative Leishmania Species. Pharmaceutics 2023, 15, 992. [Google Scholar] [CrossRef] [PubMed]
- Verma, G.; Chashoo, G.; Ali, A.; Khan, M.F.; Akhtar, W.; Ali, I.; Akhtar, M.; Alam, M.M.; Shaquiquzzaman, M. Synthesis of pyrazole acrylic acid based oxadiazole and amide derivatives as antimalarial and anticancer agents. Bioorganic Chem. 2018, 77, 106–124. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.E.; Lechuga, G.; Lara, L.S.; Souto, B.A.; Viganó, M.G.; Bourguignon, S.C.; Calvet, C.M.; Oliveira, F.O.R., Jr.; Alves, C.R.; Souza-Silva, F.; et al. Synthesis, structure-activity relationship and trypanocidal activity of pyrazole-imidazoline and new pyrazole-tetrahydropyrimidine hybrids as promising chemotherapeutic agents for Chagas disease. Eur. J. Med. Chem. 2019, 182, 111610. [Google Scholar] [CrossRef] [PubMed]
- Tochowicz, A.; Mckerrow, J.H.; Craik, C.S. Crystal Structure Analysis of Cruzain with Fragment 1 (N-(1H-benimidazole-2-yl)-1,3-dimethyl-pyrazole-4-carboxamide). Available online: https://www.wwpdb.org/pdb?id=pdb_00004w5b (accessed on 31 May 2025).
- Orlando, L.M.R.; Lechuga, G.C.; Lara, L.S.; Ferreira, B.S.; Pereira, C.N.; Silva, R.C.; Dos Santos, M.S.; Pereira, M.C.S. Structural Optimization and Biological Activity of Pyrazole Derivatives: Virtual Computational Analysis, Recovery Assay and 3D Culture Model as Potential Predictive Tools of Effectiveness against Trypanosoma cruzi. Molecules 2021, 26, 6742. [Google Scholar] [CrossRef] [PubMed]
- Lara, L.D.S.; Lechuga, G.C.; Orlando, L.M.R.; Ferreira, B.S.; Souto, B.A.; Dos Santos, M.S.; Pereira, M.C.S. Bioactivity of Novel Pyrazole-Thiazolines Scaffolds against Trypanosoma cruzi: Computational Approaches and 3D Spheroid Model on Drug Discovery for Chagas Disease. Pharmaceutics 2022, 14, 995. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.P.; Orlando, L.M.R.; Lara, L.D.S.; Paes, V.B.; Dutra, L.P.; Dos Santos, M.S.; Pereira, M.C.S. Synthesis and Anti-Trypanosoma cruzi Activity of New Pyrazole-Thiadiazole Scaffolds. Molecules 2024, 29, 3544. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.N.; Rosa, J.O.; Lara, L.S.; Orlando, L.M.R.; Figueiredo, N.S.; Pereira, M.C.S.; Nobuyasu, J.R.S.; Dos Santos, M.S. Synthesis by microwave irradiation of new pyrazole-imidazoline-pyrimidine analogs: Physicochemical and photophysical properties and their biological activity against Trypanosoma cruzi. J. Mol. Struct. 2023, 1290, 135899. [Google Scholar] [CrossRef]
- Rosa, G.S.; Souto, B.A.; Pereira, C.N.; Teixeira, B.C.; Dos Santos, M.S. A convenient synthesis of pyrazole-imidazoline derivatives by microwave irradiation. J. Heterocycl. Chem. 2019, 56, 1825. [Google Scholar] [CrossRef]
- Dos Santos, M.S.; Oliveira, M.L.; Bernardino, A.M.R.; De Léo, R.M.; Amaral, V.F.; De Carvalho, F.T.; Leon, L.L.; Canto-Cavalheiro, M.M. Synthesis and antileishmanial evaluation of 1-aryl-4-(4,5-dihydro-1H-imidazol-2-yl)-1H-pyrazole derivatives. Bioorganic Med. Chem. Lett. 2011, 21, 7451. [Google Scholar] [CrossRef] [PubMed]
- Sharifian, G.M. Recent Experimental Developments in Studying Passive Membrane Transport of Drug Molecules. Mol. Pharm. 2021, 18, 2122–2141. [Google Scholar] [CrossRef] [PubMed]
- Picchio, V.; Gaetani, R.; Chimenti, I. Recent Advances in 3D Cultures. Int. J. Mol. Sci. 2024, 25, 4189. [Google Scholar] [CrossRef] [PubMed]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Mamoshina, P.; Rodriguez, B.; Bueno-Orovio, A. Toward a broader view of mechanisms of drug cardiotoxicity. Cell Rep. Med. 2021, 2, 100216. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Li, T.; Liu, Z.; Li, D.; Tong, W. DICTrank: The largest reference list of 1318 human drugs ranked by risk of drug-induced cardiotoxicity using FDA labeling. Drug Discov. Today 2023, 28, 103770. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.A.G.; Xavier, C.P.R.; Pereira, R.F.; Petrikaitė, V.; Vasconcelos, M.H. 3D Cell Culture Models as Recapitulators of the Tumor Microenvironment for the Screening of Anti-Cancer Drugs. Cancers 2021, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Arez, F.; Rebelo, S.P.; Fontinha, D.; Simão, D.; Martins, T.R.; Machado, M.; Fischli, C.; Oeuvray, C.; Badolo, L.; Carrondo, M.J.T.; et al. Flexible 3D Cell-Based Platforms for the Discovery and Profiling of Novel Drugs Targeting Plasmodium Hepatic Infection. ACS Infect. Dis. 2019, 5, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Garzoni, L.R.; Adesse, D.; Soares, M.J.; Rossi, M.I.; Borojevic, R.; de Meirelles, M.N. Fibrosis and hypertrophy induced by Trypanosoma cruzi in a three-dimensional cardiomyocyte-culture system. J. Infect. Dis. 2008, 197, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Nisimura, L.M.; Ferrão, P.M.; Nogueira, A.D.R.; Waghabi, M.C.; Meuser-Batista, M.; Moreira, O.C.; Urbina, J.A.; Garzoni, L.R. Effect of Posaconazole in an in vitro model of cardiac fibrosis induced by Trypanosoma cruzi. Mol. Biochem. Parasitol. 2020, 238, 111283. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.M.R.; Lara, L.D.S.; Lechuga, G.C.; Rodrigues, G.C.; Pandoli, O.G.; de Sá, D.S.; Pereira, M.C.S. Antitrypanosomal Activity of 1,2,3-Triazole-Based Hybrids Evaluated Using in Vitro Preclinical Translational Models. Biology 2023, 12, 1222. [Google Scholar] [CrossRef] [PubMed]
- Cal, M.; Ioset, J.R.; Fügi, M.A.; Mäser, P.; Kaiser, M. Assessing anti-T. cruzi candidates in vitro for sterile cidality. Int. J. Parasitol. Drugs Drug Resist. 2016, 6, 165–170. [Google Scholar] [PubMed]
- MacLean, L.M.; Thomas, J.; Lewis, M.D.; Cotillo, I.; Gray, D.W.; De Rycker, M. Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Neglected Trop. Dis. 2018, 12, e0006612. [Google Scholar] [CrossRef] [PubMed]
- Soeiro, M.N.C.; Sales-Junior, P.A.; Pereira, V.R.A.; Vannier-Santos, M.A.; Murta, S.M.F.; Sousa, A.S.; Sangenis, L.H.C.; Moreno, A.M.H.; Boechat, N.; Branco, F.S.C.; et al. Drug screening and development cascade for Chagas disease: An update of in vitro and in vivo experimental models. Memórias Inst. Oswaldo Cruz 2024, 119, e240057. [Google Scholar]
- Sánchez-Valdéz, F.J.; Padilla, A.; Wang, W.; Orr, D.; Tarleton, R.L. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. Elife 2018, 7, e34039. [Google Scholar] [CrossRef] [PubMed]
- Jayawardhana, S.; Ward, A.I.; Francisco, A.F.; Lewis, M.D.; Taylor, M.C.; Kelly, J.M.; Olmo, F. Benznidazole treatment leads to DNA damage in Trypanosoma cruzi and the persistence of rare widely dispersed non-replicative amastigotes in mice. PLoS Pathog. 2023, 19, e1011627. [Google Scholar] [CrossRef] [PubMed]
- Alcântara, L.M.; Ferreira, T.C.S.; Gadelha, F.R.; Miguel, D.C. Challenges in drug discovery targeting TriTryp diseases with an emphasis on leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Mazzeti, A.L.; Gonçalves, K.R.; Mota, S.L.A.; Pereira, D.E.; Diniz, L.F.; Bahia, M.T. Combination therapy using nitro compounds improves the efficacy of experimental Chagas disease treatment. Parasitology 2021, 148, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Chaachouay, N. Synergy, Additive Effects, and Antagonism of Drugs with Plant Bioactive Compounds. Drugs Drug Candidates 2025, 4, 4. [Google Scholar] [CrossRef]
- Pandey, R.P.; Nascimento, M.S.; Franco, C.H.; Bortoluci, K.; Silva, M.N.; Zingales, B.; Gibaldi, D.; Barrios, L.C.; LannesVieira, J.; Cariste, L.M.; et al. Drug repurposing in Chagas disease: Chloroquine potentiates benznidazole activity against Trypanosoma cruzi in vitro and in vivo. Antimicrob. Agents Chemother. 2022, 15, 0028422. [Google Scholar] [CrossRef] [PubMed]
- Machado, Y.A.; Bahia, M.T.; Caldas, I.S.; Mazzeti, A.L.; Novaes, R.D.; Boas, R.V.B.; Santos, L.J.S.; Martins-Filho, O.A.; Marques, M.J.; Diniz, L.F. Amlodipine increases the therapeutic potential of ravuconazole upon Trypanosoma cruzi infection. Antimicrob. Agents Chemother. 2020, 64, e2497–e2519. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. The role of imidazole and benzimidazole heterocycles in Chagas disease: A review. Eur. J. Med. Chem. 2020, 206, 112692. [Google Scholar] [CrossRef] [PubMed]
- Paes, V.B.; Lara, L.S.; Orlando, L.M.R.; Lechuga, G.C.; Souza, T.P.; Ferreira, B.S.; Zago, M.O.; Santos, M.S.; Pereira, M.C.S. Insights into the Antiparasitic Activity of Pyrazole-benzimidazole against Trypanosoma cruzi. Curr. Med. Chem. 2025, 32, e09298673342932. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.C.; Ferreira, R.S. Computational approaches towards the discovery and optimisation of cruzain inhibitors. Mem. Inst. Oswaldo Cruz 2022, 117, e210385. [Google Scholar] [CrossRef] [PubMed]
- Nery, E.D.; Juliano, M.A.; Meldal, M.; Svendsen, I.; Scharfstein, J.; Walmsley, A.; Juliano, L. Characterization of the substrate specificity of the major cysteine protease (cruzipain) from Trypanosoma cruzi using a portion-mixing combinatorial library and fluorogenic peptides. Biochem. J. 1997, 323, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Duschak, V.G.; Couto, A.S. Cruzipain, the major cysteine protease of Trypanosoma cruzi: A sulfated glycoprotein antigen as a relevant candidate for vaccine development and drug target. A review. Curr. Med. Chem. 2009, 16, 3174–3202. [Google Scholar] [CrossRef] [PubMed]
- Burtoloso, A.C.; de Albuquerque, S.; Furber, M.; Gomes, J.C.; Gonçalez, C.; Kenny, P.W.; Leitão, A.; Montanari, C.A.; Quilles, J.C.J.; Ribeiro, J.F.; et al. Anti-trypanosomal activity of non-peptidic nitrile-based cysteine protease inhibitors. PLoS Negl Trop Dis. 2017, 11, e0005343. [Google Scholar] [CrossRef] [PubMed]
- Moraes, B.S.; Azeredo, F.J.; Izoton, J.C.; Amaral, M.; Barreiro, E.J.; Freddo, R.J.; Costa, T.D.; Lima, L.M.; Haas, S.E. Leishmanicidal candidate LASSBio-1736, a cysteine protease inhibitor with favorable pharmacokinetics: Low clearance and good distribution. Xenobiotica 2018, 48, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.Q.; Schaeffer, E.; da Silva, A.J.M.; Alves, C.R.; Souza-Silva, F. A Virtual Screening Approach to Evaluate the Multitarget Potential of a Chalcone Library with Binding Properties to Oligopeptidase B and Cysteine Proteinase B from Leishmania (Viannia) braziliensis. Int J Mol Sci. 2025, 26, 2025. [Google Scholar] [CrossRef] [PubMed]
- Sykes, M.L.; Avery, V.M. 3-pyridyl inhibitors with novel activity against Trypanosoma cruzi reveal in vitro profiles can aid prediction of putative cytochrome P450 inhibition. Sci. Rep. 2018, 8, 4901. [Google Scholar] [CrossRef] [PubMed]
- Fiuza, L.F.A.; Peres, R.B.; Simões-Silva, M.R.; da Silva, P.B.; Batista, D.D.G.J.; da Silva, C.F.; Nefertiti, A.S.G.; Krishna, T.R.R.; Soeiro, M.N.C. Identification of Pyrazolo[3,4-e][1,4]thiazepin based CYP51 inhibitors as potential Chagas disease therapeutic alternative: In vitro and in vivo evaluation, binding mode prediction and SAR exploration. Eur. J. Med. Chem. 2018, 149, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Strašek, N.; Lavrenčič, L.; Oštrek, A.; Slapšak, D.; Grošelj, U.; Klemenčič, M.; Brodnik, H.Ž.; Wagger, J.; Novinec, M.; Svete, J. Tetrahydro-1H,5H-pyrazolo[1,2-a]pyrazole-1-carboxylates as inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. Bioorganic Chem. 2019, 89, 102982. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, A.L.P.G.; Vegi, P.F.; Faria, J.V.; Pinto, G.S.P.; Dos Santos, M.S.; Sathler, P.C.; Saito, M.S.; Santana, M.; Dutra, T.P.P.; Rodrigues, C.R.; et al. Pyrazolyl-tetrazoles and imidazolyl-pyrazoles as potential anticoagulants and their integrated multiplex analysis virtual screening. J. Braz. Chem. Soc. 2019, 30, 33. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; DataWarrior, R.C. An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Meirelles, M.N.; de Araujo-Jorge, T.C.; Miranda, C.F.; de Souza, W.; Barbosa, H.S. Interaction of Trypanosoma cruzi with heart muscle cells: Ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur. J. Cell Biol. 1986, 41, 198–206. [Google Scholar] [PubMed]
- Henriques, C.; Henrique-Pons, A.; Meuser-Batista, M.; Ribeiro, A.S.; de Souza, W. In vivo imaging of mice infected with bioluminescent Trypanosoma cruzi unveils novel sites of infection. Parasites Vectors 2014, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Fivelman, Q.L.; Adagu, I.S.; Warhurst, D.C. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob. Agents Chemother. 2004, 48, 4097–4102. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MW | cLogP | cLogS | HBA | HBD | TPSA | Drug Likeness | RB |
|---|---|---|---|---|---|---|---|---|
| 1a | 296.16 | 1.085 | −3.63 | 5 | 2 | 68.23 | 6.54 | 2 |
| 1b | 245.26 | −0.02 | −2.74 | 5 | 2 | 68.23 | 5.2 | 2 |
| 1c | 275.74 | 0.823 | −3.23 | 5 | 2 | 68.23 | 6.46 | 2 |
| 1d | 275.74 | 0.823 | −3.23 | 5 | 2 | 68.23 | 6.46 | 2 |
| 1e | 241.29 | 0.21 | −2.5 | 5 | 2 | 68.23 | 6.45 | 2 |
| 1f | 226.28 | 0.543 | −1.9 | 4 | 1 | 42.21 | 6.24 | 2 |
| 1g | 241.29 | 0.217 | −2.5 | 5 | 2 | 68.23 | 6.45 | 2 |
| 1h | 226.28 | 0.543 | −1.9 | 4 | 1 | 42.21 | 6.24 | 2 |
| 1i | 295.26 | 0.721 | −2.93 | 5 | 2 | 68.23 | −0.64 | 3 |
| 1j | 295.26 | 0.721 | −2.93 | 5 | 2 | 68.23 | −0.64 | 3 |
| 1k | 280.25 | 1.04 | −2.33 | 4 | 1 | 42.21 | −0.83 | 3 |
| 1l | 363.26 | 1.57 | −3.71 | 5 | 2 | 68.23 | −0.64 | 4 |
| 1m | 348.24 | 1.89 | −3.11 | 4 | 1 | 42.21 | −0.83 | 4 |
| Bz | 260.25 | −0.25 | −1.62 | 7 | 1 | 92.74 | −1.71 | 5 |
| Compounds(R) | Anti-T. cruzi Activity (Mean ± SD) | Cytotoxicity (Mean ± SD) | |||||
|---|---|---|---|---|---|---|---|
| Trypomastigotes | Intracellular Amastigotes | ||||||
| IC50 (µM) | IC90 (µM) | IC50 (µM) | IC90 (µM) | SI | pIC50 | CC50 (µM) | |
| 1a (2,4-diCl) | >100 | >100 | 40.8 ± 3.0 | >100 | >12.2 | 4.12 | >500 |
| 1b (3-F) | >100 | >100 | >100 | >100 | ND | <4 | >500 |
| 1c (4-CH3) | >100 | >100 | >100 | >100 | ND | <4 | >500 |
| 1d (4-CH3) * | >100 | >100 | 33.9 ± 0.8 | >100 | >14.7 | 4.4 | >500 |
| 1e (3-Cl, 4-CH3) | 76.5 ± 10.3 | >100 | 68.9 ± 8.1 | 96.6 ± 0.2 | >7.2 | 4.1 | >500 |
| 1f (4-Cl, 2-CH3) | >100 | >100 | 76.1 ± 1.2 | >100 | >6.5 | 4.1 | >500 |
| 1g (3-CH3) | >100 | >100 | 78.9 ± 2.2 | >100 | >6.3 | 4.1 | >500 |
| 1h (3-CH3) * | >100 | >100 | 26.2 ± 3.4 | 89.7 ± 7.4 | >19.1 | 4.6 | >500 |
| 1i (3-CF3) | >100 | >100 | 51.9 ± 3.6 | >100 | >9.6 | 4.2 | >500 |
| 1j (4-CF3) | >100 | >100 | 88.1 ± 0.2 | >100 | >5.6 | 4.0 | >500 |
| 1k (4-CF3) * | 72.2 ± 10.3 | >100 | 3.3 ± 0.2 | 28.1 ± 0.4 | 73.9 | 5.4 | 243.8 ± 21.1 |
| 1l (3,5-diCF3) | >100 | >100 | 27.7 ± 2.0 | 80.1 ± 11.4 | 16.7 | 4.5 | 463.6 ± 15.2 |
| 1m (3,5-diCF3) * | >100 | >100 | 32.4 ± 5.9 | 80.7 ± 0.4 | 3.5 | 4.5 | 113.5 ± 11.9 |
| Bz | 20.5 ± 1.8 | >100 | 2.2 ± 0.4 | 10.4 ± 0.5 | >221.2 | 5.6 | >500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, E.C.; Lara, L.d.S.; Orlando, L.M.R.; Lanera, S.d.C.; de Souza, T.P.; Figueiredo, N.d.S.; Paes, V.B.; Mazzochi, A.C.; Fernandes, P.H.M.; dos Santos, M.S.; et al. Anti-Trypanosoma cruzi Potential of New Pyrazole-Imidazoline Derivatives. Molecules 2025, 30, 3082. https://doi.org/10.3390/molecules30153082
de Oliveira EC, Lara LdS, Orlando LMR, Lanera SdC, de Souza TP, Figueiredo NdS, Paes VB, Mazzochi AC, Fernandes PHM, dos Santos MS, et al. Anti-Trypanosoma cruzi Potential of New Pyrazole-Imidazoline Derivatives. Molecules. 2025; 30(15):3082. https://doi.org/10.3390/molecules30153082
Chicago/Turabian Stylede Oliveira, Edinaldo Castro, Leonardo da Silva Lara, Lorraine Martins Rocha Orlando, Sarah da Costa Lanera, Thamyris Perez de Souza, Nathalia da Silva Figueiredo, Vitoria Barbosa Paes, Ana Carolina Mazzochi, Pedro Henrique Myra Fernandes, Maurício Silva dos Santos, and et al. 2025. "Anti-Trypanosoma cruzi Potential of New Pyrazole-Imidazoline Derivatives" Molecules 30, no. 15: 3082. https://doi.org/10.3390/molecules30153082
APA Stylede Oliveira, E. C., Lara, L. d. S., Orlando, L. M. R., Lanera, S. d. C., de Souza, T. P., Figueiredo, N. d. S., Paes, V. B., Mazzochi, A. C., Fernandes, P. H. M., dos Santos, M. S., & Pereira, M. C. d. S. (2025). Anti-Trypanosoma cruzi Potential of New Pyrazole-Imidazoline Derivatives. Molecules, 30(15), 3082. https://doi.org/10.3390/molecules30153082