
Current Status of the Application of Antimicrobial Peptides and Their Conjugated Derivatives


Abstract

1. Introduction
2. Antimicrobial Peptides
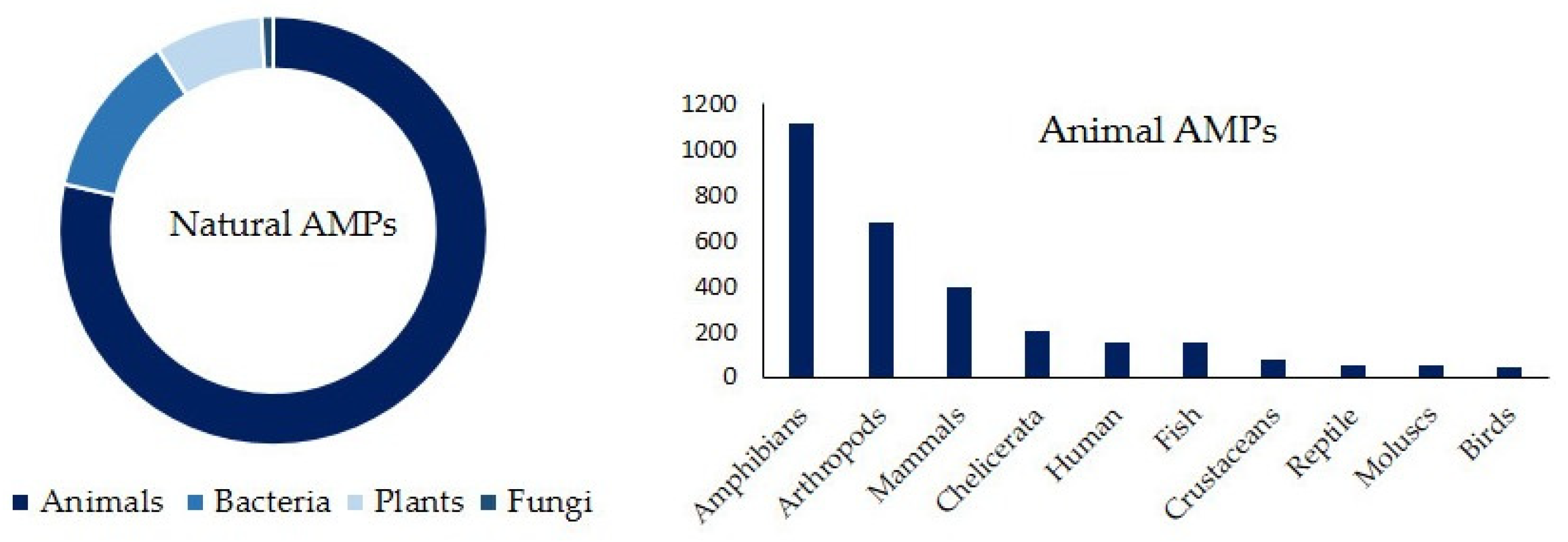
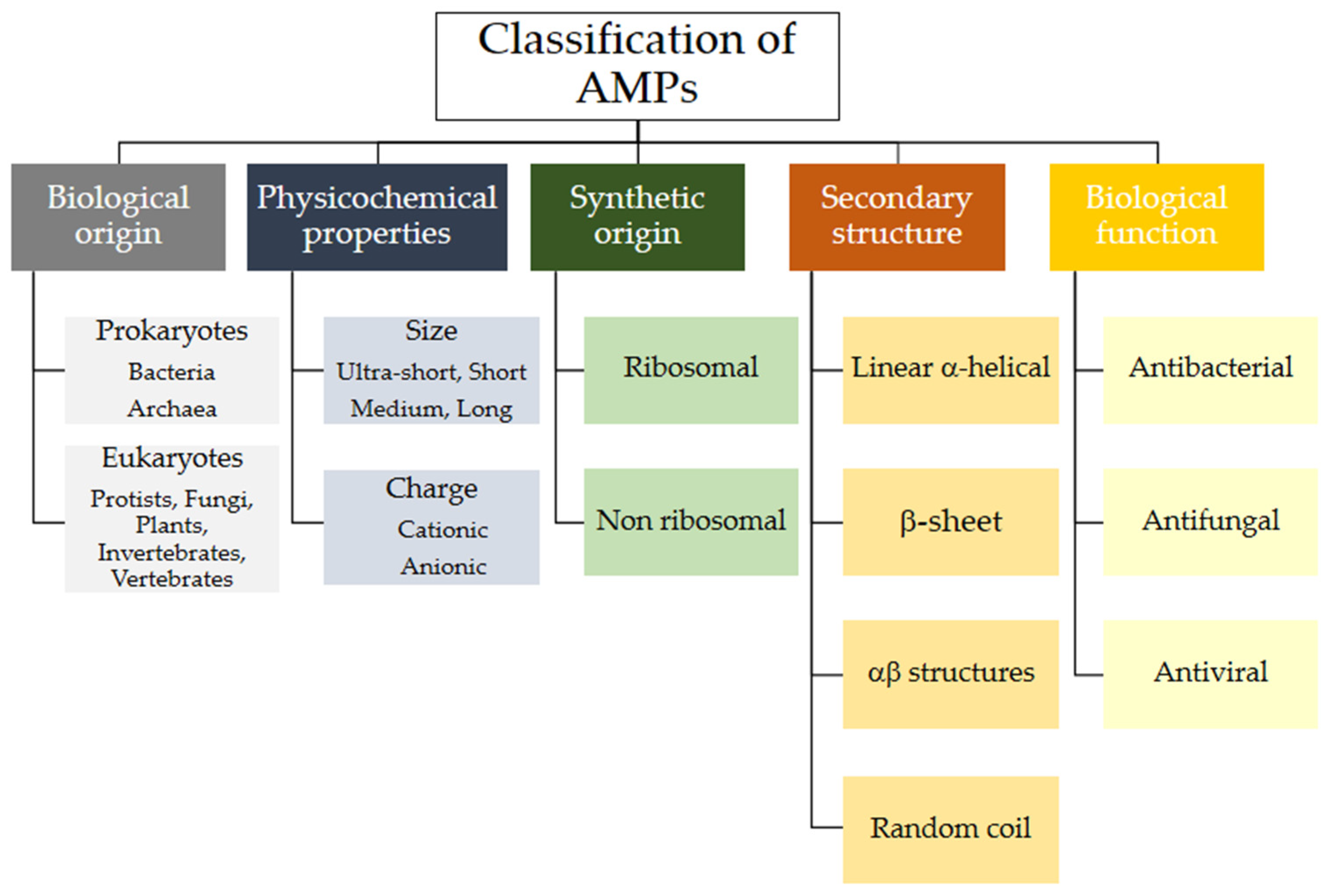
2.1. Classification of Naturally Occurring AMPs
2.1.1. Classification of AMPs by Biosynthetic Origin
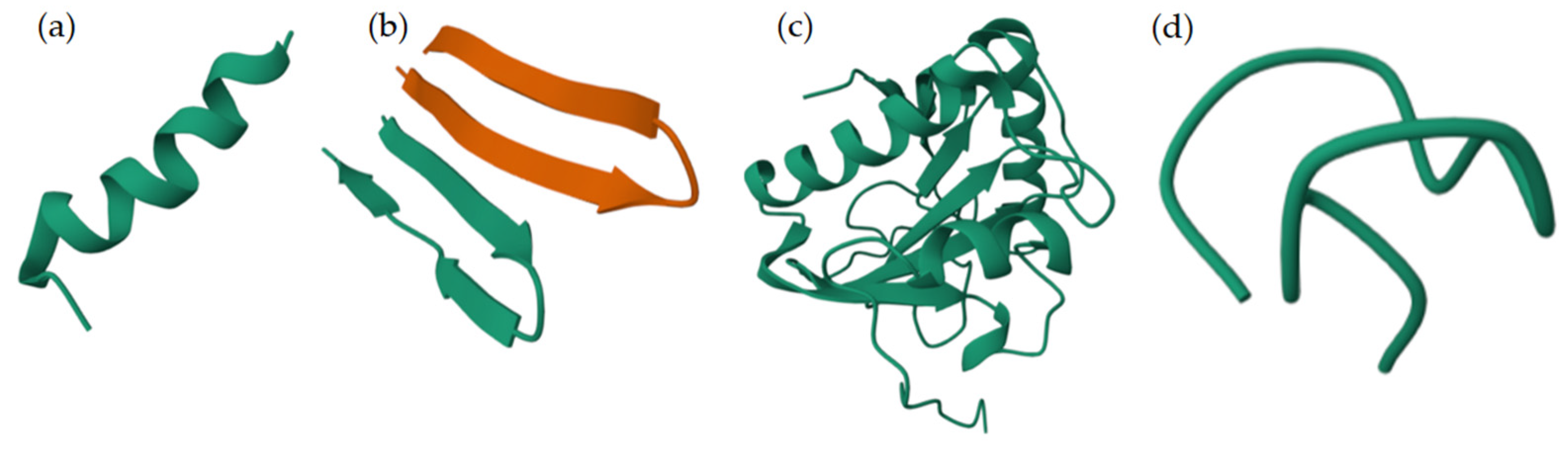
2.1.2. Classification of AMPs by Structure
2.1.3. Classification of AMPs According to Their Biological Function
2.2. Mechanism of Action of AMPs
3. Antimicrobial Peptides of Animal Origin
4. Plant AMPs
5. AMPs Produced by Bacteria
5.1. Bacteriocins or Ribosomal AMPs
5.2. Non-Ribosomal AMPs
6. Fungal AMPs
7. Bacteriophage AMPs
8. Synthetic AMPs
9. Cryptic AMPs
10. Clinical Applications of AMPs
10.1. Strategies to Improve Peptide Stability
10.2. The Economic Cost of Commercial Production of AMPs
10.3. AMPs Approved for Use
11. Strategies to Improve the Properties of AMPs
11.1. Combinations of AMPs with Other Antimicrobials
11.1.1. AMP Combinations
11.1.2. Combination of AMPs with Traditional Antibiotics
11.1.3. Combinations of AMPs and Non-Direct Antimicrobial Cationic Peptides
11.1.4. Antibiotic Adjuvants
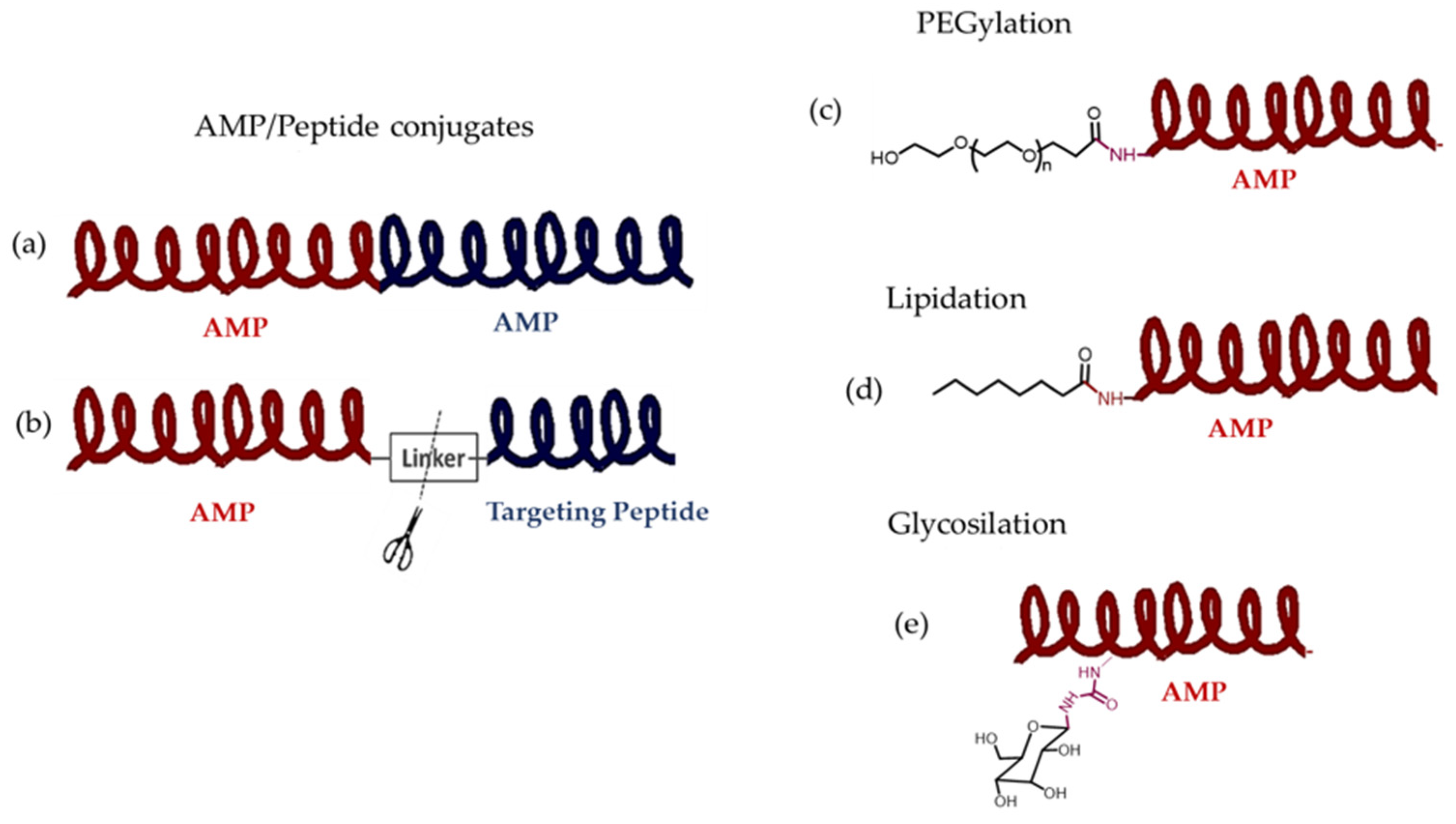
11.2. Covalent Conjugates with AMPs
11.2.1. Hybrid Drugs of Two AMPs and Covalently Conjugated AMP-Targeting Peptide
11.2.2. Covalent Conjugation of AMPs with Polymers
11.2.3. Lipidation and Glycosylation of AMPs
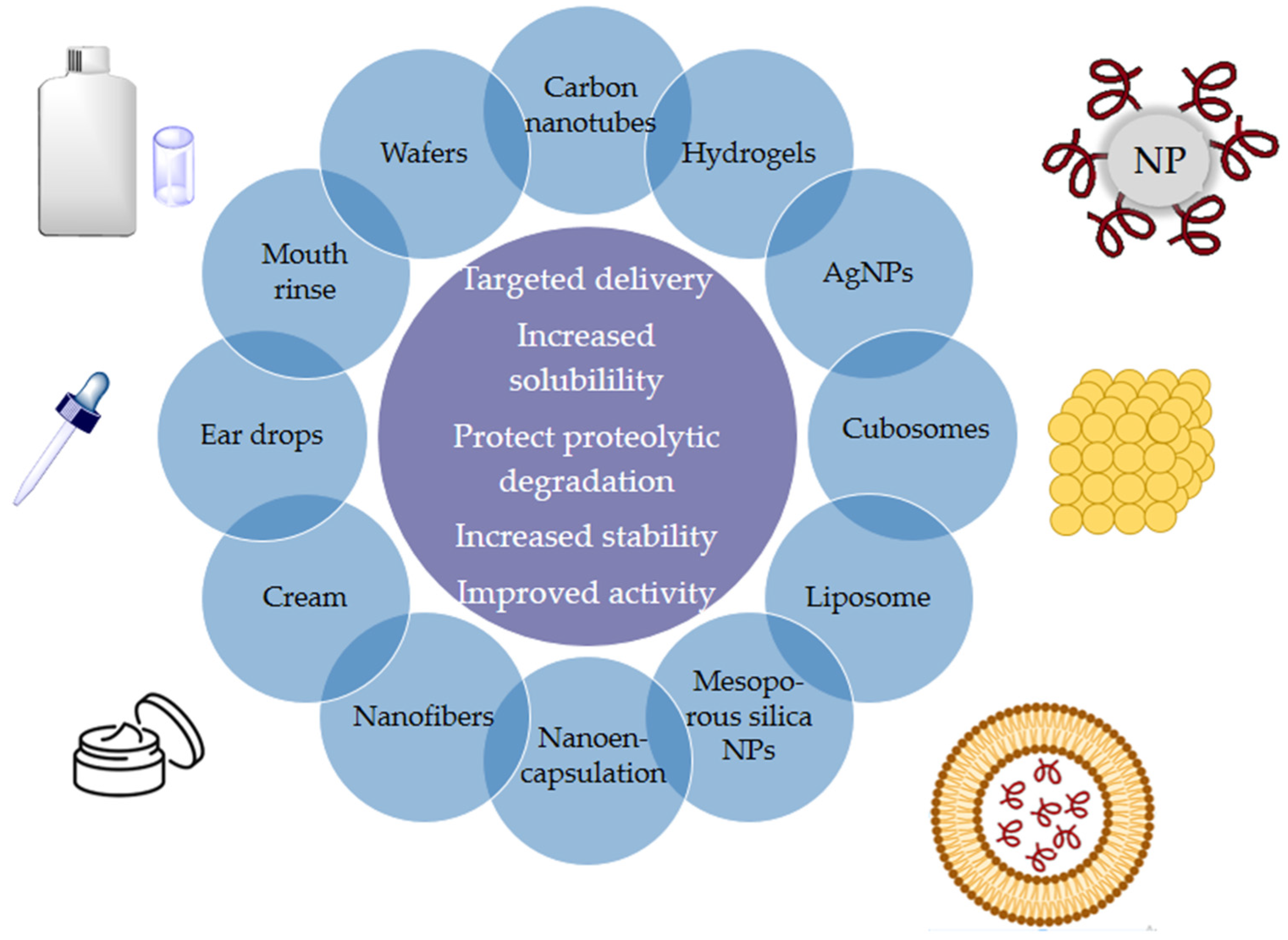
12. Nanostructures
13. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | Amino acid(s) |
| ABPs | Antibacterial peptides |
| Aib | α-aminoisobutyric acid |
| AMPs | Antimicrobial peptides |
| APD | Antimicrobial peptide database |
| cHDPs | Cryptic host defense peptides |
| EMA | European medicines agency |
| FDA | United States food and drug administration |
| FIC | Fractional inhibitory concentrations |
| HBPs | Heparin-binding proteins |
| HDPs | Host defense molecules |
| LAB | Lactic acid bacteria |
| LPS | Lipopolysaccharides |
| MATE | Multidrug and toxic compound extrusion family |
| MSF | Major facilitator superfamily |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NDACPs | Non-direct antimicrobial cationic peptides |
| NP | Nanoparticle |
| NRPs | Non-ribosomal peptides |
| NRPSs | Non-ribosomal peptide synthetases |
| PDB | Protein Data Bank |
| PrAMPs | Proline-rich AMPs |
| PTM | Post-translational modification |
| VAPGHs | Virion-associated peptidoglycan hydrolases |
| VRE | Vancomycin-resistant Enterococci |
| WHO | World Health Organization |
References
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef]
- Spagnolo, F.; Trujillo, M.; Dennehy, J.J. Why Do Antibiotics Exist? mBio 2021, 12, e01966-21. [Google Scholar] [CrossRef]
- Haq, S.U.; Ling, W.; Aqib, A.I.; Danmei, H.; Aleem, M.T.; Fatima, M.; Ahmad, S.; Gao, F. Exploring the Intricacies of Antimicrobial Resistance: Understanding Mechanisms, Overcoming Challenges, and Pioneering Innovative Solutions. Eur. J. Pharmacol. 2025, 998, 177511. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Burki, T.K. Development of New Antibacterial Agents: A Sense of Urgency Needed. Lancet Respir. Med. 2021, 9, e54. [Google Scholar] [CrossRef] [PubMed]
- Baran, A.; Kwiatkowska, A.; Potocki, L. Antibiotics and Bacterial Resistance—A Short Story of an Endless Arms Race. Int. J. Mol. Sci. 2023, 24, 5777. [Google Scholar] [CrossRef]
- Martinez, J.L. General Principles of Antibiotic Resistance in Bacteria. Drug Discov. Today Technol. 2014, 11, 33–39. [Google Scholar] [CrossRef]
- Gillings, M.R.; Paulsen, I.T.; Tetu, S.G. Genomics and the Evolution of Antibiotic Resistance. Ann. N. Y. Acad. Sci. 2017, 1388, 92–107. [Google Scholar] [CrossRef]
- Ho, C.S.; Wong, C.T.H.; Aung, T.T.; Lakshminarayanan, R.; Mehta, J.S.; Rauz, S.; McNally, A.; Kintses, B.; Peacock, S.J.; De La Fuente-Nunez, C.; et al. Antimicrobial Resistance: A Concise Update. Lancet Microbe 2025, 6, 100947. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Gomes, D.; Esteruelas, G.; Bonilla, L.; Lopez-Machado, A.L.; Galindo, R.; Cano, A.; Espina, M.; Ettcheto, M.; Camins, A.; et al. Metal-Based Nanoparticles as Antimicrobial Agents: An Overview. Nanomaterials 2020, 10, 292. [Google Scholar] [CrossRef]
- Mondal, S.K.; Chakraborty, S.; Manna, S.; Mandal, S.M. Antimicrobial Nanoparticles: Current Landscape and Future Challenges. RSC Pharm. 2024, 1, 388–402. [Google Scholar] [CrossRef]
- Luo, L.; Huang, W.; Zhang, J.; Yu, Y.; Sun, T. Metal-Based Nanoparticles as Antimicrobial Agents: A Review. ACS Appl. Nano Mater. 2024, 7, 2529–2545. [Google Scholar] [CrossRef]
- Uddin Mahamud, A.S.; Nahar, S.; Ashrafudoulla, M.; Park, S.H.; Ha, S.D. Insights into Antibiofilm Mechanisms of Phytochemicals: Prospects in the Food Industry. Crit. Rev. Food Sci. Nutr. 2024, 64, 1736–1763. [Google Scholar] [CrossRef] [PubMed]
- Murugan, S.; Senthilvelan, T.; Govindasamy, M.; Thangavel, K. A Comprehensive Review on Exploring the Potential of Phytochemicals and Biogenic Nanoparticles for the Treatment of Antimicrobial-Resistant Pathogenic Bacteria. Curr. Microbiol. 2025, 82, 90. [Google Scholar] [CrossRef]
- Tyagi, J.L.; Gupta, P.; Ghate, M.M.; Kumar, D.; Poluri, K.M. Assessing the Synergistic Potential of Bacteriophage Endolysins and Antimicrobial Peptides for Eradicating Bacterial Biofilms. Arch. Microbiol. 2024, 206, 272. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.; Purandare, K. Bacteriophage Therapy Inspired New Age Technologies to Control Antimicrobial Resistance. J. Umm Al-Qura Univ. Appll. Sci. 2025, 1–22. [Google Scholar] [CrossRef]
- Kuchay, R.A.H. Novel and Emerging Therapeutics for Antimicrobial Resistance: A Brief Review. Drug Discov. Ther. 2024, 18, 269–276. [Google Scholar] [CrossRef]
- Berger, I.; Loewy, Z.G. Antimicrobial Resistance and Novel Alternative Approaches to Conventional Antibiotics. Bacteria 2024, 3, 171–182. [Google Scholar] [CrossRef]
- La Guidara, C.; Adamo, R.; Sala, C.; Micoli, F. Vaccines and Monoclonal Antibodies as Alternative Strategies to Antibiotics to Fight Antimicrobial Resistance. Int. J. Mol. Sci. 2024, 25, 5487. [Google Scholar] [CrossRef]
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A.; et al. Antimicrobial Peptides: A New Hope in Biomedical and Pharmaceutical Fields. Front. Cell. Infect. Microbiol. 2021, 11, 668632. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Reddy, K.V.R.; Yedery, R.D.; Aranha, C. Antimicrobial Peptides: Premises and Promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T. Antimicrobial Peptides in Mammalian and Insect Host Defence. Curr. Opin. Immunol. 1999, 11, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Diana, J. The Dual Role of Antimicrobial Peptides in Autoimmunity. Front. Immunol. 2020, 11, 2077. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Kjos, M.; Nes, I.F.; Diep, D.B.; Lotfipour, F. Natural Antimicrobial Peptides from Bacteria: Characteristics and Potential Applications to Fight Against Antibiotic Resistance. J. Appl. Microbiol. 2012, 113, 723–736. [Google Scholar] [CrossRef]
- Arbulu, S.; Kjos, M. Revisiting the Multifaceted Roles of Bacteriocins: The Multifaceted Roles of Bacteriocins. Microb. Ecol. 2024, 87, 41. [Google Scholar] [CrossRef]
- Anurag Anand, A.; Amod, A.; Anwar, S.; Sahoo, A.K.; Sethi, G.; Samanta, S.K. A Comprehensive Guide on Screening and Selection of a Suitable AMP Against Biofilm-Forming Bacteria. Crit. Rev. Microbiol. 2024, 50, 859–878. [Google Scholar] [CrossRef]
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial Peptides and Their Interaction with Biofilms of Medically Relevant Bacteria. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 1044–1060. [Google Scholar] [CrossRef]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 435645. [Google Scholar] [CrossRef]
- Luong, H.X.; Ngan, H.D.; Thi Phuong, H.B.; Quoc, T.N.; Tung, T.T. Multiple Roles of Ribosomal Antimicrobial Peptides in Tackling Global Antimicrobial Resistance. R. Soc. Open Sci. 2022, 9, 211583. [Google Scholar] [CrossRef]
- Noor, H.B.; Mou, N.A.; Salem, L.; Shimul, M.F.A.; Biswas, S.; Akther, R.; Khan, S.; Raihan, S.; Mohib, M.M.; Sagor, M.A.T. Anti-Inflammatory Property of AMP-Activated Protein Kinase. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2020, 19, 2–41. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2850. [Google Scholar] [CrossRef]
- Wang, G. Unifying the Classification of Antimicrobial Peptides in the Antimicrobial Peptide Database. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2022; Volume 663; pp. 1–18. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Straus, S.K. Tryptophan- and Arginine-Rich Antimicrobial Peptides: Anti-Infectives with Great Potential. Biochim. Biophys. Acta (BBA) Biomembr. 2024, 1866, 184260. [Google Scholar] [CrossRef] [PubMed]
- Dzurová, L.; Holásková, E.; Pospíšilová, H.; Schneider Rauber, G.; Frébortová, J. Cathelicidins: Opportunities and Challenges in Skin Therapeutics and Clinical Translation. Antibiotics 2024, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Stączek, S.; Kunat-Budzyńska, M.; Cytryńska, M.; Zdybicka-Barabas, A. Proline-Rich Antimicrobial Peptides from Invertebrates. Molecules 2024, 29, 5864. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Vishnepolsky, B.; Grigolava, M.; Managadze, G. Physicochemical Features and Peculiarities of Interaction of AMP with the Membrane. Pharmaceuticals 2021, 14, 471. [Google Scholar] [CrossRef]
- Díaz, M.D.; Palomino-Schätzlein, M.; Corzana, F.; Andreu, C.; Carbajo, R.J.; Del Olmo, M.; Canales-Mayordomo, A.; Pineda-Lucena, A.; Asensio, G.; Jiménez-Barbero, J. Antimicrobial Peptides and Their Superior Fluorinated Analogues: Structure–Activity Relationships as Revealed by NMR Spectroscopy and MD Calculations. ChemBioChem 2010, 11, 2424–2432. [Google Scholar] [CrossRef]
- Sudheendra, U.S.; Dhople, V.; Datta, A.; Kar, R.K.; Shelburne, C.E.; Bhunia, A.; Ramamoorthy, A. Membrane Disruptive Antimicrobial Activities of Human β-Defensin-3 Analogs. Eur. J. Med. Chem. 2015, 91, 91–99. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Wang, G. Discovery, classification and functional diversity of antimicrobial peptides. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; Wang, G., Ed.; CABI: Boston, MA, USA, 2017; Chapter 1; pp. 1–19. [Google Scholar]
- Bourbigot, S.; Dodd, E.; Horwood, C.; Booth, V. NMR Structure of the Antimicrobial Peptide RP-1 Bound to SDS Micelles: 2rlg. Biopolymers 2008, 91, 1–13. [Google Scholar] [CrossRef]
- Shenkarev, Z.O.; Balandin, S.V.; Trunov, K.I.; Paramonov, A.S.; Sukhanov, S.V.; Barsukov, L.I.; Arseniev, A.S.; Ovchinnikova, T.V. Molecular Mechanism of Action of β-Hairpin Antimicrobial Peptide Arenicin: Oligomeric Structure in Dodecylphosphocholine Micelles and Pore Formation in Planar Lipid Bilayers. Biochemistry 2011, 50, 6255–6265. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.B.; Teyton, L.; Wilson, I.A. Crystal Structure of the Drosophila Peptidoglycan Recognition Protein (PGRP)-SA at 1.56Å Resolution. J. Mol. Biol. 2004, 340, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.; Shawon, J.; Ali, M.A.; Williams, B.; Shahinuzzaman, A.D.A.; Rupa, S.A.; Al-Adhami, T.; Jia, R.; Bourque, C.; Faddis, R.; et al. Antiviral Peptides Inhibiting the Main Protease of SARS-CoV-2 Investigated by Computational Screening and In Vitro Protease Assay. J. Pept. Sci. 2024, 30, e3553. [Google Scholar] [CrossRef]
- Erdem Büyükkiraz, M.; Kesmen, Z. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. J. Appl. Microbiol. 2022, 132, 1573–1596. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Yan, Z.-B.; Meng, Y.-M.; Hong, X.-Y.; Shao, G.; Ma, J.-J.; Cheng, X.-R.; Liu, J.; Kang, J.; Fu, C.-Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Seyfi, R.; Kahaki, F.A.; Ebrahimi, T.; Montazersaheb, S.; Eyvazi, S.; Babaeipour, V.; Tarhriz, V. Antimicrobial Peptides (AMPs): Roles, Functions and Mechanism of Action. Int. J. Pept. Res. Ther. 2020, 26, 1451–1463. [Google Scholar] [CrossRef]
- Nayab, S.; Aslam, M.A.; Rahman, S.U.; Sindhu, Z.U.; Sajid, S.; Zafar, N.; Razaq, M.; Kanwar, R. Amanullah A Review of Antimicrobial Peptides: Its Function, Mode of Action and Therapeutic Potential. Int. J. Pept. Res. Ther. 2022, 28, 46. [Google Scholar] [CrossRef]
- Francis, F.; Chaudhary, N. Antimicrobial Peptides: Features and Modes of Action. In Antimicrobial Peptides; Elsevier: Amsterdam, The Netherlands, 2023; pp. 33–65. [Google Scholar] [CrossRef]
- Zheng, S.; Tu, Y.; Li, B.; Qu, G.; Li, A.; Peng, X.; Li, S.; Shao, C. Antimicrobial Peptide Biological Activity, Delivery Systems and Clinical Translation Status and Challenges. J. Transl. Med. 2025, 23, 292. [Google Scholar] [CrossRef]
- Gagandeep, K.R.; Narasingappa, R.B.; Vyas, G.V. Unveiling Mechanisms of Antimicrobial Peptide: Actions beyond the Membranes Disruption. Heliyon 2024, 10, e38079. [Google Scholar] [CrossRef]
- León-Buitimea, A.; Garza-Cárdenas, C.R.; Garza-Cervantes, J.A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. The Demand for New Antibiotics: Antimicrobial Peptides, Nanoparticles, and Combinatorial Therapies as Future Strategies in Antibacterial Agent Design. Front. Microbiol. 2020, 11, 1669. [Google Scholar] [CrossRef] [PubMed]
- Fatima, H.; Goel, N.; Sinha, R.; Khare, S.K. Recent Strategies for Inhibiting Multidrug-Resistant and β-Lactamase Producing Bacteria: A Review. Colloids Surf. B Biointerfaces 2021, 205, 111901. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, B.-H.; Jang, Y.-S. Understanding the Roles of Host Defense Peptides in Immune Modulation: From Antimicrobial Action to Potential as Adjuvants. J. Microbiol. Biotechnol. 2023, 33, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lu, W. Defensins: A Double-Edged Sword in Host Immunity. Front. Immunol. 2020, 11, 764. [Google Scholar] [CrossRef]
- Patterson-Delafield, J.; Martinez, R.J.; Lehrer, R.I. Microbicidal Cationic Proteins in Rabbit Alveolar Macrophages: A Potential Host Defense Mechanism. Infect. Immun. 1980, 30, 180–192. [Google Scholar] [CrossRef]
- Solanki, S.S.; Singh, P.; Kashyap, P.; Sansi, M.S.; Ali, S.A. Promising Role of Defensins Peptides as Therapeutics to Combat Against Viral Infection. Microb. Pathog. 2021, 155, 104930. [Google Scholar] [CrossRef]
- Kumaresan, V.; Kamaraj, Y.; Subramaniyan, S.; Punamalai, G. Understanding the Dynamics of Human Defensin Antimicrobial Peptides: Pathogen Resistance and Commensal Induction. Appl. Biochem. Biotechnol. 2024, 196, 6993–7024. [Google Scholar] [CrossRef]
- Bin Hafeez, A.; Jiang, X.; Bergen, P.J.; Zhu, Y. Antimicrobial Peptides: An Update on Classifications and Databases. Int. J. Mol. Sci. 2021, 22, 11691. [Google Scholar] [CrossRef]
- Salnikov, E.; Adélaïde, M.; Ramos-Martín, F.; Saad, A.; Schauer, J.; Cremanns, M.; Rima, M.; Aisenbrey, C.; Oueslati, S.; Naas, T.; et al. Cathelicidin-BF: A Potent Antimicrobial Peptide Leveraging Charge and Phospholipid Recruitment Against Multidrug-Resistant Clinical Bacterial Isolates. J. Am. Chem. Soc. 2025, 147, 11199–11215. [Google Scholar] [CrossRef]
- Talapko, J.; Meštrović, T.; Juzbašić, M.; Tomas, M.; Erić, S.; Horvat Aleksijević, L.; Bekić, S.; Schwarz, D.; Matić, S.; Neuberg, M.; et al. Antimicrobial Peptides—Mechanisms of Action, Antimicrobial Effects and Clinical Applications. Antibiotics 2022, 11, 1417. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, X.; Gao, Q. Effects and Mechanisms of Histatins as Novel Skin Wound-Healing Agents. J. Tissue Viability 2021, 30, 190–195. [Google Scholar] [CrossRef]
- Fazly Bazzaz, B.S.; Seyedi, S.; Hoseini Goki, N.; Khameneh, B. Human Antimicrobial Peptides: Spectrum, Mode of Action and Resistance Mechanisms. Int. J. Pept. Res. Ther. 2021, 27, 801–816. [Google Scholar] [CrossRef]
- Luong, A.D.; Buzid, A.; Luong, J.H.T. Important Roles and Potential Uses of Natural and Synthetic Antimicrobial Peptides (AMPs) in Oral Diseases: Cavity, Periodontal Disease, and Thrush. J. Funct. Biomater. 2022, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Mechkarska, M. Host-Defense Peptides with Therapeutic Potential from Skin Secretions of Frogs from the Family Pipidae. Pharmaceuticals 2014, 7, 58–77. [Google Scholar] [CrossRef] [PubMed]
- Mohamed Abd El-Aziz, T.; Soares, A.G.; Stockand, J.D. Snake Venoms in Drug Discovery: Valuable Therapeutic Tools for Life Saving. Toxins 2019, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Anandhan Sujatha, V.; Gopalakrishnan, C.; Anbarasu, A.; Ponnusamy, C.S.; Choudhary, R.; Saravanan Geetha, S.A.; Ramalingam, R. Beyond the Venom: Exploring the Antimicrobial Peptides from Androctonus Species of Scorpion. J. Pept. Sci. 2024, 30, e3613. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Song, Y.; Liu, Z.; Tang, X.; Tan, H. LC-AMP-I1, a Novel Venom-Derived Antimicrobial Peptide from the Wolf Spider. Lycosa Coelestis. Antimicrob. Agents Chemother. 2025, 69, e00424-24. [Google Scholar] [CrossRef]
- Dutta, P.; Sahu, R.K.; Dey, T.; Lahkar, M.D.; Manna, P.; Kalita, J. Beneficial Role of Insect-Derived Bioactive Components Against Inflammation and Its Associated Complications (Colitis and Arthritis) and Cancer. Chem. Biol. Interact. 2019, 313, 108824. [Google Scholar] [CrossRef]
- Bulet, P.; Urge, L.; Ohresser, S.; Hetru, C.; Otvos, L. Enlarged Scale Chemical Synthesis and Range of Activity of Drosocin, an O-Glycosylated Antibacterial Peptide of Drosophila. Eur. J. Biochem. 1996, 238, 64–69. [Google Scholar] [CrossRef]
- Liu, F.-F.; Ding, C.; Yang, L.-L.; Li, H.; Rao, X.-J. Identification and Analysis of Two Lebocins in the Oriental Armyworm Mythimna separata. Dev. Comp. Immunol. 2021, 116, 103962. [Google Scholar] [CrossRef]
- Li, J.; Hu, S.; Jian, W.; Xie, C.; Yang, X. Plant Antimicrobial Peptides: Structures, Functions, and Applications. Bot. Stud. 2021, 62, 5. [Google Scholar] [CrossRef]
- Tam, J.; Wang, S.; Wong, K.; Tan, W. Antimicrobial Peptides from Plants. Pharmaceuticals 2015, 8, 711–757. [Google Scholar] [CrossRef] [PubMed]
- Darbandi, A.; Asadi, A.; Mahdizade Ari, M.; Ohadi, E.; Talebi, M.; Halaj Zadeh, M.; Darb Emamie, A.; Ghanavati, R.; Kakanj, M. Bacteriocins: Properties and Potential Use as Antimicrobials. Clin. Lab. Anal. 2022, 36, e24093. [Google Scholar] [CrossRef] [PubMed]
- Zimina, M.; Babich, O.; Prosekov, A.; Sukhikh, S.; Ivanova, S.; Shevchenko, M.; Noskova, S. Overview of Global Trends in Classification, Methods of Preparation and Application of Bacteriocins. Antibiotics 2020, 9, 553. [Google Scholar] [CrossRef] [PubMed]
- Soltani, S.; Hammami, R.; Cotter, P.D.; Rebuffat, S.; Said, L.B.; Gaudreau, H.; Bédard, F.; Biron, E.; Drider, D.; Fliss, I. Bacteriocins as a New Generation of Antimicrobials: Toxicity Aspects and Regulations. FEMS Microbiol. Rev. 2021, 45, fuaa039. [Google Scholar] [CrossRef]
- Negash, A.W.; Tsehai, B.A. Current Applications of Bacteriocin. Int. J. Microbiol. 2020, 2020, 4374891. [Google Scholar] [CrossRef]
- Benítez-Chao, D.F.; León-Buitimea, A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. Bacteriocins: An Overview of Antimicrobial, Toxicity, and Biosafety Assessment by in Vivo Models. Front. Microbiol. 2021, 12, 630695. [Google Scholar] [CrossRef]
- Ismael, M.; Huang, M.; Zhong, Q. The Bacteriocins Produced by Lactic Acid Bacteria and the Promising Applications in Promoting Gastrointestinal Health. Foods 2024, 13, 3887. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Karimi, A.; Fallah, F.; Akhavan, M.M. Overview of Ribosomal and Non-Ribosomal Antimicrobial Peptides Produced by Gram Positive Bacteria. Cell Mol. Biol. (Noisy-Le-Grand) 2017, 63, 20–32. [Google Scholar] [CrossRef]
- Duban, M.; Cociancich, S.; Leclère, V. Nonribosomal Peptide Synthesis Definitely Working Out of the Rules. Microorganisms 2022, 10, 577. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, C.; Chen, K.; Zhong, W.; Xu, Y.; Molnár, I. Engineering the Biosynthesis of Fungal Nonribosomal Peptides. Nat. Prod. Rep. 2023, 40, 62–88. [Google Scholar] [CrossRef]
- Bann, S.J.; Ballantine, R.D.; Cochrane, S.A. The Tridecaptins: Non-Ribosomal Peptides That Selectively Target Gram-Negative Bacteria. RSC Med. Chem. 2021, 12, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Abdel Monaim, S.A.; Somboro, A.M.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Bacteria Hunt Bacteria Through an Intriguing Cyclic Peptide. ChemMedChem 2019, 14, 24–51. [Google Scholar] [CrossRef] [PubMed]
- Nuske, M.R.; Zhong, J.; Huang, R.; Sarojini, V.; Chen, J.L.Y.; Squire, C.J.; Blaskovich, M.A.T.; Leung, I.K.H. Adjuvant Strategies to Tackle Mcr-Mediated Polymyxin Resistance. RSC Med. Chem. 2025, 16, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, C.J.; Martin, N.I. Recent Advances in the Development of Polymyxin Antibiotics: 2010–2023. ACS Infect. Dis. 2024, 10, 1056–1079. [Google Scholar] [CrossRef]
- Machushynets, N.V.; Al Ayed, K.; Terlouw, B.R.; Du, C.; Buijs, N.P.; Willemse, J.; Elsayed, S.S.; Schill, J.; Trebosc, V.; Pieren, M.; et al. Discovery and Derivatization of Tridecaptin Antibiotics with Altered Host Specificity and Enhanced Bioactivity. ACS Chem. Biol. 2024, 19, 1106–1115. [Google Scholar] [CrossRef]
- Zhu, S. Discovery of Six Families of Fungal Defensin-Like Peptides Provides Insights into Origin and Evolution of the CSαβ Defensins. Mol. Immunol. 2008, 45, 828–838. [Google Scholar] [CrossRef]
- Hou, X.; Sun, R.; Feng, Y.; Zhang, R.; Zhu, T.; Che, Q.; Zhang, G.; Li, D. Peptaibols: Diversity, Bioactivity, and Biosynthesis. Eng. Microbiol. 2022, 2, 100026. [Google Scholar] [CrossRef]
- Gavryushina, I.A.; Georgieva, M.L.; Kuvarina, A.E.; Sadykova, V.S. Peptaibols as Potential Antifungal and Anticancer Antibiotics: Current and Foreseeable Development (Review). Appl. Biochem. Microbiol. 2021, 57, 556–563. [Google Scholar] [CrossRef]
- Víglaš, J.; Dobiasová, S.; Viktorová, J.; Ruml, T.; Repiská, V.; Olejníková, P.; Gbelcová, H. Peptaibol-Containing Extracts of Trichoderma atroviride and the Fight Against Resistant Microorganisms and Cancer Cells. Molecules 2021, 26, 6025. [Google Scholar] [CrossRef]
- Talapko, J.; Škrlec, I. The Principles, Mechanisms, and Benefits of Unconventional Agents in the Treatment of Biofilm Infection. Pharmaceuticals 2020, 13, 299. [Google Scholar] [CrossRef]
- Kalelkar, P.P.; Riddick, M.; García, A.J. Biomaterial-Based Antimicrobial Therapies for the Treatment of Bacterial Infections. Nat. Rev. Mater. 2021, 7, 39–54. [Google Scholar] [CrossRef]
- Lima, P.G.; Oliveira, J.T.A.; Amaral, J.L.; Freitas, C.D.T.; Souza, P.F.N. Synthetic Antimicrobial Peptides: Characteristics, Design, and Potential as Alternative Molecules to Overcome Microbial Resistance. Life Sci. 2021, 278, 119647. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Chetia, M.; Chatterjee, S. Antimicrobial Peptides and Proteins: From Nature’s Reservoir to the Laboratory and Beyond. Front. Chem. 2021, 9, 691532. [Google Scholar] [CrossRef] [PubMed]
- Svenson, J.; Molchanova, N.; Schroeder, C.I. Antimicrobial Peptide Mimics for Clinical Use: Does Size Matter? Front. Immunol. 2022, 13, 915368. [Google Scholar] [CrossRef] [PubMed]
- Lata, M.; Telang, V.; Gupta, P.; Pant, G.; Kalyan, M.; Arockiaraj, J.; Pasupuleti, M. Synthetic Short Cryptic Antimicrobial Peptides as Templates for the Development of Novel Biotherapeutics Against WHO Priority Pathogen. Int. J. Pept. Res. Ther. 2024, 30, 57. [Google Scholar] [CrossRef]
- Yan, J.; Cai, J.; Zhang, B.; Wang, Y.; Wong, D.F.; Siu, S.W.I. Recent Progress in the Discovery and Design of Antimicrobial Peptides Using Traditional Machine Learning and Deep Learning. Antibiotics 2022, 11, 1451. [Google Scholar] [CrossRef]
- Saubi, C.; Carratalá, J.V.; Bello-Madruga, R.; López-Cano, A.; Navarro, S.; Arís, A.; Garcia-Fruitós, E. Evaluating Host Defense Peptides: A Comparative Analysis of Synthetic Peptides and Recombinant Concatemers. Biomolecules 2025, 15, 980. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, M.; Lai, R.; Zhang, Z. Chemical Modifications to Increase the Therapeutic Potential of Antimicrobial Peptides. Peptides 2021, 146, 170666. [Google Scholar] [CrossRef]
- Qi, Y.-K.; Zheng, J.-S.; Liu, L. Mirror-Image Protein and Peptide Drug Discovery Through Mirror-Image Phage Display. Chem 2024, 10, 2390–2407. [Google Scholar] [CrossRef]
- Xu, S.; Tan, P.; Tang, Q.; Wang, T.; Ding, Y.; Fu, H.; Zhang, Y.; Zhou, C.; Song, M.; Tang, Q.; et al. Enhancing the Stability of Antimicrobial Peptides: From Design Strategies to Applications. Chem. Eng. J. 2023, 475, 145923. [Google Scholar] [CrossRef]
- Jin, K.; Sam, I.; Po, K.H.L.; Lin, D.A.; Ghazvini Zadeh, E.H.; Chen, S.; Yuan, Y.; Li, X. Total synthesis of teixobactin. Nat. Commun. 2016, 7, 12394. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.K.; Tang, X.; Wei, N.N.; Pang, C.J.; Du, S.S.; Wang, K. Discovery, synthesis, and optimization of teixobactin, a novel antibiotic without detectable bacterial resistance. J. Pept. Sci. 2022, 28, e3428. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, D.; Zhao, C.-X. Recent Achievements and Perspectives for Large-Scale Recombinant Production of Antimicrobial Peptides. Appl. Microbiol. Biotechnol. 2019, 103, 659–671. [Google Scholar] [CrossRef]
- Shanmugaraj, B.; Bulaon, C.J.I.; Malla, A.; Phoolcharoen, W. Biotechnological Insights on the Expression and Production of Antimicrobial Peptides in Plants. Molecules 2021, 26, 4032. [Google Scholar] [CrossRef]
- Mazurkiewicz-Pisarek, A.; Baran, J.; Ciach, T. Antimicrobial Peptides: Challenging Journey to the Pharmaceutical, Biomedical, and Cosmeceutical Use. Int. J. Mol. Sci. 2023, 24, 9031. [Google Scholar] [CrossRef]
- Zhang, Q. Antimicrobial Peptides: From Discovery to Developmental Applications. Appl. Environ. Microbiol. 2025, 91, e02115-24. [Google Scholar] [CrossRef]
- Asif, F.; Zaman, S.U.; Arnab, M.K.; Hasan, M.; Islam, M.M. Antimicrobial Peptides as Therapeutics: Confronting Delivery Challenges to Optimize Efficacy. Microbe 2024, 2, 100051. [Google Scholar] [CrossRef]
- Arsene, M.M.; Jorelle, A.B.; Sarra, S.; Viktorovna, P.I.; Davares, A.K.; Ingrid, N.K.; Steve, A.A.; Andreevna, S.L.; Vyacheslavovna, Y.N.; Carime, B.Z. Short Review on the Potential Alternatives to Antibiotics in the Era of Antibiotic Resistance. J. Appl. Pharm. Sci. 2022, 12, 29–40. [Google Scholar] [CrossRef]
- Divyashree, M.; Mani, M.K.; Reddy, D.; Kumavath, R.; Ghosh, P.; Azevedo, V.; Barh, D. Clinical Applications of Antimicrobial Peptides (AMPs): Where Do We Stand Now? Protein Pept. Lett. 2020, 27, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O. FDA-Approved Antibacterials and Echinocandins. Antibiotics 2025, 14, 166. [Google Scholar] [CrossRef] [PubMed]
- Baindara, P.; Kumari, S.; Dinata, R.; Mandal, S.M. Antimicrobial Peptides: Evolving Soldiers in the Battle against Drug-Resistant Superbugs. Mol. Biol. Rep. 2025, 52, 432. [Google Scholar] [CrossRef] [PubMed]
- Duong, L.; Gross, S.P.; Siryaporn, A. Developing Antimicrobial Synergy with AMPs. Front. Med. Technol. 2021, 3, 640981. [Google Scholar] [CrossRef]
- Mhlongo, J.T.; Waddad, A.Y.; Albericio, F.; De La Torre, B.G. Antimicrobial Peptide Synergies for Fighting Infectious Diseases. Adv. Sci. 2023, 10, 2300472. [Google Scholar] [CrossRef]
- Zharkova, M.S.; Orlov, D.S.; Golubeva, O.Y.; Chakchir, O.B.; Eliseev, I.E.; Grinchuk, T.M.; Shamova, O.V. Application of Antimicrobial Peptides of the Innate Immune System in Combination with Conventional Antibiotics—A Novel Way to Combat Antibiotic Resistance? Front. Cell. Infect. Microbiol. 2019, 9, 128. [Google Scholar] [CrossRef]
- Kampshoff, F.; Willcox, M.D.P.; Dutta, D. A Pilot Study of the Synergy between Two Antimicrobial Peptides and Two Common Antibiotics. Antibiotics 2019, 8, 60. [Google Scholar] [CrossRef]
- Taheri-Araghi, S. Synergistic Action of Antimicrobial Peptides and Antibiotics: Current Understanding and Future Directions. Front. Microbiol. 2024, 15, 1390765. [Google Scholar] [CrossRef]
- Xiao, G.; Li, J.; Sun, Z. The Combination of Antibiotic and Non-Antibiotic Compounds Improves Antibiotic Efficacy Against Multidrug-Resistant Bacteria. Int. J. Mol. Sci. 2023, 24, 15493. [Google Scholar] [CrossRef]
- Dhanda, G.; Acharya, Y.; Haldar, J. Antibiotic Adjuvants: A Versatile Approach to Combat Antibiotic Resistance. ACS Omega 2023, 8, 10757–10783. [Google Scholar] [CrossRef]
- Ye, Z.; Fu, L.; Li, S.; Chen, Z.; Ouyang, J.; Shang, X.; Liu, Y.; Gao, L.; Wang, Y. Synergistic Collaboration between AMPs and Non-Direct Antimicrobial Cationic Peptides. Nat. Commun. 2024, 15, 7319. [Google Scholar] [CrossRef]
- Li, J.; Fernández-Millán, P.; Boix, E. Synergism between Host Defence Peptides and Antibiotics Against Bacterial Infections. Curr. Top. Med. Chem. 2020, 20, 1238–1263. [Google Scholar] [CrossRef]
- Shemyakin, I.G.; Firstova, V.V.; Fursova, N.K.; Abaev, I.V.; Filippovich, S.Y.; Ignatov, S.G.; Dyatlov, I.A. Next-Generation Antibiotics, Bacteriophage Endolysins, and Nanomaterials for Combating Pathogens. Biochem. Mosc. 2020, 85, 1374–1388. [Google Scholar] [CrossRef]
- Hemmati, S.; Saeidikia, Z.; Seradj, H.; Mohagheghzadeh, A. Immunomodulatory Peptides as Vaccine Adjuvants and Antimicrobial Agents. Pharmaceuticals 2024, 17, 201. [Google Scholar] [CrossRef]
- Panjla, A.; Kaul, G.; Chopra, S.; Titz, A.; Verma, S. Short Peptides and Their Mimetics as Potent Antibacterial Agents and Antibiotic Adjuvants. ACS Chem. Biol. 2021, 16, 2731–2745. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Loya-Perez, V.; Franzen, J.; Denton, P.W.; Conda-Sheridan, M.; Rodrigues De Almeida, N. Designing Peptide Amphiphiles as Novel Antibacterials and Antibiotic Adjuvants Against Gram-Negative Bacteria. Bioorganic Med. Chem. 2023, 94, 117481. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Zheng, X.; Mao, F.; Cao, Q.; Cao, Q.; Zhu, J.; Li, X.; Lan, L.; Li, B.; Li, J. Novel Niclosamide-Derived Adjuvants Elevating the Efficacy of Polymyxin B Against MDR Pseudomonas Aeruginosa DK2. Eur. J. Med. Chem. 2022, 236, 114318. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Han, H.; Wu, C.; Li, Q.; Hu, H.; Liu, W.; Shi, D.; Chen, F.; Lan, L.; Li, J.; et al. Discovery of a Novel Polymyxin Adjuvant Against Multidrug-Resistant Gram-Negative Bacteria Through Oxidative Stress Modulation. Acta Pharm. Sin. B 2025, 15, 1680–1695. [Google Scholar] [CrossRef]
- Taylor, S.D.; Palmer, M. The Action Mechanism of Daptomycin. Bioorg. Med. Chem. 2016, 24, 6253–6268. [Google Scholar] [CrossRef]
- Lev, K.; Kunz Coyne, A.J.; Kebriaei, R.; Morrisette, T.; Stamper, K.; Holger, D.J.; Canfield, G.S.; Duerkop, B.A.; Arias, C.A.; Rybak, M.J. Evaluation of Bacteriophage-Antibiotic Combination Therapy for Biofilm-Embedded MDR Enterococcus faecium. Antibiotics 2022, 11, 392. [Google Scholar] [CrossRef]
- Silva, A.R.P.; Guimarães, M.S.; Rabelo, J.; Belén, L.H.; Perecin, C.J.; Farías, J.G.; Santos, J.H.P.M.; Rangel-Yagui, C.O. Recent advances in the design of antimicrobial peptide conjugates. J. Mater. Chem. B. 2022, 10, 3587–3600. [Google Scholar] [CrossRef]
- Sun, H.; Hong, Y.; Xi, Y.; Zou, Y.; Gao, J.; Du, J. Synthesis, Self-Assembly, and Biomedical Applications of Antimicrobial Peptide–Polymer Conjugates. Biomacromolecules 2018, 19, 1701–1720. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.-H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef] [PubMed]
- Alkhzem, A.H.; Woodman, T.J.; Blagbrough, I.S. Design and Synthesis of Hybrid Compounds as Novel Drugs and Medicines. RSC Adv. 2022, 12, 19470–19484. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, C.H.; Luchian, T.; Park, Y. Antimicrobial Peptide CMA3 Derived from the CA-MA Hybrid Peptide: Antibacterial and Anti-Inflammatory Activities with Low Cytotoxicity and Mechanism of Action in Escherichia Coli. Antimicrob. Agents Chemother. 2016, 60, 495–506. [Google Scholar] [CrossRef]
- Kim, H.; Jang, J.H.; Kim, S.C.; Cho, J.H. Development of a Novel Hybrid Antimicrobial Peptide for Targeted Killing of Pseudomonas aeruginosa. Eur. J. Med. Chem. 2020, 185, 111814. [Google Scholar] [CrossRef]
- Kim, H.; Jang, J.H.; Kim, S.C.; Cho, J.H. Enhancement of the Antimicrobial Activity and Selectivity of GNU7 Against Gram-Negative Bacteria by Fusion with LPS-Targeting Peptide. Peptides 2016, 82, 60–66. [Google Scholar] [CrossRef]
- Cui, Z.; Luo, Q.; Bannon, M.S.; Gray, V.P.; Bloom, T.G.; Clore, M.F.; Hughes, M.A.; Crawford, M.A.; Letteri, R.A. Molecular Engineering of Antimicrobial Peptide (AMP)–Polymer Conjugates. Biomater. Sci. 2021, 9, 5069–5091. [Google Scholar] [CrossRef]
- Li, C.; Li, T.; Tian, X.; An, W.; Wang, Z.; Han, B.; Tao, H.; Wang, J.; Wang, X. Research Progress on the PEGylation of Therapeutic Proteins and Peptides (TPPs). Front. Pharmacol. 2024, 15, 1353626. [Google Scholar] [CrossRef]
- Bellotto, O.; Semeraro, S.; Bandiera, A.; Tramer, F.; Pavan, N.; Marchesan, S. Polymer Conjugates of Antimicrobial Peptides (AMPs) with d-Amino Acids (d-Aa): State of the Art and Future Opportunities. Pharmaceutics 2022, 14, 446. [Google Scholar] [CrossRef]
- Patrulea, V.; Gan, B.-H.; Perron, K.; Cai, X.; Abdel-Sayed, P.; Sublet, E.; Ducret, V.; Nerhot, N.P.; Applegate, L.A.; Borchard, G.; et al. Synergistic Effects of Antimicrobial Peptide Dendrimer-Chitosan Polymer Conjugates Against Pseudomonas aeruginosa. Carbohydr. Polym. 2022, 280, 119025. [Google Scholar] [CrossRef]
- Zhang, L.; Bulaj, G. Converting Peptides into Drug Leads by Lipidation. Curr. Med. Chem. 2012, 19, 1602–1618. [Google Scholar] [CrossRef]
- Bellavita, R.; Braccia, S.; Galdiero, S.; Falanga, A. Glycosylation and Lipidation Strategies: Approaches for Improving Antimicrobial Peptide Efficacy. Pharmaceuticals 2023, 16, 439. [Google Scholar] [CrossRef]
- Rounds, T.; Straus, S.K. Lipidation of Antimicrobial Peptides as a Design Strategy for Future Alternatives to Antibiotics. Int. J. Mol. Sci. 2020, 21, 9692. [Google Scholar] [CrossRef] [PubMed]
- Myšková, A.; Sýkora, D.; Kuneš, J.; Maletínská, L. Lipidization as a Tool Toward Peptide Therapeutics. Drug Deliv. 2023, 30, 2284685. [Google Scholar] [CrossRef] [PubMed]
- Makowska, M.; Kosikowska-Adamus, P.; Zdrowowicz, M.; Wyrzykowski, D.; Prahl, A.; Sikorska, E. Lipidation of Naturally Occurring α-Helical Antimicrobial Peptides as a Promising Strategy for Drug Design. Int. J. Mol. Sci. 2023, 24, 3951. [Google Scholar] [CrossRef]
- Tortorella, A.; Leone, L.; Lombardi, A.; Pizzo, E.; Bosso, A.; Winter, R.; Petraccone, L.; Del Vecchio, P.; Oliva, R. The Impact of N-Glycosylation on the Properties of the Antimicrobial Peptide LL-III. Sci. Rep. 2023, 13, 3733. [Google Scholar] [CrossRef]
- Yang, Z.; He, S.; Wu, H.; Yin, T.; Wang, L.; Shan, A. Nanostructured Antimicrobial Peptides: Crucial Steps of Overcoming the Bottleneck for Clinics. Front. Microbiol. 2021, 12, 710199. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Araújo, P.M. Antimicrobial Polymer−Based Assemblies: A Review. Int. J. Mol. Sci. 2021, 22, 5424. [Google Scholar] [CrossRef]
- Skwarecki, A.S.; Milewski, S.; Schielmann, M.; Milewska, M.J. Antimicrobial Molecular Nanocarrier–Drug Conjugates. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 2215–2240. [Google Scholar] [CrossRef]
- Copling, A.; Akantibila, M.; Kumaresan, R.; Fleischer, G.; Cortes, D.; Tripathi, R.S.; Carabetta, V.J.; Vega, S.L. Recent Advances in Antimicrobial Peptide Hydrogels. Int. J. Mol. Sci. 2023, 24, 7563. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zou, L.; Wang, J.; Jin, Q.; Ji, J. Stimuli-responsive Nanoplatforms for Antibacterial Applications. WIREs Nanomed. Nanobiotechnol. 2022, 14, e1775. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, M.; Filho, D.; Ayala, A.N.; Rafael, D.; Andrade, F.; Marican, A.; Vijayakumar, S.; Durán-Lara, E.F. Hydrogel-Antimicrobial Peptide Association: A Novel and Promising Strategy to Combat Resistant Infections. Colloids Surf. B Biointerfaces 2025, 247, 114451. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, Y.; Li, H.; Niu, X.; Zhang, D.; Wang, K. Antimicrobial Hydrogels: Potential Materials for Medical Application. Small 2024, 20, 2304047. [Google Scholar] [CrossRef]
- Borro, B.C.; Nordström, R.; Malmsten, M. Microgels and Hydrogels as Delivery Systems for Antimicrobial Peptides. Colloids Surf. B Biointerfaces 2020, 187, 110835. [Google Scholar] [CrossRef]
- Pulat, G.; Çelebi, N.N.; Bilgiç, E. The Effect of Immobilization Methods of P9-4 Antimicrobial Peptide onto Gelatin Methacrylate on Multidrug-Resistant Bacteria: A Comparative Study. Macromol. Biosci. 2025, 25, 2400324. [Google Scholar] [CrossRef]
- Sharmin, S.; Rahaman, M.M.; Sarkar, C.; Atolani, O.; Islam, M.T.; Adeyemi, O.S. Nanoparticles as Antimicrobial and Antiviral Agents: A Literature-Based Perspective Study. Heliyon 2021, 7, e06456. [Google Scholar] [CrossRef]
- Gakiya-Teruya, M.; Palomino-Marcelo, L.; Pierce, S.; Angeles-Boza, A.M.; Krishna, V.; Rodriguez-Reyes, J.C.F. Enhanced Antimicrobial Activity of Silver Nanoparticles Conjugated with Synthetic Peptide by Click Chemistry. J. Nanopart. Res. 2020, 22, 90. [Google Scholar] [CrossRef]
- Zharkova, M.S.; Golubeva, O.Y.; Orlov, D.S.; Vladimirova, E.V.; Dmitriev, A.V.; Tossi, A.; Shamova, O.V. Silver Nanoparticles Functionalized with Antimicrobial Polypeptides: Benefits and Possible Pitfalls of a Novel Anti-Infective Tool. Front. Microbiol. 2021, 12, 750556. [Google Scholar] [CrossRef]
- Zheng, K.; Setyawati, M.I.; Lim, T.-P.; Leong, D.T.; Xie, J. Antimicrobial Cluster Bombs: Silver Nanoclusters Packed with Daptomycin. ACS Nano 2016, 10, 7934–7942. [Google Scholar] [CrossRef]
- Gao, J.; Na, H.; Zhong, R.; Yuan, M.; Guo, J.; Zhao, L.; Wang, Y.; Wang, L.; Zhang, F. One Step Synthesis of Antimicrobial Peptide Protected Silver Nanoparticles: The Core-Shell Mutual Enhancement of Antibacterial Activity. Colloids Surf. B Biointerfaces 2020, 186, 110704. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, Y.; Wang, H.; Zhu, M.; Feng, W.; Liang, G. Enhanced Antibacterial and Anti-Biofilm Activities of Antimicrobial Peptides Modified Silver Nanoparticles. Int. J. Nanomed. 2021, 16, 4831–4846. [Google Scholar] [CrossRef] [PubMed]
- Boge, L.; Hallstensson, K.; Ringstad, L.; Johansson, J.; Andersson, T.; Davoudi, M.; Larsson, P.T.; Mahlapuu, M.; Håkansson, J.; Andersson, M. Cubosomes for topical delivery of the antimicrobial peptide LL-37. Eur. J. Pharm. Biopharm. 2019, 134, 60–67. [Google Scholar] [CrossRef]
- Yi, R.; Shi, Y.; Cao, X.; Pan, C. Actinomycetes: Treasure Trove for Discovering Novel Antibiotic Candidates. Eur. J. Med. Chem. 2025, 286, 117317. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
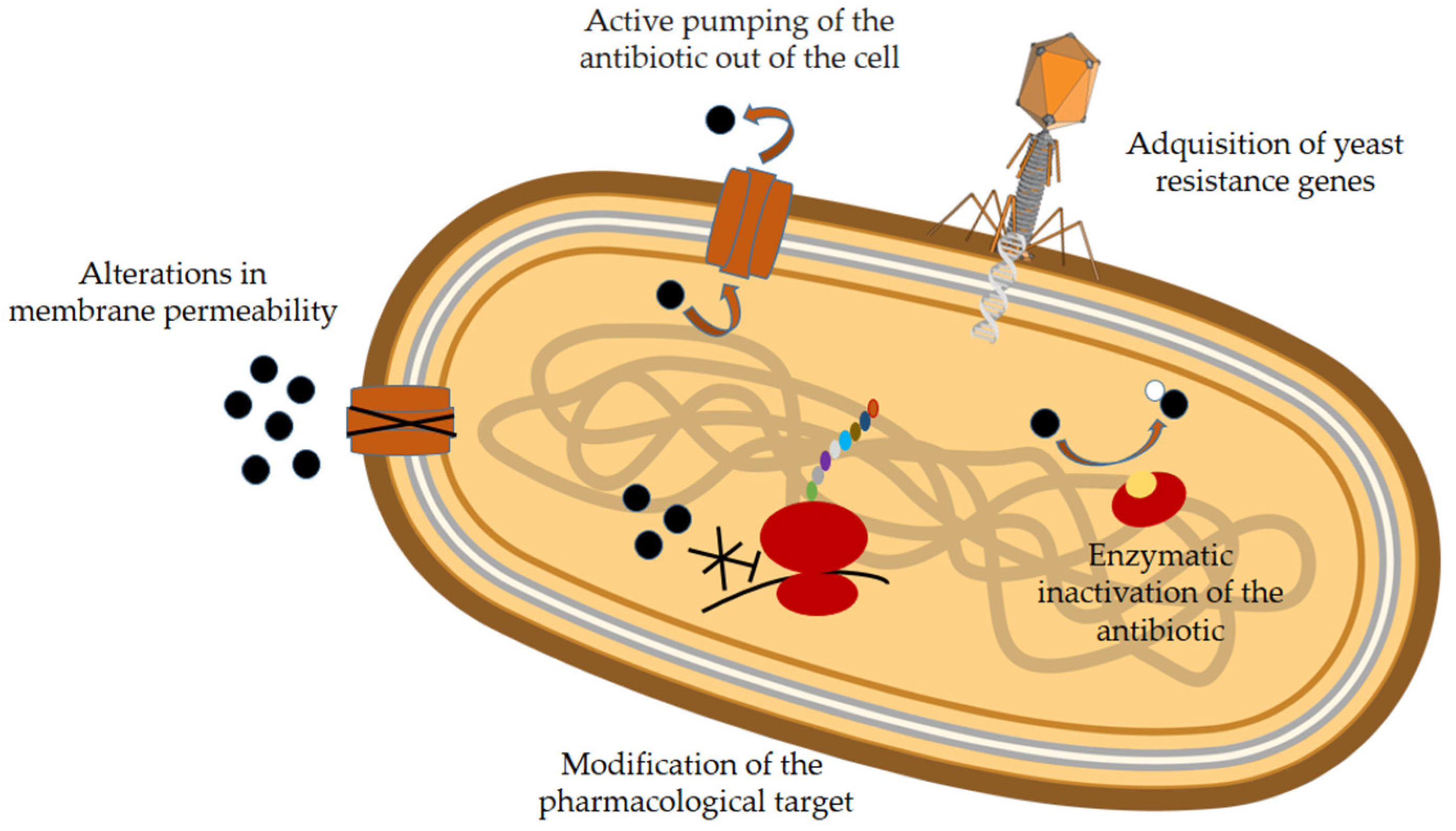
| Mechanism | Example |
|---|---|
| Modification of the pharmacological target | Modification of the structure of penicillin-binding proteins, which allow for the synthesis of peptidoglycan, the component that makes up the cell wall (in MRSA) |
| Modification of the last amino acid of peptidoglycan (in glycopeptide-resistant bacteria) | |
| Remodeling of the chemical structure of bacterial gyrase and/or topoisomerase IV (in fluoroquinolone-resistant bacteria) | |
| Expression of vanA and vanB genes products, which modify cell wall precursors and decrease vancomycin binding (in glycopeptide-resistant bacteria) | |
| Expression of ermb gene, which codifies ribosome methylase enzyme. This enzyme modifies rRNA and impedes drug binding to the ribosome (in macrolide and tetracycline-resistant bacteria) | |
| Alterations in membrane permeability that prevent antibiotic penetration into the cell | Decreased sensitivity of porins to beta-lactam antibiotics and fluoroquinolones. |
| Active pumping of the antibiotic out of the cell | MSF provides resistance to fluoroquinolones, macrolides, linezolid, … (in strains of S. aureus and Escherichia coli) |
| MATE provides resistance to fluoroquinolones and some aminoglucosides (in Neisseria gonorrhoeae and S. aureus) | |
| Enzymatic inactivation of the antibiotic | Modification of macrolides by esterases that hydrolyze their lactone ring (in Salmonella enterica, Pseudomonas spp., Vibrio cholera, and Klebsiella spp.) |
| Phosphorylation, adenylation or acetylation of aminoglycosides (in kanamycin, neomycin, and paromomycin-resistant bacteria) |
| Compound | Via | Application | Target Species | Year of Approval |
|---|---|---|---|---|
| Anidulafungin (semisynthetic lipopeptide) | Intravenous | Treatment of invasive candidiasis | Fungi (mainly Candida) | 2006 |
| 1 Atazanavir (azapeptide) | Oral | HIV-infection | HIV-1 | 2014 |
| 1 Bacitracin (Cyclic peptide) | Topical | Pneumonia, localized skin and eye infections and wound infections | Gram-positive | 1948 |
| 1 Boceprevir | Oral | Chronic hepatitis C genotype 1 | Hepatitis C virus | 2011 |
| 1 Bulevirtide | Subcutaneous | Chronic hepatitis D | Hepatitis D virus | 2015 (EMA) |
| 3 Carfilzomib | Intravenous | Multiple myeloma cells | 2012 | |
| Caspofungin (semisynthetic lipopetide) | Intravenous | Fungal infections | Candida and Aspergillus spp. and other fungi | 2001 |
| 2 Colistin (lipopeptide) | Auricular (otic) | Infections caused by Gram-negative bacteria resistant to other antibiotics | P. aeruginosa and some Gram-negative bacilli | 1959 |
| Dalbavancin (semisynthetic lipoglycopeptide) | Intravenous | Acute bacterial skin infections | S. aureus S. pyogenes Streptococcus agalactiae | 2014 |
| 2 Daptomycin (lipopeptide) | Intravenous | Particularly complicated infections of the skin and its structures | S.aureus | 2003 |
| 1 Enfuvirtide | Subcutaneous | Infections produced by HIV-1 | HIV-1 | 2003 |
| 1 Glecaprevir | Oral | Chronic hepatitis C genotype 1–6 | Hepatitis C virus | 2017 |
| 2 Gramicidin D | Topical | Infected surface wounds, as well as eye, nose, and throat infections | Most Gram-positive and some Gram-negative bacteria | 1955 |
| 1 Invinavir | Oral | Treatment of HIV infection | HIV-1 | 1996 |
| 3 Liraglutide | Subcutaneous | Improve blood sugar, reduce the risk of major cardiovascular events | 2010 | |
| 1 Lopinavir | Oral | HIV infections | HIV-1 | 2000 |
| 2 Interferon alfa 2B (leucocyte fraction of human blood) | Oral | Hepatitis B infections and cancer | Hepatitis B virus | 1986 |
| Micafungin (Semisynthetic lipopeptide) | Intravenous | Fungi (main Candida) | 2005 | |
| 1 Nelfinavir | Oral | HIV infections | HIV-1 | 1997 |
| 2 Nisin (polycyclic peptide) | Food preservative | Gram-positive bacteria and their spores | 1988 | |
| Oritavancin (Semisynthetic glycopeptide) | Intravenous | Skin and skin structure infections | S. aureus | 2014 |
| Rezafungin (Semisynthetic lipopeptide) | Intravenous | Fungal infections | Candida, Aspergillus, and Pneumocystis spp. | 2023 |
| 1 Ritonavir (Biomimetic peptide) | Oral | HIV infection | HIV-1 | 2015 |
| 3 Romidepsin (naturally occurring bicyclic depsipeptide) | Intravenous | Cutaneous T-cell lymphoma | 2009 | |
| 1 Saquinavir | Oral | HIV infection | HIV-1 | 1995 |
| Teicoplanin (Semisynthetic glycopeptide) | Intravenous | Life threatening Gram-positive bacterial infections | E. faecalis and MRSA | 1988 (EMA) |
| Telaprevir (Semisynthetic linear peptide) | Oral | Chronic hepatitis C | Hepatitis C virus | 2011 |
| Telavancin (Semisynthetic lipoglycopeptide, derivative of vancomycin) | Intravenous | Complicated infections of skin and skin structures | S. aureus, S. pyogenes, and vancomycin-susceptible E. faecalis | 2009 |
| 1 Tesamorelin | Subcutaneous | Reduce the amount of abdominal fat that is excessive and keep it from returning | HIV-1 | 2010 |
| 2 Vancomycin (glycopeptide) | Intravenous, oral | MRSA infections | MRSA | 1954 |
| Polymer Architecture: Properties of the AMP–Polymer Conjugates and Antimicrobial Performance | |
| 1 Linear AMP— polymer conjugates 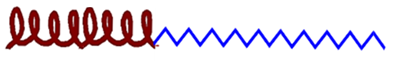 | A single AMP is attached to a linear polymer chain. Improved stability and biocompatibility. The antimicrobial activity and cytotoxicity depend on the size of the polymer: Conjugation to polymers decreases a peptide’s cytotoxic effects and antibacterial activity because the peptide is likely buried within the polymer. Generally, shortening the polymer increases its antimicrobial effect. |
| 1 Comb/brush AMP— polymer conjugates 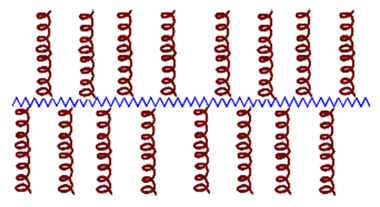 | Macromolecular (polymeric or peptide) pendent groups on a main chain, which can be a polymer or a peptide. The local concentration of AMPs in a single polymer molecule generates potent bactericidal conjugates. The bactericidal activity and cytotoxic effects of these conjugates can be modulated by modifying the density, the length and the orientation of the AMPs and the length of the polymer chain. In general, conjugating AMPs to this scaffold reduces cytotoxicity while preserving or enhancing their bactericidal activity. |
| 1 Star-shaped AMP— polymer conjugates 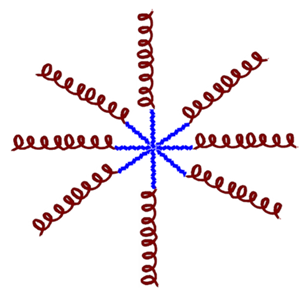 | Multiple polymer chains (arms) emanating from a central core. The balance of bactericidal activity, mammalian cell toxicity, protease resistance, and conjugate aggregation is influenced by the number and length of the arms, as well as their composition. They exhibit a high local concentration of AMP and potent antimicrobial activity. |
| 1 Hyperbranched AMP– polymer conjugates 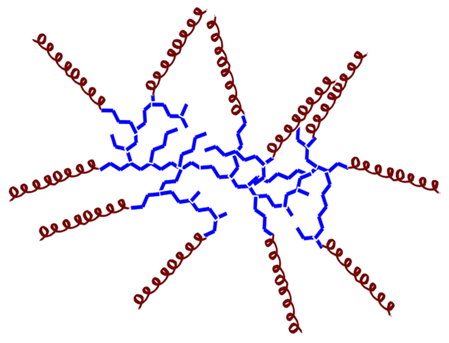 | There are not many studies of conjugates with this architecture. Generally, they exhibit reduced antimicrobial activity compared to AMP alone. However, they also demonstrate lower toxicity to mammalian cells and improved compatibility with blood components. These properties can be modulated by hyperbranched architecture, composition, degree of branching, molecular weight, and terminal group functionality. Decreasing the molecular weight of the hyperbranched polymer is related to enhanced antimicrobial activity. |
| Supramolecular Assembly of AMP–Polymer Conjugates | |
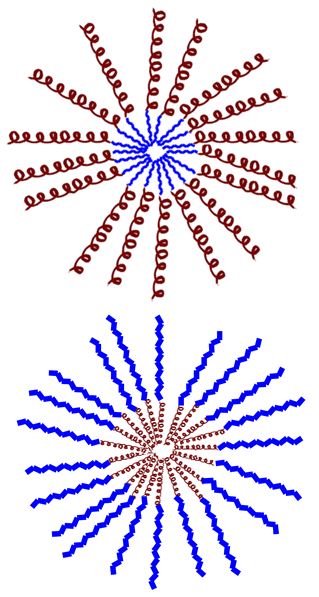 | 1 Micelles formed by the assembly of a hydrophilic AMP attached to a hydrophobic polymer. Increased antimicrobial activity. Reduced toxicity. 1 Micelles formed by the assembly of a hydrophobic AMP attached to a hydrophilic polymer. They exhibit lower cytotoxicity and protection against degradation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Olmo, M.; Andreu, C. Current Status of the Application of Antimicrobial Peptides and Their Conjugated Derivatives. Molecules 2025, 30, 3070. https://doi.org/10.3390/molecules30153070
del Olmo M, Andreu C. Current Status of the Application of Antimicrobial Peptides and Their Conjugated Derivatives. Molecules. 2025; 30(15):3070. https://doi.org/10.3390/molecules30153070
Chicago/Turabian Styledel Olmo, Marcel·lí, and Cecilia Andreu. 2025. "Current Status of the Application of Antimicrobial Peptides and Their Conjugated Derivatives" Molecules 30, no. 15: 3070. https://doi.org/10.3390/molecules30153070
APA Styledel Olmo, M., & Andreu, C. (2025). Current Status of the Application of Antimicrobial Peptides and Their Conjugated Derivatives. Molecules, 30(15), 3070. https://doi.org/10.3390/molecules30153070