2.1. Study of the De-Alkoxylation Reaction in the Presence of Ketones
The de-alkoxylation reaction in the presence of acetone was investigated using model alkoxysilanes, such as PhMe2SiOMe and PhMe2SiOEt, in the presence of catalytic amounts of Cp*Ge+ B(C6F5)4−. 1H NMR and 29Si NMR spectroscopy, as well as GC and GC/MS, were used as the main analytical techniques to identify the substrate conversion and product formation.
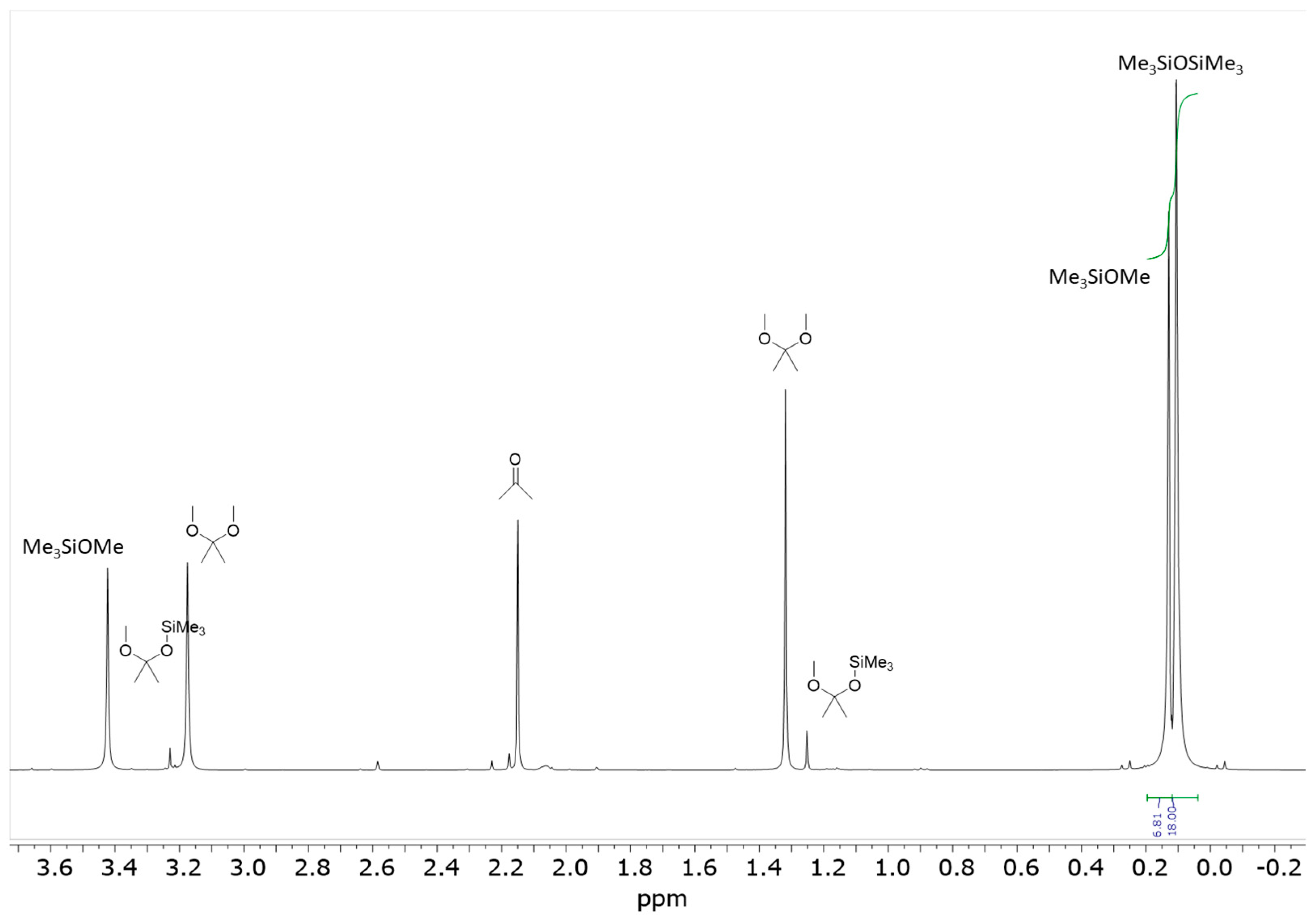
The reaction between 2 equivalents of PhMe
2SiOMe and 1 equivalent acetone in the presence of 0.02 equivalent of Cp*Ge
+ B(C
6F
5)
4− did not result in the quantitative conversion of methoxy silane to the corresponding disiloxane, as previously observed in the analogous reaction with propionaldehyde [
1]. The
1H NMR spectrum of the reaction mixture recorded after 24 h indicates an approximately 60% conversion of PhMe
2SiOMe, as evidenced by two singlets at δ = 0.44 ppm (SiCH
3) and 3.49 ppm (SiOCH
3), to the corresponding disiloxane (singlet at δ = 0.42 ppm (SiCH
3)). Additionally, acetone (singlet at δ = 2.17 ppm (CH
3)) underwent a conversion to the 2,2-dimethoxypropane, as characterized by signals at δ = 1.36 ppm (CH
3) and 3.22 ppm (OCH
3) (
Figure 1). Moreover, two other singlets observed at δ = 1.28 ppm (CH
3) and δ = 3.26 ppm (OCH
3) were assigned to a mixed ketal 2-methoxy-2-dimethylphenylsiloxypropane. The concentration of this mixed ketal was approximately 15 mol% relative to 2,2-dimethoxypropane. The presence of these two types of ketals was further confirmed by a GC/MS analysis of the reaction mixture (
Figures S1 and S2). The
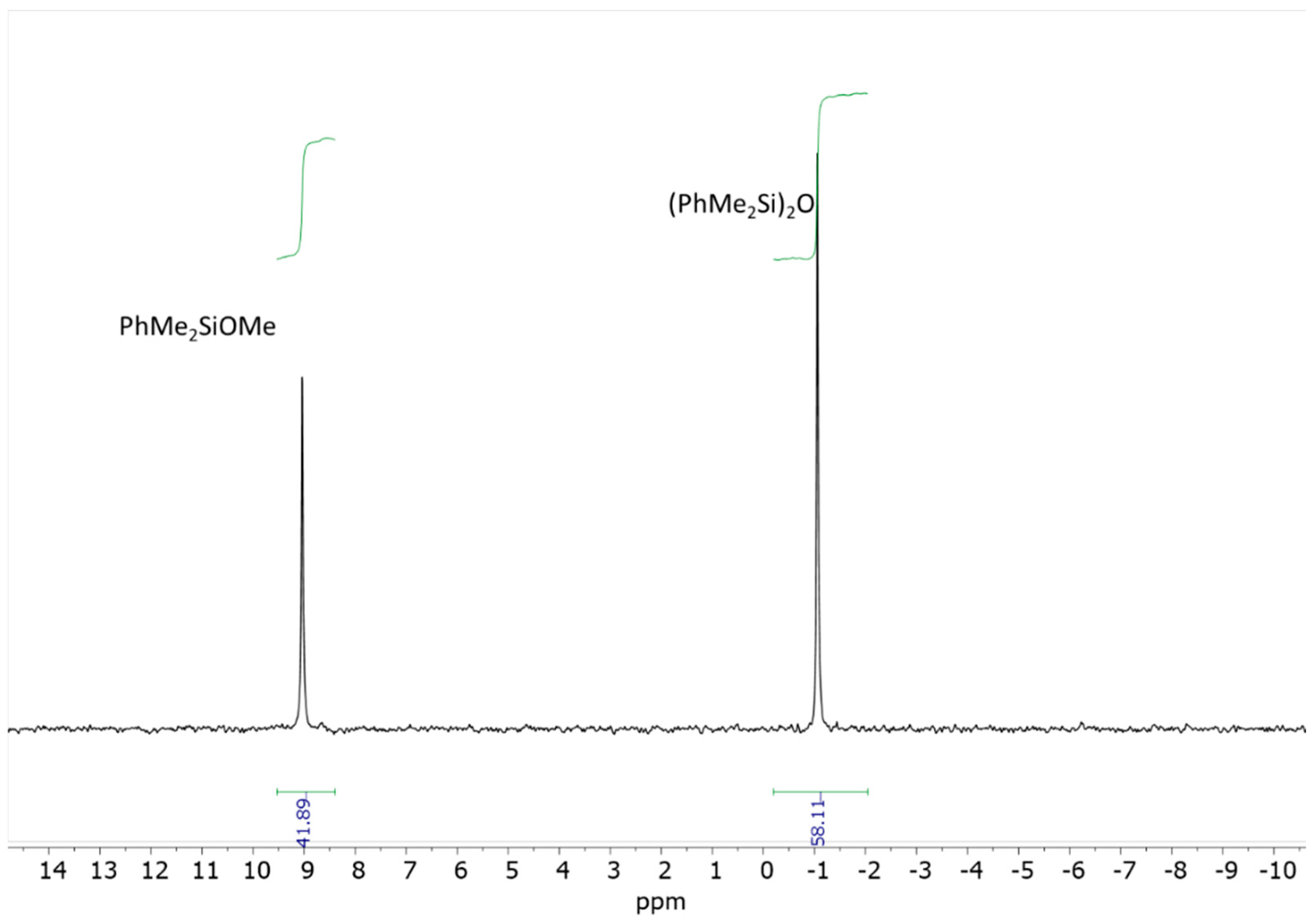
29Si NMR spectrum recorded after 3 days confirmed the presence of two major compounds: approximately 40 mol% of the starting PhMe
2SiOMe (singlet at δ = 9.42 ppm) and the corresponding disiloxane (singlet at δ = −1.14 ppm) (
Figure 2) [
14,
15].
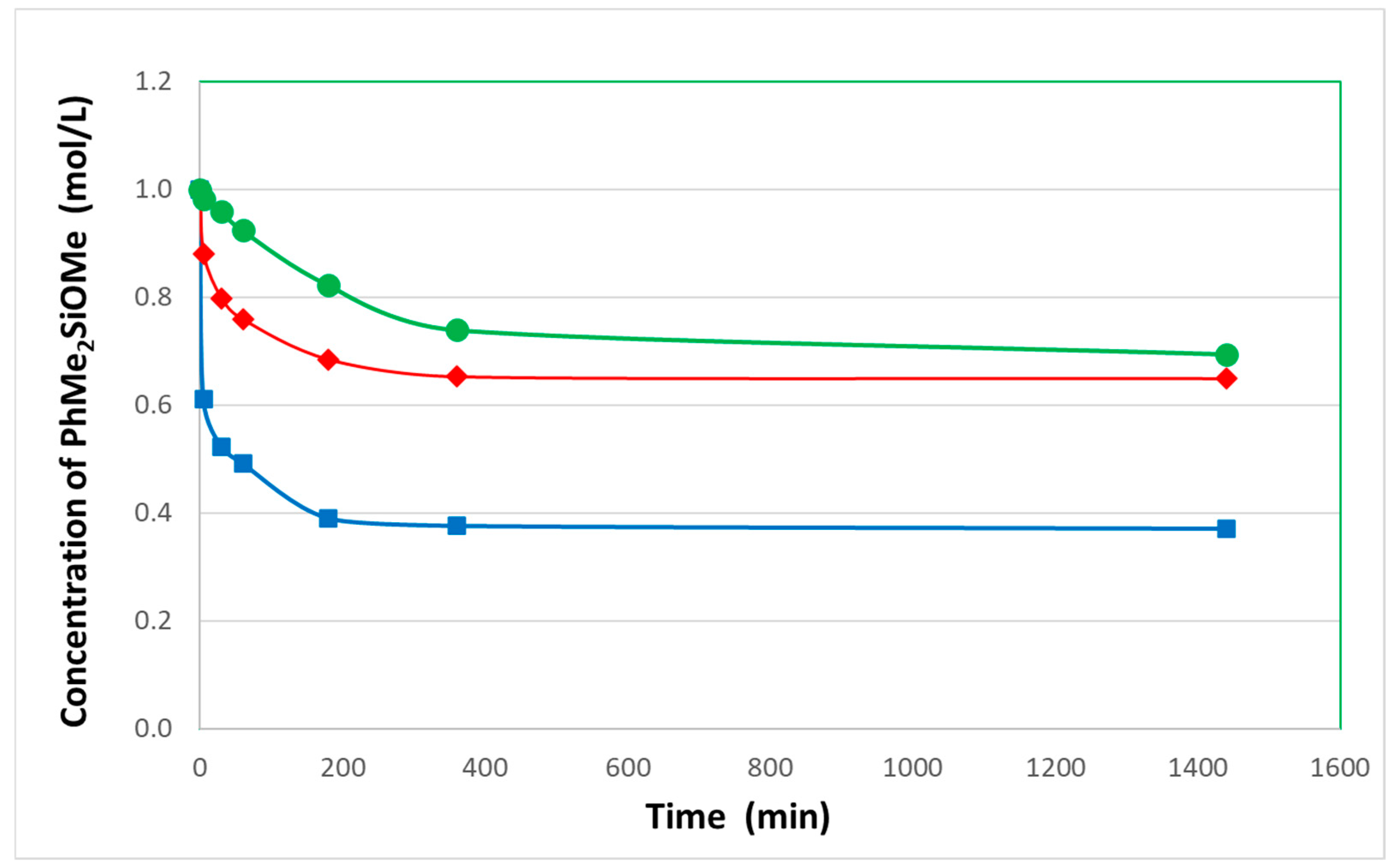
The quantitative analysis of the reaction progress over time for the 2 equivalents of PhMe
2SiOMe with 1 equivalent of acetone indicated that, indeed, the conversion of both reagents, PhMe
2SiOMe and acetone, proceeded at the same rate. However, in the case of acetone, unlike the propionaldehyde [
1], the de-alkoxylation process reached a plateau after about 6 h at approximately 60% conversion of methoxysilane (
Figure 3). This phenomenon can be explained by the fact that the studied reaction in the presence of acetone is reversible and leads to equilibrium between reagents and products (Equation (1)). Based on the NMR data, the equilibrium constant for this process was estimated to be around 2.8.
The reaction of the ethoxy-functional silane (PhMe
2SiOEt) with acetone in the presence of 1 mol% Cp*Ge
+ B(C
6F
5)
4− proceeded similarly to its methyl-functional counterpart. However, the conversion of PhMe
2SiOEt to the corresponding disiloxane (diphenyltetramethyldisiloxane) proceeded more slowly, probably due to steric hindrance imposed by the ethyl group. Nevertheless, the steric effect of the ethyl group did not affect the equilibrium position between the alkoxysilane and the disiloxane. Like the reaction of PhMe
2SiOMe with acetone, the conversion of PhMe
2SiOEt to disiloxane reached about 60% at equilibrium, although over a longer period of about 48 h (
Figure S3).
The influence of the ketone structure on its reactivity in the de-alkoxylation reaction of PhMe
2SiOMe was investigated for acetone, acetophenone, and benzophenone. Replacing a methyl group with a phenyl substituent significantly decreased the de-alkoxylation rate of the alkoxysilane (
Figure 4). Furthermore, the phenyl-containing ketones exhibited a notably lower equilibrium conversion of alkoxysilane.
The observed effect of the phenyl substituent on the reactivity of the ketones in the series acetone, acetophenone, and benzophenone in the de-alkoxylation reaction can be explained by a combination of electronic and steric effects. The phenyl group contributes to resonance stabilization by delocalizing the electron density to the carbonyl system, thereby reducing the electrophilicity of the carbonyl carbon. As a result, ketones such as acetophenone and benzophenone exhibit lower reactivity compared with acetone. Moreover, the steric hindrance of the phenyl groups further inhibits nucleophilic attack, making the nucleophilic substitution at the carbonyl carbon in acetophenone and benzophenone slower than in acetone [
16,
17].
Quantum chemical CBS-QB3 calculations for the de-alkoxylation reaction of Me
3SiOMe in the presence of aldehydes and ketones in DCM revealed that the process of de-alkoxylation with an aldehyde exhibited a significantly negative Gibbs free energy change (ΔG = −6.6 kcal/mol), which corresponded to an equilibrium constant in the order of 10
4–10
5. In contrast, the analogous reaction with acetone had a higher ΔG of −0.9 kcal/mol, which corresponded to an equilibrium constant of approximately 4 (
Scheme 1,
Table 1).
These results align with experimental findings, confirming that the de-alkoxylation condensation of alkoxysilanes in the presence of acetone is a reversible process that establishes equilibrium between alkoxysilane and siloxane. They also support a two-step reversible mechanism for this reaction, involving the formation of a disiloxane and its corresponding dialkyl ketal, as illustrated in
Scheme 2. The proposed mechanism of de-alkoxylation condensation is discussed in detail based on theoretical calculations in
Section 2.4.
2.2. Reaction of Hexamethyldisiloxane with 2,2-Dimethoxypropane in the Presence of Cp*Ge+ B(C6F5)4−
For the reaction of PhMe
2SiOMe with acetone to be a reversible process, the resulting ketal (2,2-dimethoxypropane (DMP)) must undergo the reverse reaction with disiloxane. To prove this hypothesis, the reaction of 1 equivalent of DMP with 1 equivalent of hexamethyldisiloxane (MM) was investigated in the presence of 1 mol% of Cp*Ge
+ B(C
6F
5)
4−. After 24 h, when the reaction had reached equilibrium, the
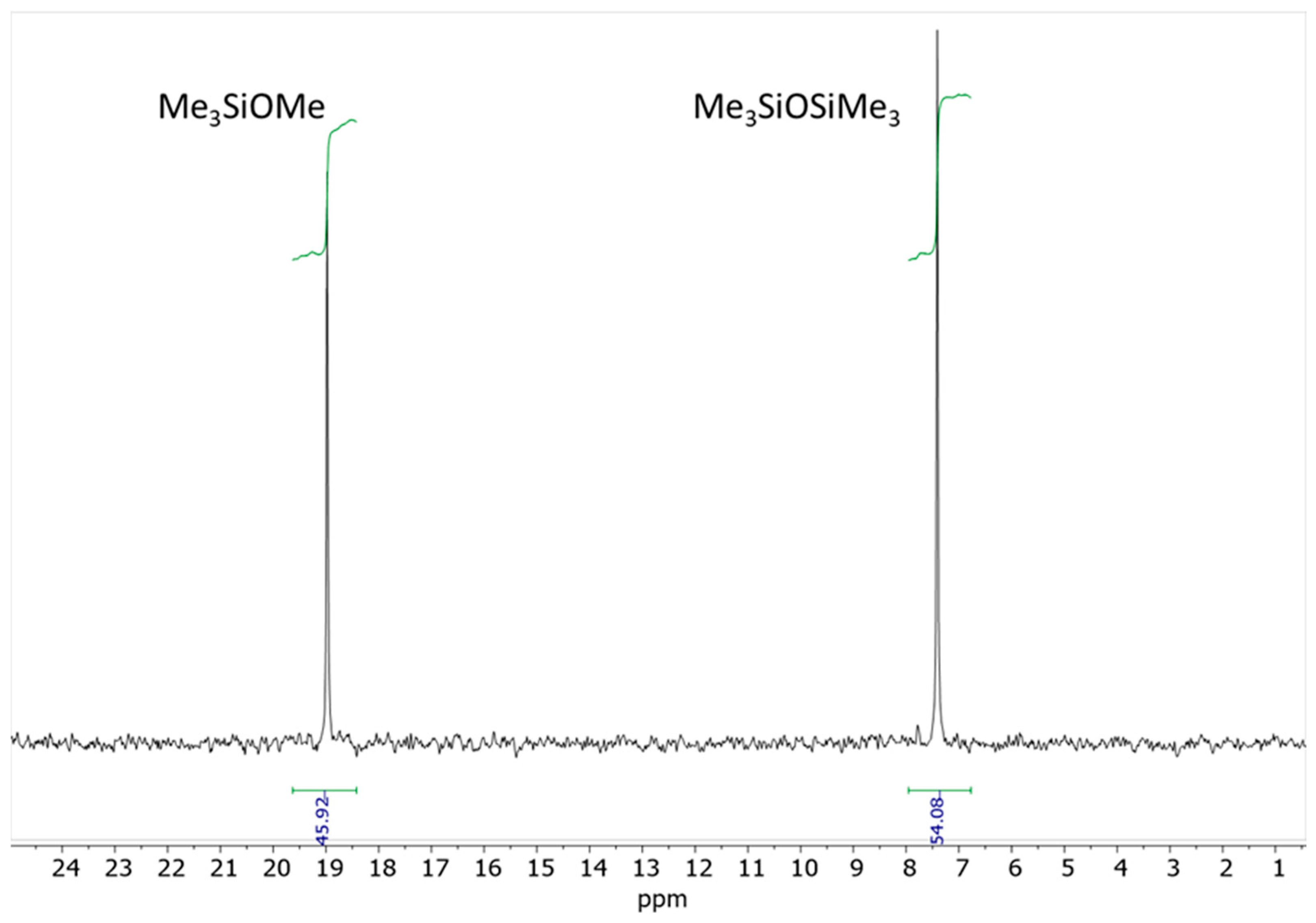
1H NMR spectrum of the mixture revealed the presence of MM, Me
3SiOMe, DMP, and acetone. Additionally, small amounts of a mixed ketal, (MeO)(Me
3SiO)C(CH
3)
2, were detected (
Figure 5). At this point, the conversion of DMP was approximately 44%. The
29Si NMR spectrum recorded after 4 days of reaction displayed two distinct singlets: one at δ = 7.22 ppm, which was assigned to MM, and another at δ = 18.87 ppm, which corresponded to the Me
3SiOMe [
14,
15]. The integrated signal intensities indicate a molar ratio of Me
3SiOMe to MM of approximately 0.85 to 1 (
Figure 6).
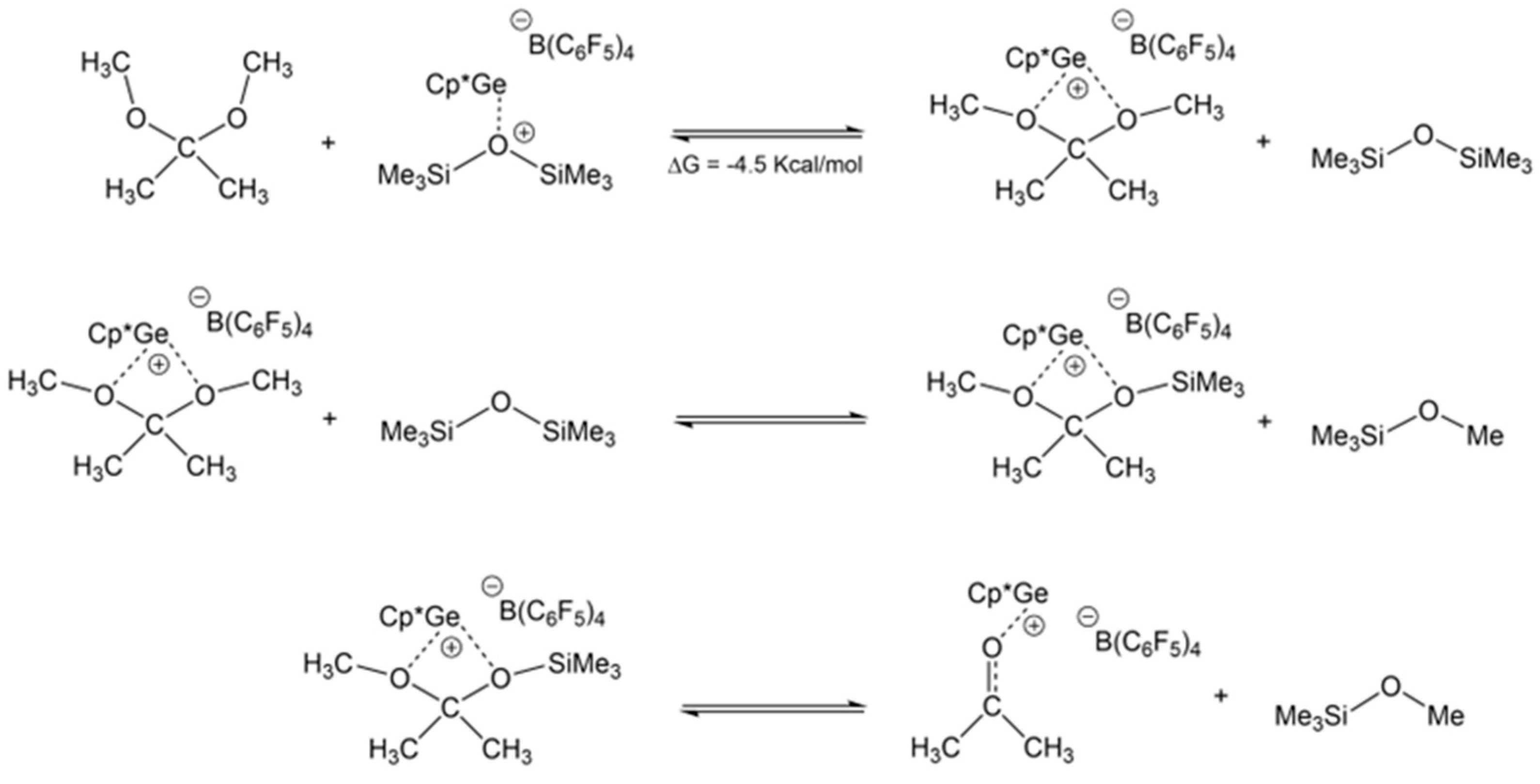
The results above confirm that the de-alkoxylation reaction that involved the acetone was a reversible process and that DMP in the presence of Cp*Ge
+ B(C
6F
5)
4− could break the siloxane bond. It must have been a two-step process (
Scheme 3). Quantum chemical calculations indicated that most of the Cp*Ge
+ cation interacted with DMP molecules in the MM and DMP mixture, with ΔG = −4.5 kcal/mol. In the thus formed Cp*Ge
+-DMP complex, the ketal carbon of DMP was activated for nucleophilic attack by siloxane oxygen, leading to the formation of a mixed ketal and Me
3SiOMe. The resulting mixed ketal underwent an intramolecular elimination reaction, which yielded acetone and an additional Me
3SiOMe molecule (
Scheme 3).
2.3. Reaction of Trimethylsilyl-Terminated Oligosiloxanes with 2,2-Dimethoxypropane (DMP) in the Presence of Cp*Ge+ B(C6F5)4−
To gain deeper insight into the cleavage of siloxane bonds by DMP, its reactions with a series of trimethylsilyl-terminated oligosiloxanes Me
3SiO(SiMe
2O)
xSiMe
3 (where x = 1, 2, 3, 4) were investigated in the presence of Cp*Ge
+ B(C
6F
5)
4−. In this manuscript, shorthand notations of siloxane oligomers were introduced to simplify their formulas, which are commonly used by silicone chemists (
Scheme 4) [
18]. Accordingly, Me
3SiO(SiMe
2O)
xSiMe
3 was abbreviated as MD
xM.
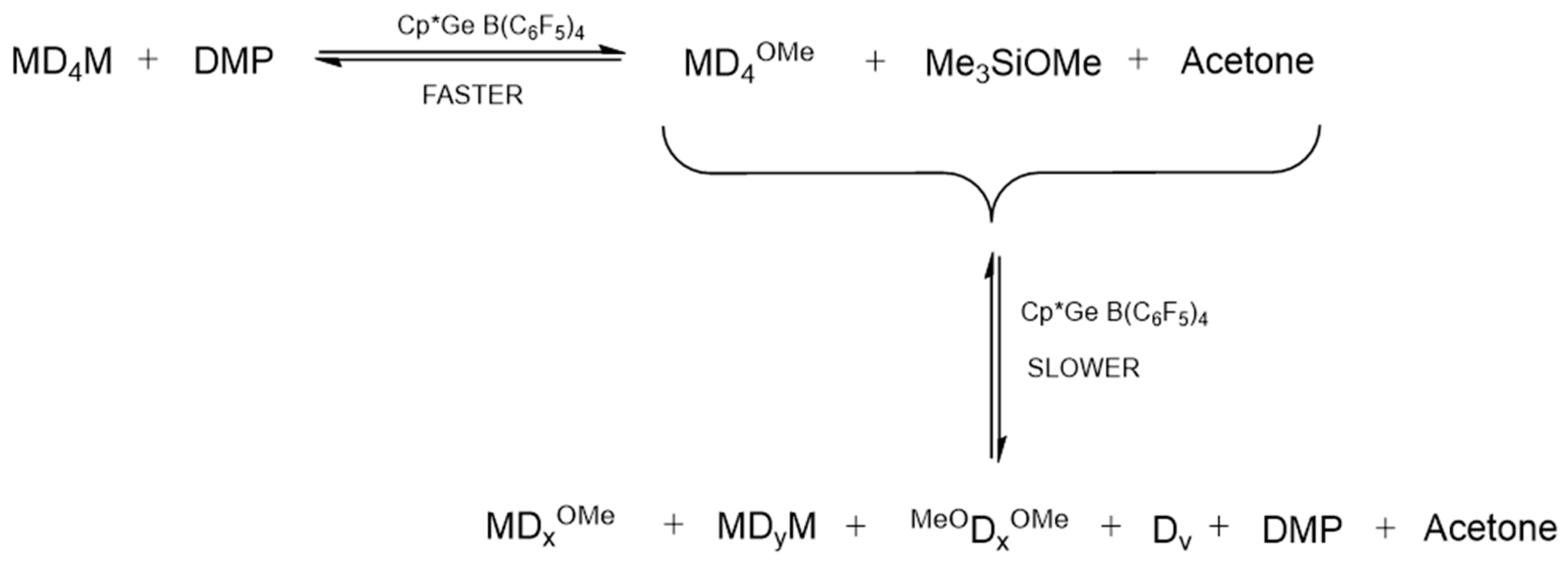
In the first experiment, the reaction of Me
3SiO(SiMe
2O)
4SiMe
3 (MD
4M) with an equimolar concentration of DMP in the presence of 0.8 mol% of Cp*Ge
+ B(C
6F
5)
4− was studied. The conversion of MD
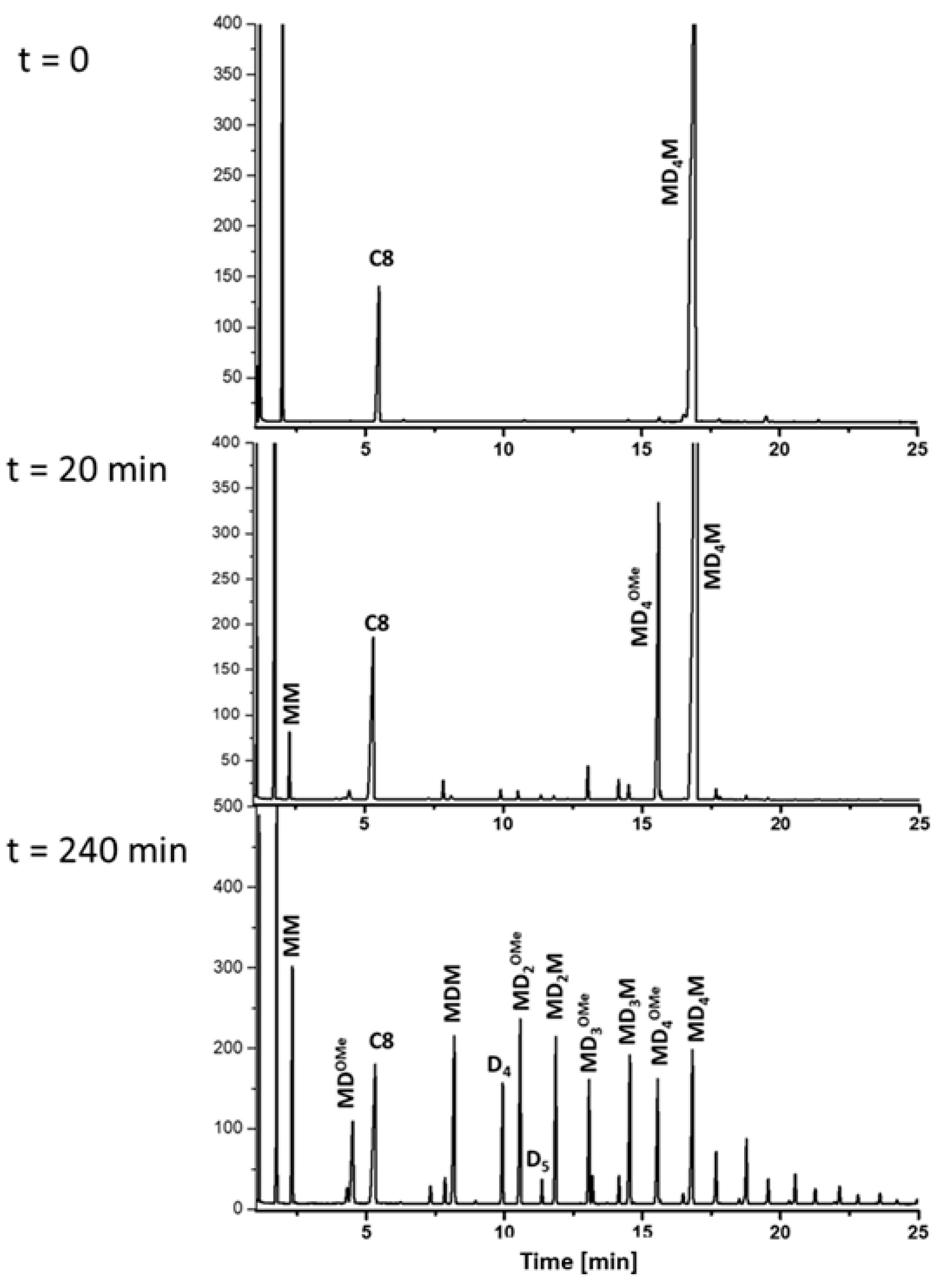
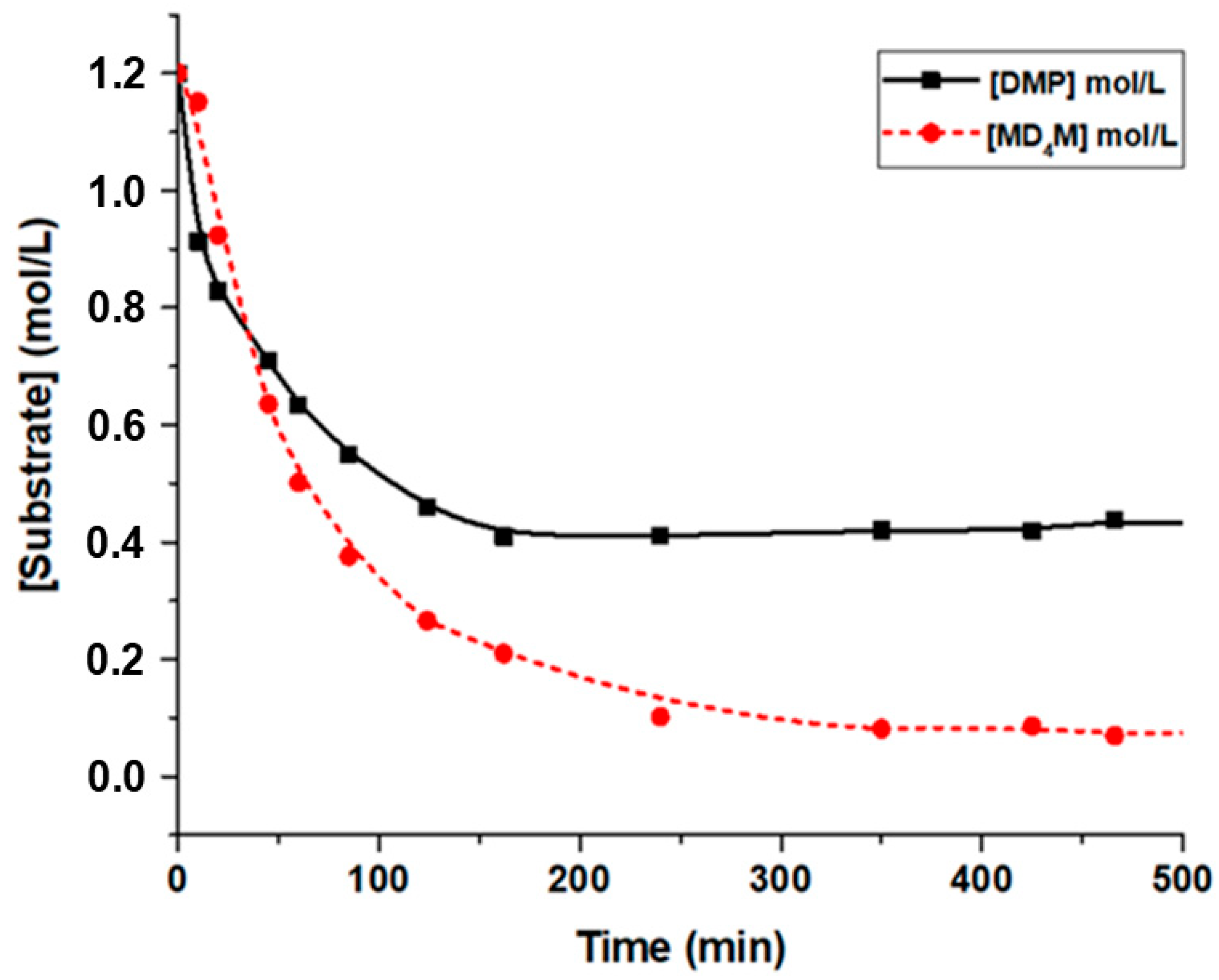
4M and DMP and the formation of oligomeric siloxanes were monitored by GC (
Figure 7). The chemical identity of the observed peaks was confirmed in a separate GC/MS experiment (
Figure S4). The process occurred in two distinct stages. During the first stage, the conversion rates of both reagents were approximately similar. However, after approximately 60 min, the conversion rate of DMP slowed significantly, where they eventually reached a plateau at around 0.5 mol/L (60% conversion) after 200 min. Meanwhile, the concentration of MD
4M continued to decrease until it stabilizes at an equilibrium concentration of approximately 0.1 mol/L (
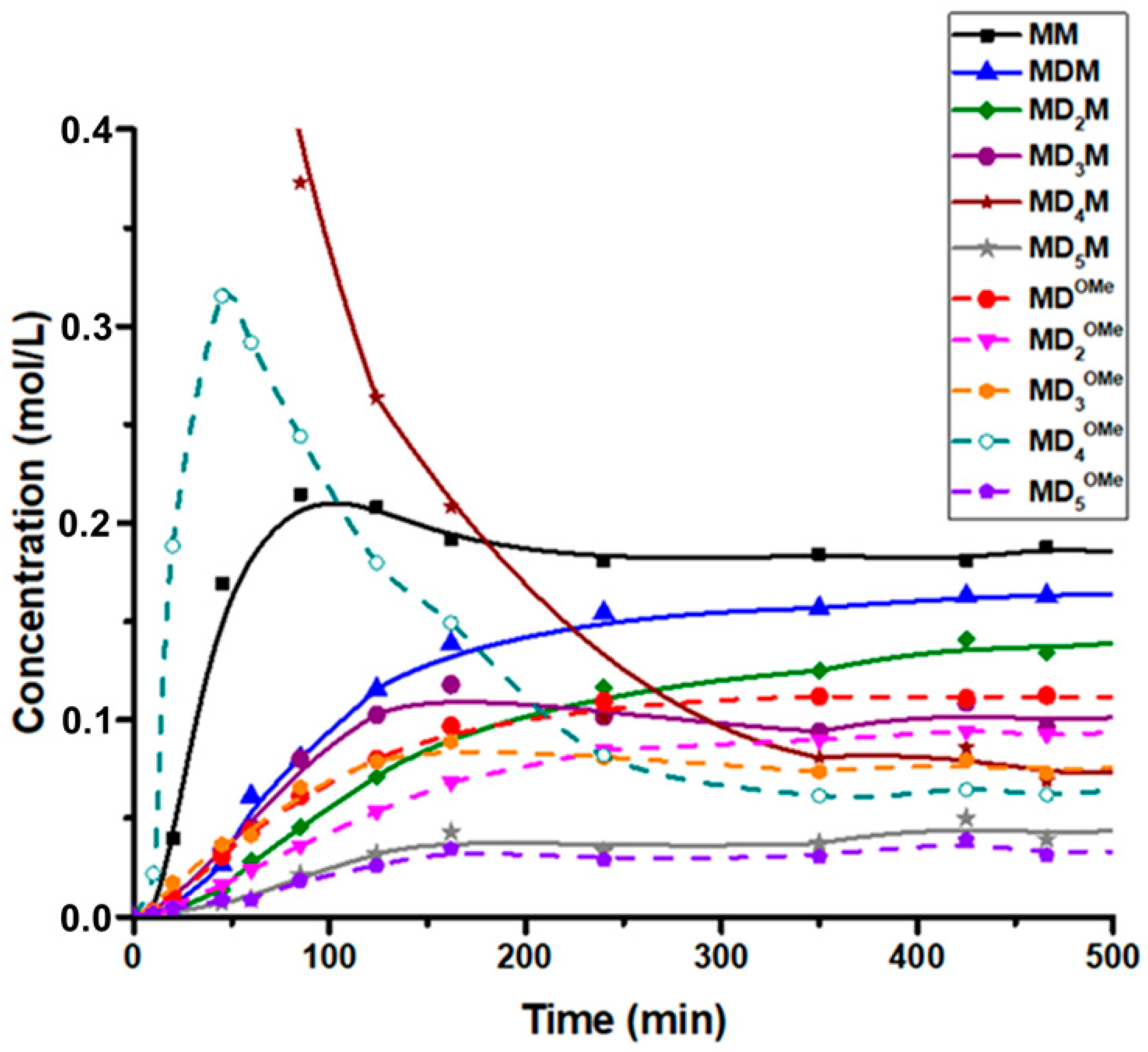
Figure 8). Interestingly, under these conditions, the preferential formation of MD
4OMe and Me
3SiOSiMe
3 (MM) was observed during the initial stage of the process (
Figure 9). These results indicate that the cleavage of the terminal siloxane bond in MD
4M by DMP, which led to the formation of MD
4OMe, Me
3SiOMe (M
OMe), and acetone, was the fastest reaction in this system. The resulting M
OMe appeared to be highly reactive under these conditions and rapidly underwent acetone-assisted condensation to form MM and other M-stopped siloxanes. However, after reaching the maximum concentration at about 60 min of reaction, the concentration of the monoalkoxy oligosiloxane (MD
4OMe) started to decline. This process was accompanied by the formation of various oligomeric alkoxy-functional and M-stopped siloxanes. The reaction observed seemed to reach an equilibrium after about 8 h. The predominant products were two series of linear oligosiloxanes, MD
xM and MD
xOMe. Additionally, cyclic siloxanes (mostly D
4 and D
5) and dimethoxy-stopped oligosiloxanes (
MeOD
xOMe) were formed, though their concentrations remained lower. The
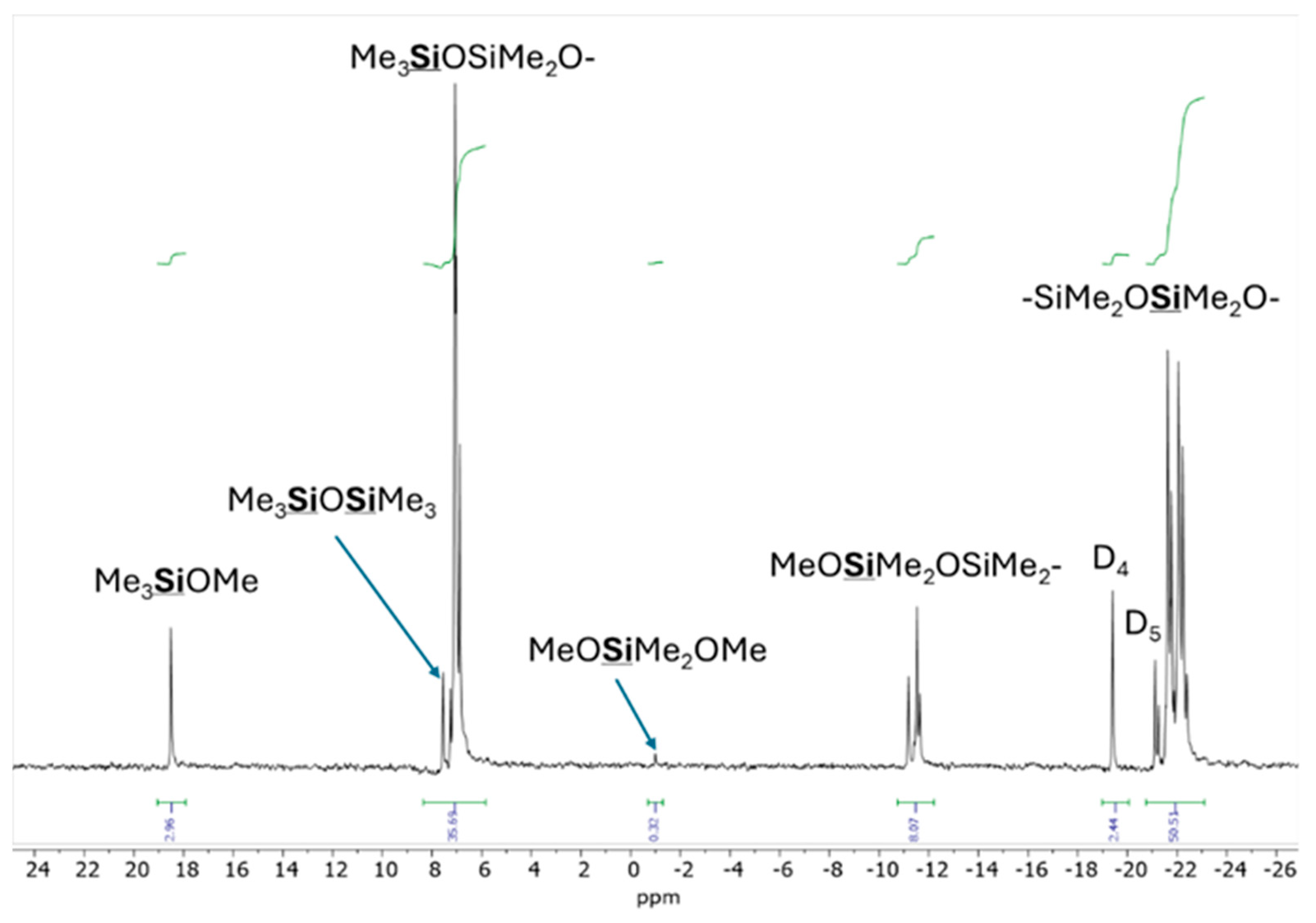
29Si NMR spectrum recorded after 7 days confirmed the presence of several products (
Figure 10). The singlet at δ = 18.53 ppm was assigned to Me
3SiOMe, while the singlet at δ = 7.55 ppm corresponded to Me
3SiOSiMe
3. An unresolved multiplet centered at δ = 7.10 ppm was attributed to the Me
3SiO groups connected to various oligosiloxanes, whereas another unresolved multiplet at δ = −11.4 ppm was associated with the MeOSiMe
2 groups within different oligosiloxanes. Additionally, distinct singlets at δ = −19.4 ppm and δ = −21.11 ppm were assigned to cyclic D
4 and D
5, respectively, and an unresolved multiplet centered at δ = −21.8 ppm corresponded to various -SiMe
2O- units [
15]. The formation of various MD
xM, MD
xOMe, and cyclic oligosiloxanes can be explained by a siloxane bond disproportionation process, which was also catalyzed by Cp*Ge
+ B(C
6F
5)
4− (
Scheme 5).
Thus, in the studied reaction of DMP with MD
4M catalyzed by Cp*Ge
+ B(C
6F
5)
4−, a fast cleavage reaction of the terminal siloxane bond by DMP took place, which led to the formation of monoalkoxy oligosiloxane MD
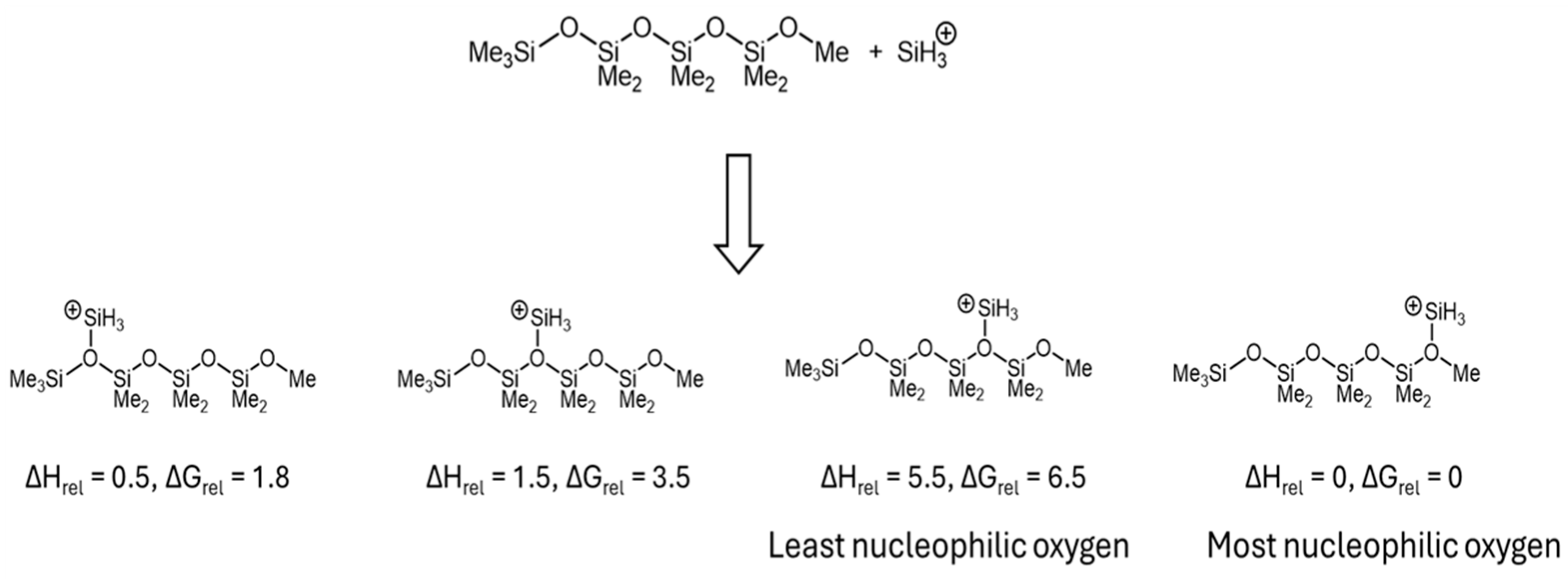
4OMe. To explain this phenomenon, the relative nucleophilicities of oxygen atoms in the short trimethylsilyl-terminated PDMS chains (Me
3Si(OSiMe
2)
3OMe) were evaluated by DFT calculations using SiH
3+ as the reference Lewis acid (
Scheme 6).
The density function theory (DFT) calculations revealed that the relative nucleophilicities of oxygen atoms in short linear oligosiloxanes of the formula Me
3Si(OSiMe
2)
xOMe (MD
xOMe) were not equal. The oxygen adjacent to the terminal Me
3Si group exhibited higher nucleophilicity compared with the oxygen atom bonded to two SiMe
2O units (
Scheme 6). Similarly, in MD
4M, the oxygen atom linked to the terminal trimethylsilyl group was expected to be the most nucleophilic in the nucleophilic substitution reaction at the activated carbon center of DMP, which was complexed by Cp*Ge
+ B(C
6F
5)
4−. This nucleophilic attack was anticipated to result in cleavage of the siloxane bond, which led to the formation of MD
4OMe, Me
3SiOMe, and acetone, as illustrated in
Scheme 5. The enhanced reactivity of the terminal (M-stopped) siloxane bond was previously observed in the cationic oligomerization of linear siloxanes [
19]. The resulting Me
3SiOMe was likely the most reactive alkoxy-functional compound, where it underwent rapid conversion to MM via a de-alkoxylation process in the presence of acetone, as experimentally observed (
Figure 9). However, even at equilibrium, a significant amount of Me
3SiOMe remained in the reaction mixture (
Figure 10). The MD
4OMe oligomer, initially formed in excess, subsequently underwent siloxane bond disproportionation reactions and acetone assisted de-alkoxylation condensation, which led to the formation of various linear siloxane oligomers with the general formulas MD
xM and MD
xOMe (
Scheme 5).
The scope of these studies was further expanded to investigate the reactions between DMP and other M-stopped short linear siloxanes, including MDM, MD
2M, and MD
3M. The reactions were carried out at a 1:1 molar ratio of DMP to MD
xM, where x = 1, 2, 3, and 1 mol% of Cp*Ge
+ B(C
6F
5)
4− served as the catalyst. These transformations followed the same pathway as the reaction between DMP and MD
4M, as illustrated in
Figures S5–S7. The cleavage of the terminal siloxane bond was the fastest step in these reactions, irrespective of the number of D units. This process resulted in the formation of excess MD
OMe in the reaction with MDM, MD
2OMe in the reaction with MD
2M, and MD
3OMe in the reaction with MD
3M. As previously observed in the reaction of DMP with MD
4M, the concentration of the initially formed in excess mono-alkoxy siloxane gradually decreased as the reaction progressed. Meanwhile, other M-stopped and alkoxy-functional siloxanes emerged and eventually reached equilibrium concentrations after several hours.
2.4. Mechanistic Study of Ketone-Assisted De-Alkoxylation via DFT and CBS-QB3 Calculations
The experimental findings support the proposed mechanism for the de-alkoxylation reaction in the presence of a ketone, as outlined in
Scheme 2. To further elucidate this process, DFT calculations were performed. It seemed reasonable to assume that the mechanism of de-alkoxylation involving ketones would closely resemble that reported for aldehydes [
1]. Therefore, the geometries of stationary points found for the reaction with propionaldehyde, after necessary modification, were used as starting points for searching the stationary structures in the reaction with acetone. To simplify the DFT calculations, the presence of the weakly nucleophilic anion B(C
6F
5)
4− was omitted. For the same reason, the methyl groups in the cyclopentadiene ligand were replaced by hydrogens. However, a comparison between the DFT calculations and experimental data revealed a significant discrepancy. The DFT calculations predicted the Gibbs free energy change (ΔG) for the acetone-assisted de-alkoxylation reaction of PhMe
2SiOMe to be approximately 5.6 kcal/mol, which corresponded to an equilibrium constant of about 10
−4. However, experimental results indicate a much higher equilibrium constant of approximately 2.8, corresponding to a ΔG of ca. −0.6 kcal/mol.
To verify the DFT calculation results, the CBS-QB3 composite method was employed to recalculate the thermochemistry for key reactions and equilibria occurring in the reaction system. This robust and cost-effective approach offers high accuracy across various chemical applications (often within 1–2 kcal/mol of experimental values) by integrating multiple calculation steps and empirical corrections [
20]. The comparison of the results obtained using the DFT and CBS-QB3 methods are presented in
Table 1.
The above results show that the DFT calculations were in some cases (for example, for the thermodynamics of the overall reactions with EtCHO (reaction A) and Me2CO (reaction B)) significantly less accurate than the CBS-QB3 calculations. Thus, when the thermochemistry of chemical equilibria is discussed, we usually refer to the CBS-QB3 results. Furthermore, the comparison of the CBS-QB3 results for the gas phase and for the DCM solution showed that the solvent effect could be estimated as 1–2.5 kcal/mol. To most accurately represent the real reaction system, we used the values obtained for the reaction in solution. However, the free energy profile for the proposed condensation mechanism was calculated by the DFT method for two reasons. First, CBS-QB3 calculations are significantly more computationally expensive; second, we aimed to preserve compatibility with our previous report to enable a direct comparison between aldehyde and ketone.
The DFT calculations for the gas-phase conditions indicate that CpGe
+ interacts more strongly with methoxy silane than with propionaldehyde, which yielded an equilibrium constant (K
eq) of 2.7 for reaction C (
Table 1). The CBS-QB3 method predicted an even larger K
eq for this equilibrium in DCM. Given that acetone is a stronger nucleophile than propionaldehyde due to the electron-donating effects of its two methyl groups, one would expect the equilibrium of reaction D (
Table 1) to shift toward the CpGe
+–acetone complex. Indeed, the gas-phase DFT calculations confirmed this expectation, where they yielded a K
eq value of 0.2. Consequently, the concentration of the CpGe
+–acetone complex was approximately 5 times higher than that of the CpGe
+ complex with Me
3SiOMe. However, the CBS-QB3 method predicted a significantly larger K
eq value of 7 for this system in DCM, indicating that only approximately 30 mol% of CpGe
+ interacted with acetone. It seems that the energy of the CpGe
+–acetone interaction was somewhat underestimated due either to the deficiency of the CBS-QB3 method itself or to inaccuracy in the solvent effect estimation.
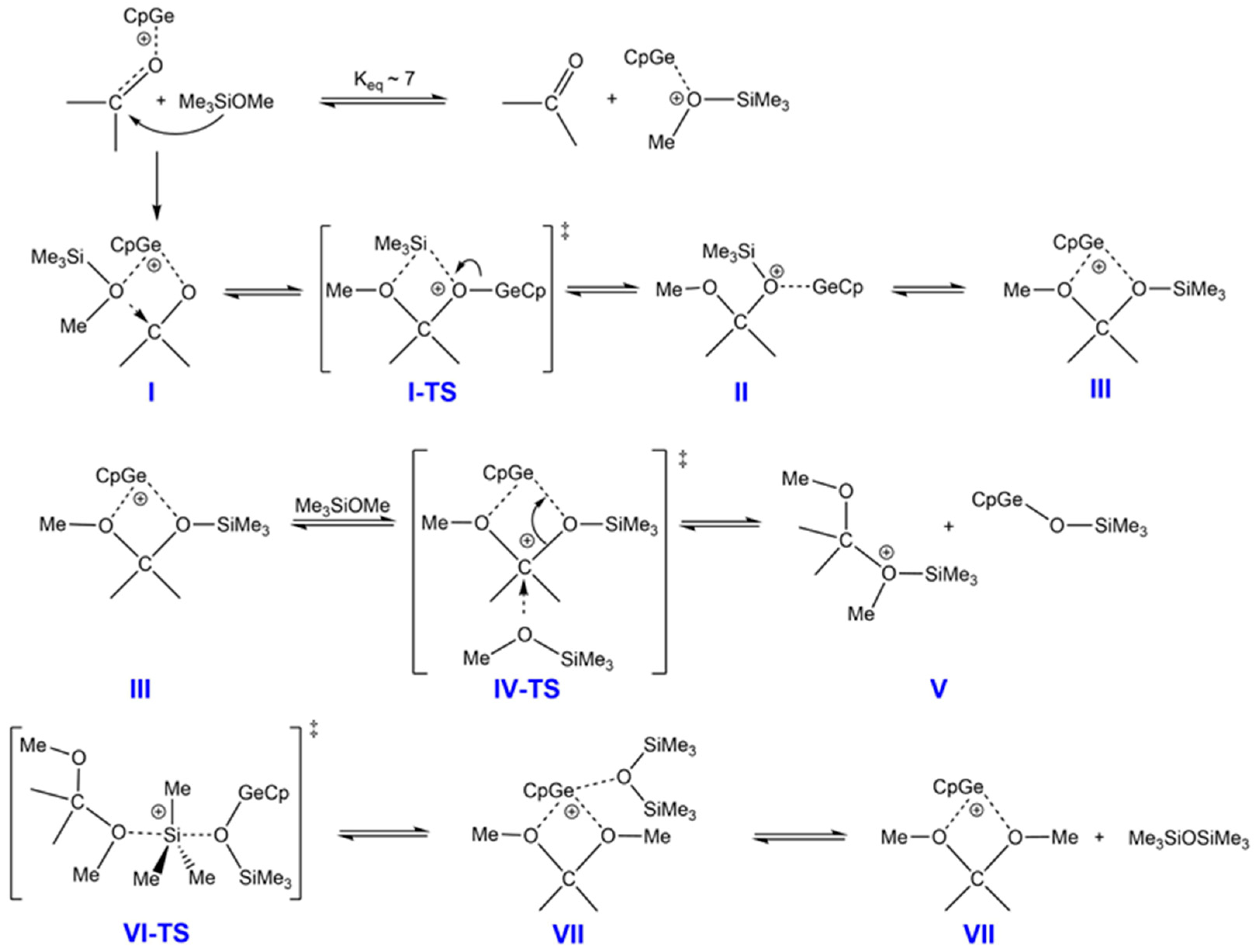
Based on the DFT results for reaction D, further investigation was conducted into the mechanism of the de-alkoxylation process. This reaction follows a widely accepted mechanism in the literature in which a nucleophile—specifically an alkoxy silane—attacks the carbon center of acetone [
17], which is activated by its interaction with the cationic CpGe(II)
+ complex (
Scheme 7).
The initial condensation step involves the nucleophilic attack of methoxysilane on the carbonyl carbon, which is activated by a germanium cation (I). This species is in equilibrium with a complex of methoxysilane and the CpGe
+ catalyst. Although this latter complex is more abundant, calculations suggest that the attack of acetone on silicon, when activated by the germanium cation, involves a higher energy barrier. Consequently, the silyl group is transferred to the carbonyl oxygen (forming intermediate II) leading to the formation of a mixed methoxy–siloxy ketal (intermediate III). Subsequently, another methoxy silane molecule attacks, substituting for the trimethylsiloxy–germanium intermediate (V). This intermediate then undergoes a rearrangement, leading to the cleavage of a trimethylsilyl group and the formation of the Ge mixed complex (VII), followed by the final formation of disiloxane and the germanium–DMP complex (VIII). The corresponding Gibbs free energy profile for the stationary points on the reaction pathway is presented in
Figure 11 and the corresponding relative enthalpies and Gibbs free energies are shown in
Table 2. The calculated structures of the stationary points and transition states are presented in
Figure S8. The formation of the CpGe
+–DMP complex was further supported by a
1H NMR experiment. The
1H NMR spectrum of a reaction mixture that contained 1 equivalent of CpGe
+ B(C
6F
5)
4− and 100 equivalents of DMP exhibited a slight upfield chemical shift of the methyl protons on the Cp* ring, which is consistent with complex formation (
Figure S9).
It is apparent that the de-alkoxylation reaction with aldehyde exhibited lower energy barriers than the reaction with ketone and, consequently, was predicted to be considerably faster (
Figure 11). It was also more thermodynamically favorable (see the CBS-QB3 values for a more precise evaluation), which confirmed that the reaction proceeded practically to completion, in contrast to the reaction with ketone.









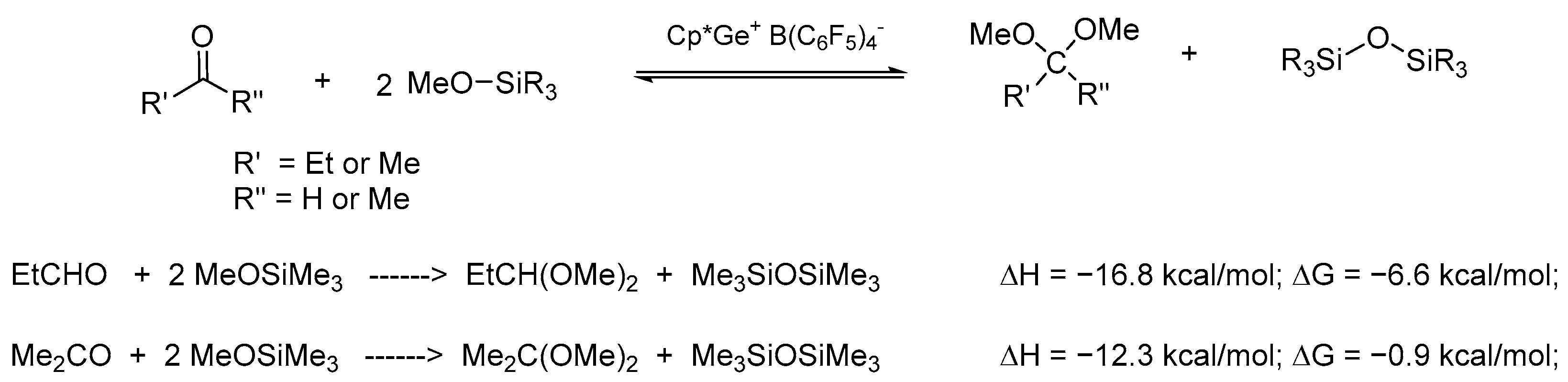



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}