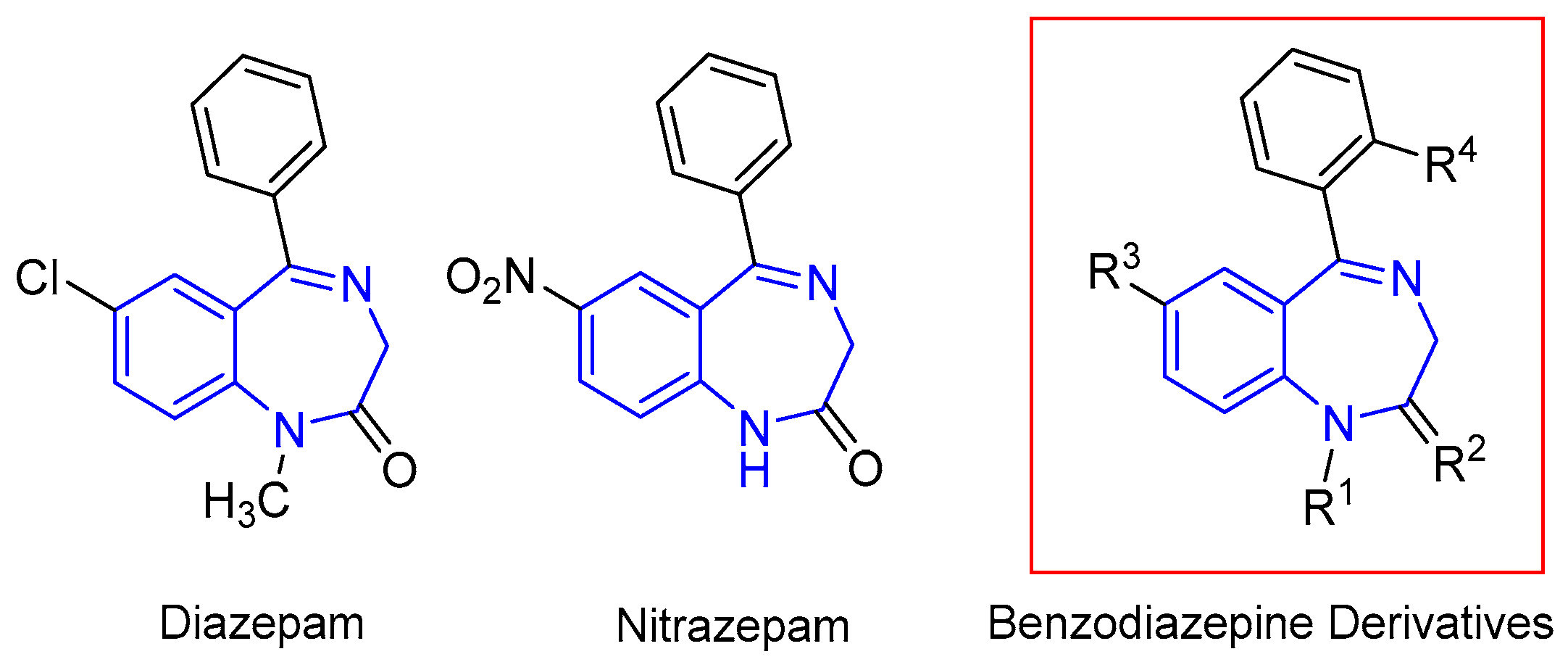
2. Results and Discussion
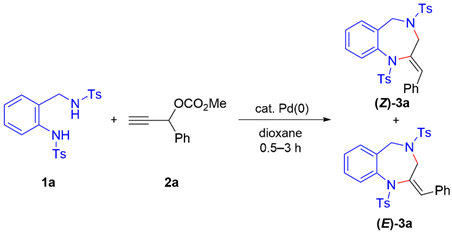
We began our investigation by examining the reaction between
N-tosyl-disubstituted 2-aminobenzylamine
1a and phenyl-substituted propargylic carbonate
2a under palladium catalysis. When the substrates were treated with 5 mol% Pd
2(dba)
3·CHCl
3 and 20 mol% bis(diphenylphosphino)methane (DPPM) in dioxane at 50 °C for 2 h, the desired 1,4-benzodiazepine
(Z)-3a and its geometric isomer
(E)-3a were obtained in 21% combined yield with a
Z:
E ratio of 3:1 (
Table 1, entry 1). Based on the examination with various bidentate ligands (entries 2–5), the use of 1,5-bis(diphenylphosphino)pentane (DPPPent) afforded the cyclized products in a combined yield of 51%. Furthermore, in the presence of the monodentate palladium complex Pd(PPh
3)
4, the yield was significantly improved to 98% (entry 6). Careful investigation of the reaction temperature (entries 7–9) revealed that at 25 °C, the cyclized products
(Z)-3a and
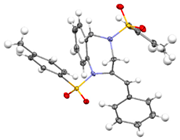
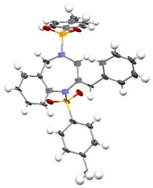
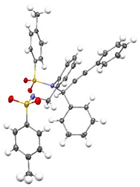
(E)-3a were obtained in a Z/E ratio of 3:1 and in 99% combined yield (entry 9). The structures of
(Z)-3a and
(E)-3a were determined by single-crystal X-ray diffraction analysis (CCDC 2454097 for
(Z)-3a, CCDC 2455059 for
(E)-3a).
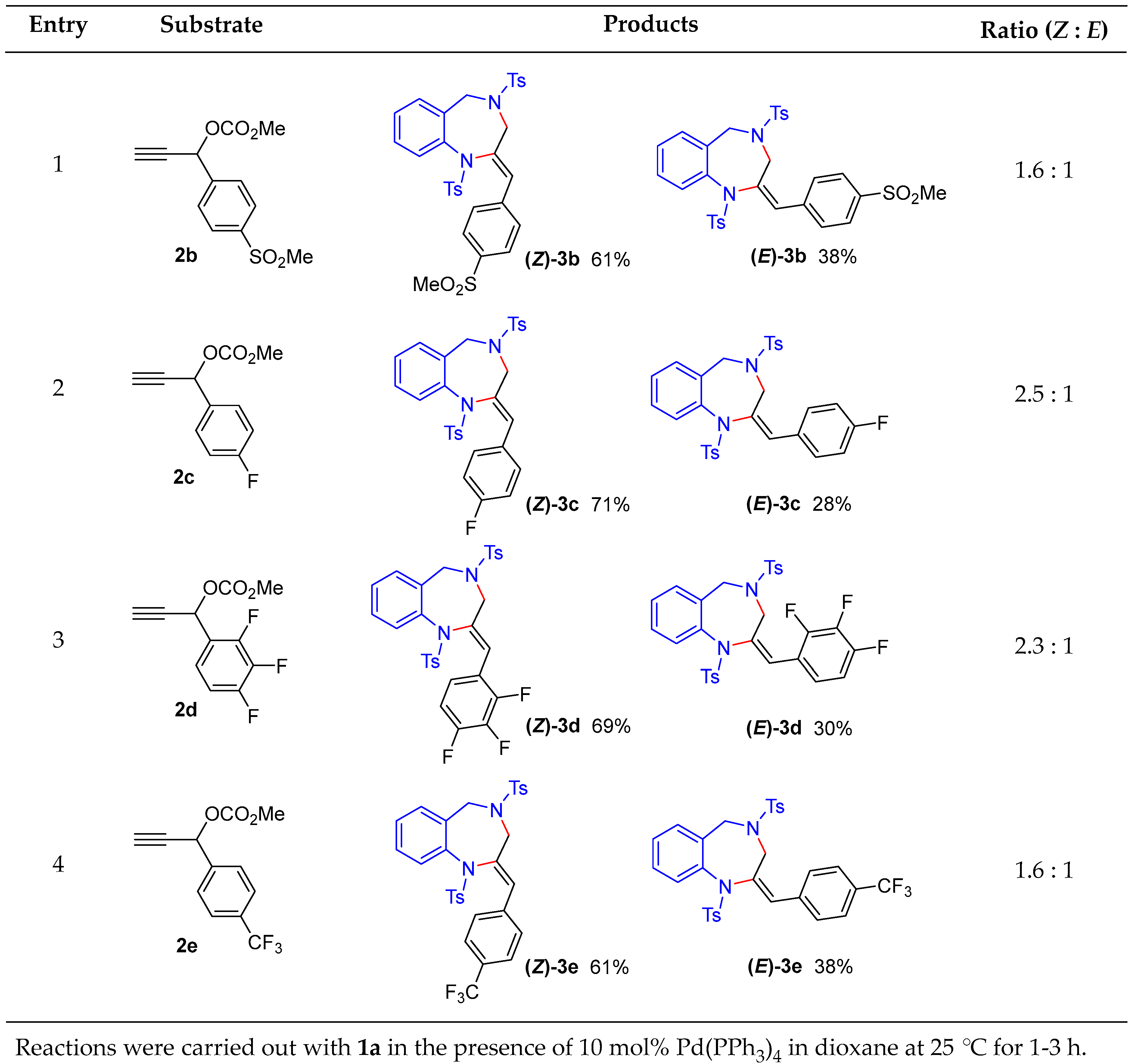
We next examined a series of aryl-substituted propargylic carbonates
2b–2e under the optimized conditions (
Figure 2). Propargylic carbonate
2b, bearing a 4-methanesulfonyl group as an electron-withdrawing substituent, afforded the corresponding benzodiazepine isomers
(Z)-3b and
(E)-3b in 61% and 38% yields, respectively, with a
Z:
E ratio of 1.6:1 (entry 1). Substrate
2c, possessing a 4-fluoro group, gave
(Z)-3c in 71% yield and
(E)-3c in 28% yield, with a
Z:
E ratio of 2.5:1 (entry 2). Substrate
2d, having a 2,3,4-trifluoro group, afforded
(Z)-3d and
(E)-3d in 69% and 30% yields, respectively, with a
Z:
E ratio of 2.3:1 (entry 3). Substrate
2e, which features a 4-trifluoromethyl group as an electron-withdrawing substituent, provided
(Z)-3e and
(E)-3e in 61% and 38% yields, respectively, with a
Z:
E ratio of 1.6:1 (entry 4). In all cases, the (
Z)-isomer was preferentially formed. The desired benzodiazepine products were obtained in nearly quantitative yields across all entries, underscoring the high efficiency of this palladium-catalyzed cyclization.
As shown in
Scheme 3, the reaction of substrate
2f, bearing a phenyl group at the terminal alkyne position, was investigated. Interestingly, the use of this substrate led to the formation of the same cyclized products
(Z)-3a and
(E)-3a as those obtained from substrate
2a, in which the substitution occurs at the propargylic position of the carbonate.
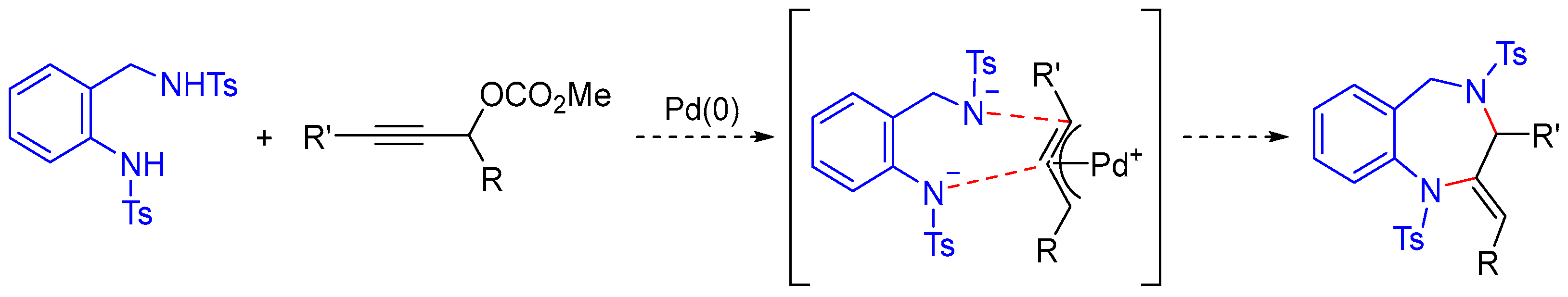
A plausible mechanism for the formation of 1,4-benzodiazepines is depicted in
Scheme 4. Upon coordination with palladium, propargylic carbonate
2a undergoes decarboxylation to form the corresponding π-propargylpalladium complex. At this stage, the tosyl anilide unit, being a relatively soft nucleophile, selectively attacks the central carbon of the π-propargylpalladium species, leading to the formation of π-allylpalladium intermediates
4 and
5. These two species are considered to be in a π–σ–π equilibrium, and the intramolecular nucleophilic attack by the benzyl amide anion is proposed to proceed predominantly from intermediate
4, likely due to reduced steric hindrance compared to intermediate
5, resulting in the formation of the cyclized product
(Z)-3a. The observation that the reaction of propargylic carbonate
2f also affords the same products,
(Z)-3a and
(E)-3a, supports the notion that both reactions proceed via the same π-allylpalladium intermediates
4 and
5.
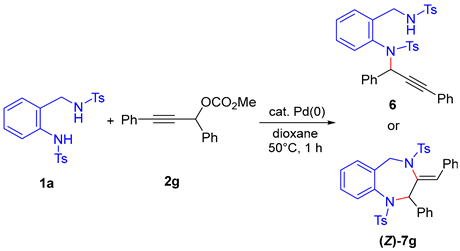
The results of reactions using diphenyl-substituted propargylic carbonate
2g are summarized in
Table 2. When
1a was initially reacted with
2g in the presence of Pd(PPh
3)
4, the product
6, in which the tosyl anilide moiety had substituted the propargylic position, was obtained in 97% yield (entry 1). Subsequent examination of other catalytic systems revealed that the use of Pd
2(dba)
3·CHCl
3 along with DPPM as the ligand led to the formation of
(Z)-7g, a 1,4-benzodiazepine derivative, in 45% yield (entry 2). Interestingly, the product
(Z)-7g is a positional isomer of the previously obtained cyclized products
3, suggesting that it was formed via a distinct reaction pathway. Further examination of different bidentate phosphine ligands showed improved yields. When 1,2-bis(diphenylphosphino)ethane (DPPE) or 1,3-bis(diphenylphosphino)propane (DPPP) was employed, the yields of
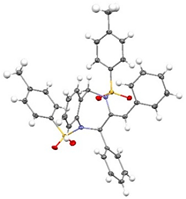
(Z)-7g increased significantly (entries 3 and 4), with the reaction in the presence of DPPP affording the product in an excellent 99% yield (entry 4). The structures of products
6 and
(Z)-7g were unambiguously confirmed by single-crystal X-ray diffraction analysis (CCDC 2454077 for
6, CCDC 2454078 for
(Z)-7g).
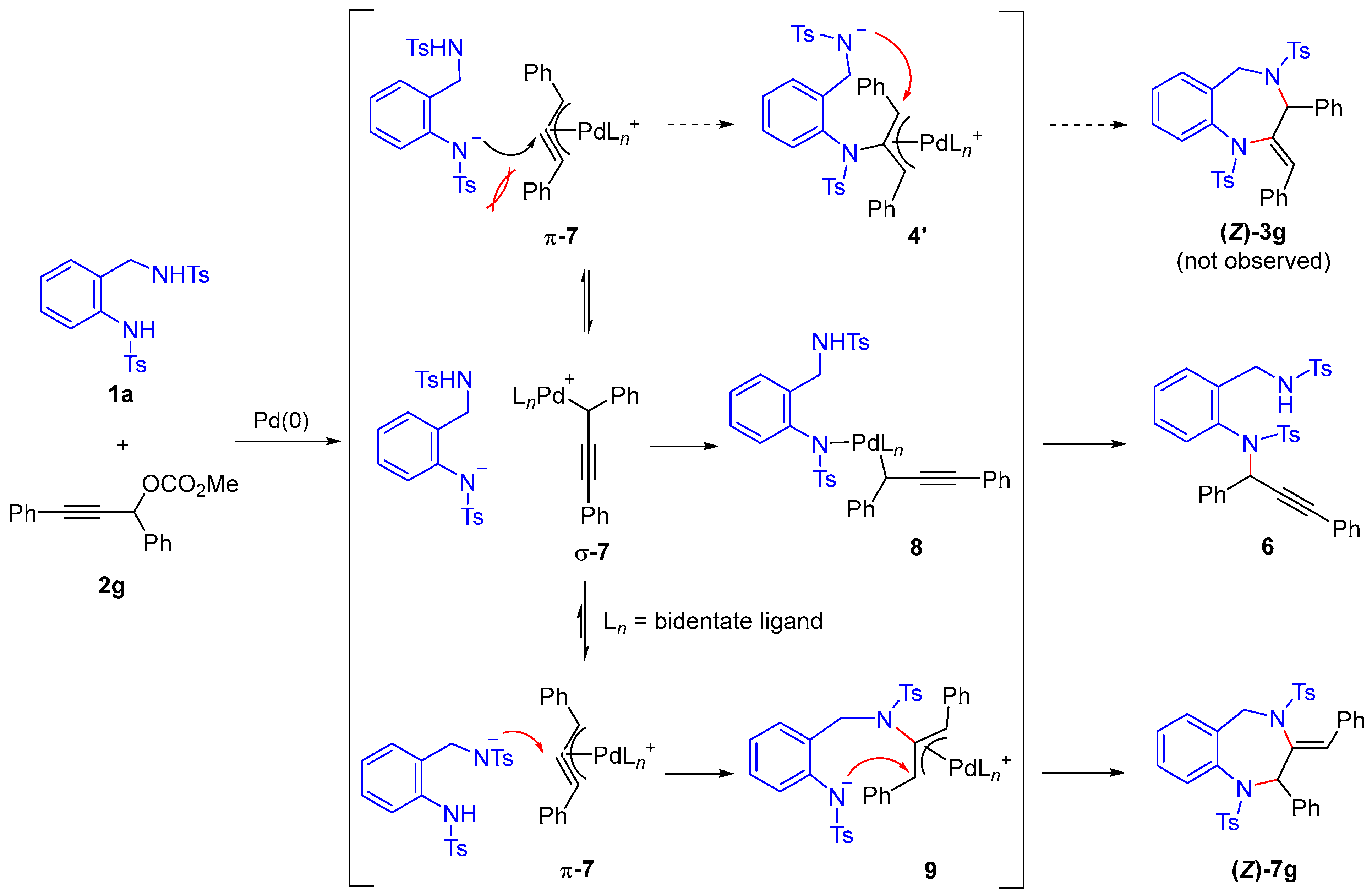
A proposed mechanism for the formation of compounds
6 and
(Z)-7g is shown in
Scheme 5. Upon reaction with a palladium catalyst, propargylic carbonate
2 is transformed into the corresponding π-propargylpalladium complex
π-7. In the case of diphenyl-substituted
π-7, nucleophilic attack by the tosyl anilide nitrogen atom of
1a is sterically hindered, and thus the expected cyclized product
(Z)-3g was not formed. Instead,
π-7 is presumed to be in equilibrium with its σ-propargylpalladium isomer
σ-7, which allows coordination of the tosyl anilide nitrogen to the palladium center to form intermediate
8. Subsequent reductive elimination from this intermediate leads to the formation of the propargylic substitution product
6. Under conditions employing bidentate phosphine ligands, this equilibrium is likely shifted toward the π-complex
π-7 [
41,
42], facilitating nucleophilic attack by the
N-benzyl sulfonamide nitrogen—which experiences less steric hindrance—leading to the formation of the π-allylpalladium intermediate
9 [
43]. This intermediate then undergoes intramolecular cyclization, resulting in the selective formation of 1,4-benzodiazepine derivative
(Z)-7g.
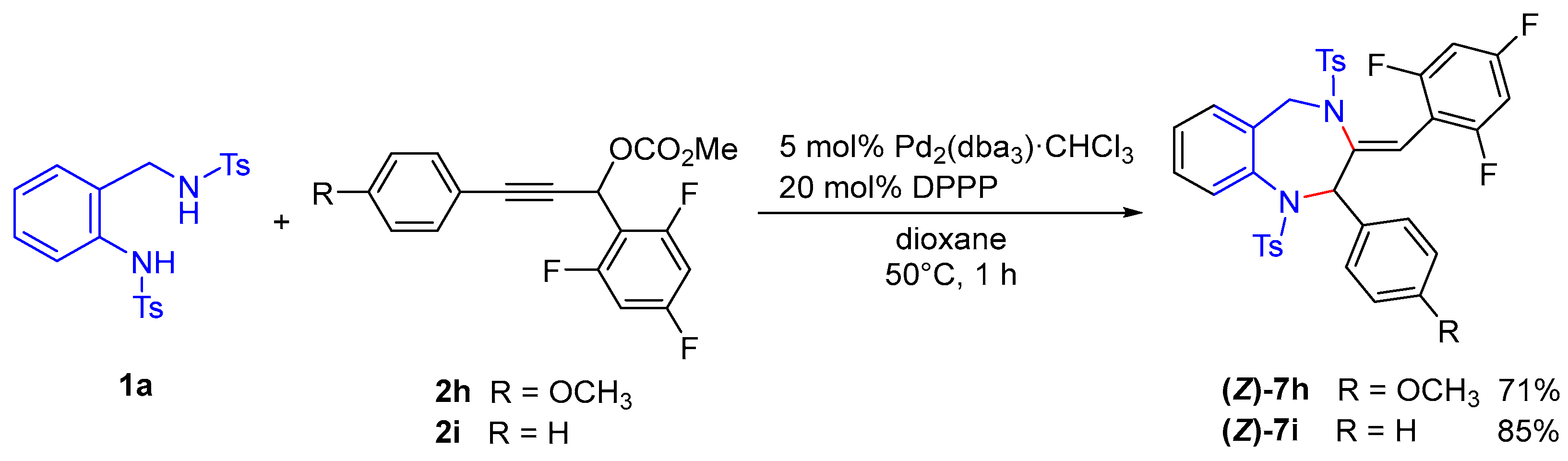
We next examined the reactions using propargylic carbonates
2h and
2i, each bearing two different aryl groups (
Scheme 6). Treatment of
2h, which has a 2,4,6-trifluorophenyl group at the propargylic position and a 4-methoxyphenyl group at the alkyne terminus, with
1a proceeded smoothly to afford the 1,4-benzodiazepine product
(Z)-7h with high regioselectivity in 71% yield. Similarly, when
2i, bearing a phenyl group at the alkyne terminus, was employed, the corresponding cyclized product
(Z)-7i was selectively obtained in 85% yield. The structure of
(Z)-7i was confirmed by X-ray crystallographic analysis (CCDC 2454080) (
Figure 3). The observed regioselectivity in these reactions is likely due to nucleophilic attack occurring preferentially at the more cationic carbon of the π-allylpalladium intermediate, which is substituted with the more electron-rich aryl group, as compared to the aryl group bearing an electron-withdrawing fluoro substituent. This electronic effect is believed to determine the selective formation of
(Z)-7h and
(Z)-7i.
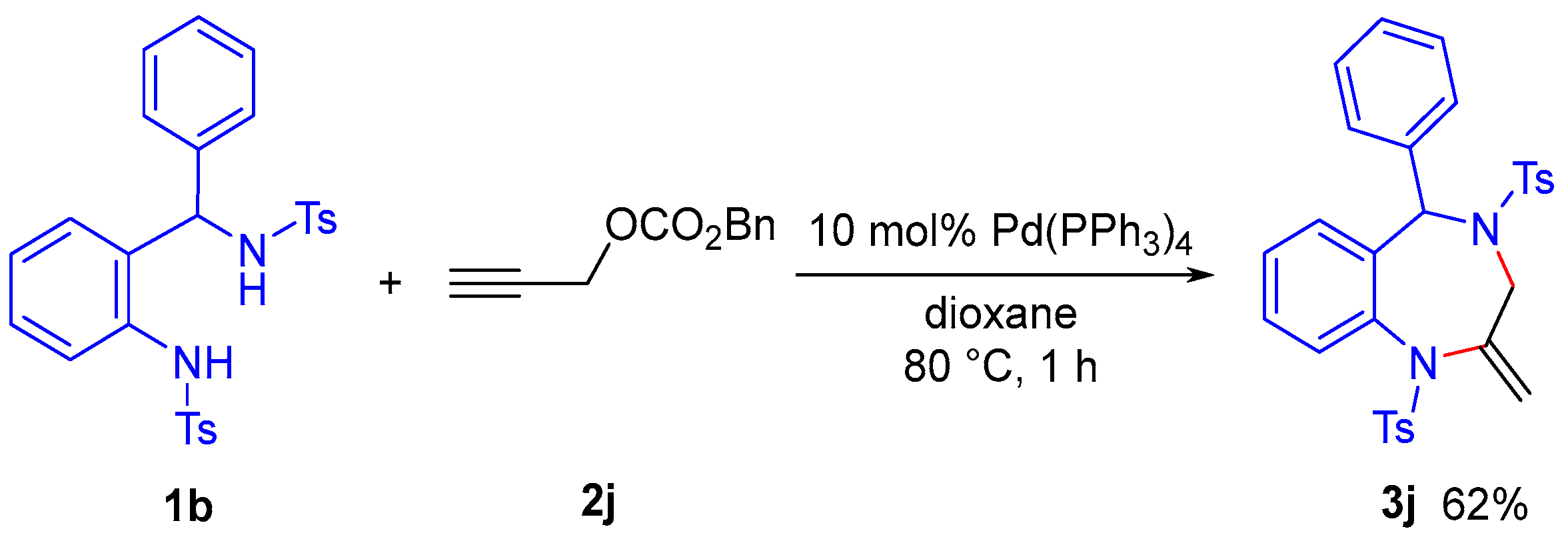
Finally, we attempted the construction of the core skeleton of benzodiazepine-based drugs (
Scheme 7). When propargylic carbonate
2j was treated with substrate
1b, which bears a phenyl group at the benzyl position, in the presence of a palladium catalyst, the expected cyclization proceeded smoothly to afford the cyclized product
3j in 62% yield. The product
3j contains the fundamental structure of benzodiazepine-class drugs, suggesting that this transformation could serve as an approach for synthesizing derivatives of such compounds.
3. Materials and Methods
All commercially available reagents were used without further purification. All reactions were performed in glassware equipped with a septum under positive argon pressure. The reaction mixture was magnetically stirred. Concentration was performed under reduced pressure. The heating experiments were conducted using an oil bath as a heat source. The reactions were monitored by TLC. TLC was performed on pre-coated plates (0.25 mm, silica gel 60F245, Merck & Co., Inc., Kenilworth, NJ, USA). Spots were visualized by exposure to UV light or by immersion in a solution of 10% phosphomolybdic acid in ethanol, followed by heating at ca. 200 °C. Column chromatography was performed on silica gel (40–50 μm, Kanto Chemical Co., Ltd., Nihonbashi, Tokyo, Japan). NMR spectra were recorded on a Bruker AVANCED III HD-500 (1H: 500 MHz, 13C: 125 MHz) spectrometer (Bruker Corporation, Billerica, USA) using tetramethylsilane (1H NMR at 0.00 ppm), CDCl3 (13C NMR at 77.16 ppm) and C6F6 (19F-NMR at −164.9 ppm) as a reference standard. Chemical shifts were reported in ppm. The following abbreviations were used to denote peak multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; sept, septet; m, multiplet; br, broadened. Mass spectra and high-resolution mass spectra were recorded on JEOL JMS-700 mass spectrometers (double-focusing magnetic sector) (JEOL Ltd., Tokyo, Japan).
N-Tosyl-disubstituted 2-aminobenzylamine
1a [
44] and propargylic carbonates
2a [34],
2c [38],
2e [45],
2f [
38],
2g [34], and
2j [46] were prepared according to procedures described in the literature.
3.1. 4-Methyl-N-(2-(((4-methylphenyl)sulfonamido)(phenyl)methyl)phenyl)benzenesulfonamide (1b)
To a stirred solution of
N-(2-benzoylphenyl)-4-methylbenzenesulfonamide [
47] (858 mg, 2.44 mmol) and
p-toluenesulfonamide (502 mg, 2.93 mmol) in 1,2-dichloroethane (20 mL) were added TiCl
4 (0.16 mL, 1.46 mmol) and Et
3N (0.68 mL, 4.88 mmol) dropwise. The reaction mixture was stirred under reflux for 13 h. After cooling to room temperature, the mixture was filtered through Celite and concentrated under reduced pressure. The residue was dissolved in EtOH (25 mL), and to the stirred solution was added NaBH
4 (185 mg, 4.88 mmol) at 0 °C. The mixture was stirred for 7 h at the same temperature, then quenched with water and extracted with CH
2Cl
2. The combined organic layers were washed with brine, dried over anhydrous Na
2SO
4, and concentrated under reduced pressure. The crude product was recrystallized from CHCl
3 and hexane to give
N-tosyl-disubstituted 2-aminobenzylamine
1b [
48] as a white solid (805 mg, 1.59 mmol, 65% yield over two steps). Mp 145–151 °C. IR (ATR): 3272, 1597, 1487 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 2.35 (3H, s), 2.42 (3H, s), 5.02 (1H, d,
J = 8.5 Hz), 5.39 (1H, d,
J = 8.5 Hz), 6.63 (2H, d,
J = 7.5 Hz), 6.72 (1H, dd,
J = 1.5, 8.0 Hz), 6.99 (1H, td,
J = 7.5, 1.0 Hz), 7.09 (4H, m), 7.14 (1H, m), 7.20 (2H, m), 7.27 (1H, m), 7.44 (1H, dd,
J = 8.0, 1.0 Hz), 7.51 (2H, d,
J = 8.5 Hz), 7.71 (2H, d,
J = 8.5 Hz);
13C-NMR (125 MHz, CDCl
3) δ 21.6 (CH
3), 21.7(CH
3), 56.8 (CH), 126.3 (CH), 126.4 (CH), 126.5 (CH), 127.0 (CH), 127.3 (CH), 127.5 (CH), 127.7 (CH), 128.6 (CH), 128.9 (CH), 129.1 (CH), 129.6 (CH), 129.9 (CH), 134.3 (Cq), 134.6 (Cq), 136.4 (Cq), 137.2 (Cq), 143.8 (Cq), 144.0 (Cq); HRMS (ES)
m/z calcd for C
27H
26N
2O
4S
2 [M]
+ 506.1334, found 506.1333.
3.2. Methyl 1-[4-(Methylsulfonyl)phenyl]prop-2-yn-1-yl Carbonate (2b)
To a stirred solution of trimethylsilylacetylene (2.08 mL, 15.0 mmol) in THF (90 mL), n-BuLi (2.8 M in THF, 5.4 mL, 15.0 mmol) was added dropwise at −78 °C. The mixture was stirred for 0.5 h at the same temperature. A solution of 4-(methylsulfonyl)benzaldehyde (1.88 g, 10.0 mmol) in THF (15 mL) was then added dropwise at −78 °C, and stirring was continued for 1 h. Subsequently, methyl chloroformate (2.32 mL, 30.0 mmol) was added dropwise at −78 °C, and then the temperature was allowed to rise to room temperature over 1 h. The reaction mixture was diluted with water and extracted with AcOEt. The combined organic layers were washed with brine, dried, and concentrated. The residue was dissolved in THF (75 mL), and to the stirred solution were added TBAF (1 M in THF, 25.0 mL, 25.0 mmol) and AcOH (2.86 mL, 50.0 mmol). The mixture was stirred for 1 h at room temperature, then diluted with water and extracted with AcOEt. The combined organic layers were washed with brine, dried, and concentrated. The crude product was purified by column chromatography on silica gel (hexane/AcOEt = 75:25 v/v) to give propargylic carbonate 2b as a colorless oil (2.54 g, 9.47 mmol, 95% yield in two steps). IR (ATR): 3262, 3020, 1749, 1598 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.78 (1H, d, J = 2.0 Hz) 3.06 (3H, s), 3.85 (3H, s), 6.35 (1H, d, J = 2.0 Hz), 7.76 (2H, d, J = 8.5 Hz), 7.99 (2H, d, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ 44.6 (CH3), 55.6 (CH3), 68.3 (CH), 77.6 (CH), 78.6 (Cq), 128.1 (CH), 128.6 (CH), 141.5 (Cq), 141.8 (Cq), 154.7 (Cq); HRMS (ES) m/z calcd for C12H12O5S [M]+ 268.0405, found 268.0402.
3.3. Methyl 1-(2,3,4-Trifluorophenyl)prop-2-yn-1-yl Carbonate (2d)
Propargylic carbonate 2d was prepared from 2,3,4-trifluorobenzaldehyde by following the same procedure described for 2b, yielding 1.12 g (6.99 mmol, 78%) of a colorless oil over two steps. IR (ATR): 3296, 1752, 1514, 1486 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.74 (1H, d, J = 2.0 Hz), 3.84 (3H, s), 6.50 (1H, d, J = 2.0 Hz), 7.00–7.06 (1H, m), 7.41–7.46 (1H, m); 13C-NMR (125 MHz, CDCl3) δ 55.6 (CH3), 62.8 (CH, d, JC-F = 4.0 Hz), 77.2 (CH), 77.9 (Cq), 112.6 (CH, dd, JC-F = 3.0, 17.2 Hz), 121.1 (Cq, dd, JC-F = 3.9, 10.8 Hz), 123.3 (CH, quintet, 6.9, 4.0 Hz), 138.9 (Cq, dt, JC-F = 14.8, 251 Hz), 149.5 (Cq, ddd, JC-F = 3.0, 10.8 and 254 Hz), 152.1 (Cq, ddd, JC-F = 3.0, 9.9 and 251 Hz), 154.5 (Cq); 19F-NMR (376 MHz, CDCl3) δ −134.7–134.8 (m, 1F), −140.2–140.3 (m, 1F), −162.4–162.5 (m, 1F); HRMS (ES) m/z calcd for C11H7F3O3 [M]+ 244.0347, found 240.0344.
3.4. 3-(4-Methoxyphenyl)-1-(2,4,6-trifluorophenyl)prop-2-yn-1-yl Methyl Carbonate (2h)
To a stirred solution of p-ethynylanisole (0.61 mL, 4.68 mmol) in THF (16 mL), n-BuLi (2.8 M in THF, 1.80 mL, 4.68 mmol) was added dropwise at −78 °C. The mixture was stirred for 0.5 h at the same temperature. A solution of 2,4,6-trifluorobenzaldehyde (500 mg, 3.12 mmol) in THF (5 mL) was then added dropwise at −78 °C, and stirring was continued for 1 h. Subsequently, methyl chloroformate (0.72 mL, 9.36 mmol) was added dropwise at −78 °C, and then the temperature was allowed to rise to room temperature over 1 h. The reaction mixture was diluted with water and extracted with AcOEt. The combined organic layers were washed with brine, dried, and concentrated. The crude product was purified by column chromatography on silica gel (hexane/AcOEt = 75:25 v/v) to give propargylic carbonate 2h as a colorless oil (1.02 g, 2.91 mmol, 93% yield). IR (ATR): 2960, 2226, 1751, 1604 cm−1; 1H-NMR (500 MHz, CDCl3) δ 3.80 (3H, s), 3.82 (3H, s), 6.72 (2H, t, J = 8.5 Hz), 6.78 (1H, s), 6.83 (2H, d, J = 8.5 Hz), 7.39 (2H, d, J = 8.5 Hz), 13C-NMR (125 MHz, CDCl3) δ 55.4 (CH3), 60.1 (CH3), 81.7 (Cq), 87.4 (Cq), 101.0 (CH, td, J = 2.9, 26.1 Hz), 110.5 (Cq, td, J = 4.9, 16.8 Hz), 113.8 (Cq), 114.1 (CH), 133.8 (CH), 154.7 (Cq), 160.3 (Cq, m), 160.4 (Cq), 162.4 (Cq, dd, J = 8.9, 13.8 Hz), 164.4 (Cq, t, J = 15.0 Hz); 19F-NMR (376 MHz, CDCl3) δ −108.2–108.3 (m, 1F), −112.0 (t, J = 6.9 Hz, 2F); HRMS (ES) m/z calcd for C18H13F3O4 [M]+ 350.0766, found 350.0769.
3.5. Methyl 3-Phenyl-1-(2,4,6-trifluorophenyl)prop-2-yn-1-yl Carbonate (2i)
By following the same procedure described for 2h, propargylic carbonate 2i was prepared from 2,3,4-trifluorobenzaldehyde in 99% yield (997 mg, 3.11 mmol) as a colorless oil. IR (ATR): 3088, 2960, 1752, 1635 cm−1; 1H-NMR (500 MHz, CDCl3) δ 3.83 (3H, s), 6.71 (2H, t, J = 8.0 Hz), 6.80 (1H, s), 7.29–7.36 (3H, m), 7.45 (2H, dd, J = 2.0, 8.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 55.4 (CH3), 59.9 (CH), 82.9 (Cq), 87.2 (Cq), 100.8 (CH, td, J = 3.0, 25.6 Hz), 110.2 (Cq, dd, J = 4.9, 16.8 Hz), 121.8 (Cq), 128.4 (CH), 129.2 (CH), 132.2 (CH), 154.7 (Cq), 160.3 (Cq, dd, J = 8.9, 15.3 Hz), 162.4 (Cq, m), 164.4 (Cq, t, J = 14.8 Hz); 19F-NMR (376 MHz, CDCl3) δ −110.0–110.1 (m, 1F), −113.0 (t, J = 6.9 Hz, 2F); HRMS (ES) m/z calcd for C17H11F3O3 [M]+ 320.0660, found 320.0655.
3.6. Procedure for the Synthesis of 1,4-Benzodiazepines—Reaction of 1a and 2a (Table 1, Entry 9)
To a stirred solution of propargylic carbonate 2a (29.9 mg, 123 µmol) in dioxane (0.5 mL) were added N-tosyl-disubstituted 2-aminobenzylamine 1a (40.5 mg, 94.2 µmol) and Pd(PPh3)4 (10.9 mg, 9.4 µmol) at 25 °C. The reaction mixture was stirred for 3 h at the same temperature. After filtration through a small amount of silica gel and concentration under reduced pressure, the crude residue was purified by column chromatography on silica gel (hexane/AcOEt = 5:1 v/v) to afford the 1,4-benzodiazepines (Z)-3a (38.0 mg, 69.8 µmol, 74%) and (E)-3a (12.8 mg, 23.6 µmol, 25%) as colorless crystals.
3.7. (Z)-2-Benzylidene-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(Z)-3a]
Yield: 74% (38.0 mg, 69.8 µmol); colorless quadrilaterals (AcOEt, mp. 147–150 °C); IR (ATR): 2855, 1597, 1489 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 2.25 (3H, s), 2.35 (3H, s), 3.59 (1H, d,
J = 15.0 Hz), 4.17 (1H, d,
J = 15.0 Hz), 4.42 (1H, d,
J = 15.0 Hz)–4.47 (1H, d,
J = 15.0 Hz), 6.45 (1H, s), 7.01 (2H, d,
J = 8.0 Hz), 7.05 (2H, d,
J = 8.0 Hz), 7.17–7.22 (4H, m), 7.24–7.29 (7H, m), 7.36 (2H, d,
J = 8.5 Hz);
13C-NMR (125 MHz, CDCl
3) δ 21.5 (CH
3), 21.7 (CH
3), 51.8 (CH
2), 55.8 (CH
2), 144.1 (Cq), 143.5 (Cq), 138.3 (Cq), 136.8 (Cq), 136.1 (CH), 136.0 (Cq), 133.5 (Cq), 131.0 (Cq), 130.7 (CH), 129.6 (CH), 129.4 (CH), 129.0 (CH), 129.0 (CH), 128.5 (CH), 128.3 (CH), 128.2 (CH), 127.8 (CH), 127.6 (CH); HRMS (EI)
m/z calcd for C
30H
28N
2O
4S
2 [M]
+ 544.1490, found 544.1496. For single-crystal data, see
Supplementary Materials.
3.8. (E)-2-Benzylidene-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(E)-3a]
Yield: 25% (12.8 mg, 23.6 µmol); colorless quadrilaterals (AcOEt, mp. 165–171 °C); IR (ATR): 2364, 1597, 1490 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 2.38 (3H, s), 2.45 (3H, s), 4.29–4.31 (4H, m), 6.71 (1H, s), 7.07 (2H, d,
J = 8.0 Hz), 7.23–7.40 (13H, m), 7.70 (2H, d,
J = 8.0 Hz);
13C-NMR (125 MHz, CDCl
3) δ 49.8 (CH
2), 51.91 (CH
2), 127.4 (CH), 127.8 (CH), 127.9 (CH), 128.3 (CH), 128.6 (CH), 128.7 (CH), 129.1 (CH), 129.3 (CH), 129.5 (CH), 129.8 (CH), 130.4 (CH), 133.1 (Cq), 133.8 (Cq), 135.3 (Cq), 135.7 (Cq), 137.3 (CH), 137.7 (Cq), 139.9 (Cq), 143.4 (Cq), 144.2 (Cq); HRMS (EI)
m/z calcd for C
30H
28N
2O
4S
2 [M]
+ 544.1490, found 544.1497. For single-crystal data, see
Supplementary Materials.
3.9. (Z)-2-(4-(Methylsulfonyl)benzylidene)-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(Z)-3b]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (Z)-3b was prepared from 1a and 2b, with a yield of 61% (26.6 mg, 42.7 µmol) as a colorless oil. IR (ATR): 2925, 2855, 1597, 1489 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.29 (3H, s), 2.38 (3H, s), 3.08 (3H, s), 3.52 (1H, d, J = 15.5 Hz), 4.14 (1H, d, J = 11.5 Hz), 4.44–4.49 (2H, m), 6.53 (1H, s), 7.05 (2H, d, J = 8.0 Hz), 7.09 (2H, d, J = 8.0 Hz), 7.14 (2H, d, J = 8.0 Hz), 7.25–7.28 (4H, m), 7.39 (2H, d, J = 8.0 Hz), 7.44 (2H, d, J = 8.0 Hz), 7.86 (2H, d, J = 8.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.6 (CH3), 21.7 (CH3), 44.6 (CH3), 51.9 (CH2), 55.4 (CH2), 127.3 (CH), 127.6 (CH), 127.7 (CH), 128.8 (CH), 129.3 (CH), 129.4 (CH), 129.5 (CH), 129.8 (CH), 130.9 (CH), 134.0 (CH), 134.3 (Cq), 136.1 (Cq), 136.4 (Cq), 137.9 (Cq), 139.2 (Cq), 140.1 (Cq), 143.7 (Cq), 144.7 (Cq); HRMS (EI) m/z calcd for C31H30N2O6S3 [M]+ 622.1266, found 622.1271.
3.10. (E)-2-(4-(Methylsulfonyl)benzylidene)-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(E)-3b]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (E)-3b was prepared from 1a and 2b, with a yield of 38% (16.6 mg, 26.6 µmol), as a colorless oil. IR (ATR): 2924 2852, 1597, 1490 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.41 (3H, s), 2.44 (3H, s), 3.08 (3H, s), 4.15–4.16 (3H, m), 6.80 (1H, s), 7.17 (2H, d, J = 8.5 Hz), 7.25–7.29 (5H, m), 7.36 (3H, d, J = 8.0 Hz), 7.46 (2H, d, J = 8.0 Hz), 7.64 (2H, d, J = 8.5 Hz), 7.95 (2H, d, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.7 (CH3), 21.8 (CH3), 44.6 (CH3), 49.7 (CH2), 51.9 (CH2), 127.4 (CH), 127.8 (CH), 128.3 (CH), 128.8 (CH), 129.6 (CH), 129.8 (CH), 130.0 (CH), 130.0 (CH), 130.5 (CH), 134.4 (CH), 135.0 (Cq), 135.5 (Cq), 136.2 (Cq), 137.4 (Cq), 139.4 (Cq), 139.6 (Cq),140.3 (Cq), 143.9 (Cq), 144.5 (Cq); HRMS (EI) m/z calcd for C31H30N2O6S3 [M]+ 622.1266, found 622.1269.
3.11. (Z)-2-(4-Fluorobenzylidene)-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(Z)-3c]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (Z)-3c was prepared from 1a and 2c, with a yield of 71% (28.0 mg, 50.0 µmol), as a colorless solid (AcOEt, mp. 151–155 °C). IR (ATR): 3023, 1596, 1510, 1488 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.27 (3H, s), 2.37 (3H, s), 3.49 (1H, d, J = 15.5 Hz), 4.14 (1H, d, J = 14.0 Hz, 1H), 4.38 (2H, d, J = 14.0 Hz), 4.44 (2H, d, J = 15.5 Hz), 6.40 (1H, s), 6.97 (2H, t, J = 8.5 Hz), 7.03 (2H, d, J = 8.5 Hz, 2H), 7.08 (2H, d, J = 8.5 Hz), 7.16–7.32 (8H, m), 7.36 (2H, d, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.5 (CH3), 21.7 (CH3), 51.8 (CH2), 55.6 (CH2), 115.4 (CH), 127.6 (CH), 127.7 (CH), 128.4 (CH, d, JC-F = 7.9 Hz), 129.1 (CH, d, JC-F = 14.8 Hz), 129.5 (CH), 129.5 (Cq, d, JC-F = 3.0 Hz), 129.6 (CH), 130.7 (CH), 130.9 (CH, d, JC-F = 7.9 Hz), 131.0 (Cq), 134.9 (CH), 135.9 (Cq), 136.1 (Cq), 136.5 (Cq), 138.1 (Cq), 143.5 (Cq), 144.3 (Cq), 162.8 (Cq, d, JC-F = 248 Hz); 19F-NMR (376 MHz, CDCl3) δ −115.4 (s, 1F); HRMS (ESI) m/z calcd for C30H27FN2O4S2 [M]+ 562.1396, found 562.1403.
3.12. (E)-2-(4-Fluorobenzylidene)-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(E)-3c]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (E)-3c was prepared from 1a and 2c, with a yield of 28% (11.0 mg, 19.6 µmol), as a colorless solid (AcOEt, mp. 165–169 °C). IR (ATR): 2922, 1601, 1508, 1490 cm−1; 1H-NMR (500 MHz, CDCl3) δ2.38 (3H, s), 2.43 (3H, s), 4.19–4.22 (4H, m), 6.66 (1H, s), 7.06 (2H, t, J = 8.5 Hz), 7.11 (2H, d, J = 8.0 Hz), 7.20–7.24 (4H, m), 7.27–7.33 (6H, m), 7.66 (2H, d, J = 8.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.7 (CH3), 21.8 (CH3), 49.9 (CH2), 52.0 (CH2), 115.8 (CH, d, JC-F = 21.8 Hz), 127.4 (CH), 127.8 (CH), 127.9 (CH), 128.5 (CH), 129.4 (CH), 129.7 (CH), 129.9 (CH), 130.5 (CH), 131.0 (CH, d, JC-F = 7.9 Hz), 133.4 (Cq), 135.2 (Cq), 135.7 (Cq), 136.3 (Cq), 137.7 (Cq), 140.0 (Cq), 143.6 (Cq), 144.3(Cq), 162.8 (Cq, d, JC-F = 249.7 Hz); 19F-NMR (376 MHz, CDCl3) δ −115.4–115.5 (m, 1F); HRMS (ES) m/z calcd for C30H27FN2O4S2 [M]+ 562.1396, found 562.1388.
3.13. (Z)-1,4-Ditosyl-2-(2,3,4-trifluorobenzylidene)-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(Z)-3d]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (Z)-3d was prepared from 1a and 2d, with a yield of 69% (33.4 mg, 55.9 µmol), as colorless needles (AcOEt, mp. 175–179 °C). IR (ATR): 2925, 1598, 1471 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.27 (3H, s), 2.38 (3H, s), 3.62 (1H, m), 4.40 (1H, m), 4.43 (2H, m), 6.51 (1H, s), 6.91 (1H, q, J = 7.5 Hz), 7.05 (2H, d, J = 8.0 Hz), 7.11 (2H, d, J = 8.0 Hz), 7.21–7.30 (5H, m), 7.36–7.40 (4H, m); 13C-NMR (125 MHz, CDCl3) δ 21.5 (CH3), 21.6 (CH3), 51.7 (CH2), 55.4 (CH2), 111.7 (CH, dd, JC-F 3.0, 17.8 Hz), 119.2 (Cq, dd, JC-F = 4.0, 9.9 Hz), 123.9 (CH, m), 126.7 (CH), 127.5 (CH), 127.7 (CH), 128.5 (CH), 129.1 (CH), 129.5 (CH), 129.7 (CH), 130.8 (CH), 134.6 (Cq), 135.7 (Cq), 135.9 (Cq), 136.3 (Cq), 138.0 (Cq), 139.8 (Cq, dt, JC-F = 15.9, 251.6 Hz), 143.6 (Cq), 144.6 (Cq), 149.2 (Cq, ddd, JC-F = 4.0, 9.9, 252.6 Hz), 151.3 (Cq, ddd, JC-F = 3.0, 8.9, 252.6 Hz); 19F-NMR (376 MHz, CDCl3) δ −136.2–136.3 (m, 1F), −137.4 (m, 1F), −163.7–163.8 (m, 1F); HRMS (EI) m/z calcd for C30H25F3N2O4S2 [M]+ 598.1208, found 598.1212.
3.14. (E)-1,4-Ditosyl-2-(2,3,4-trifluorobenzylidene)-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(E)-3d]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (E)-3d was prepared from 1a and 2d, with a yield of 30% (14.5 mg, 24.3 µmol), as colorless needles (AcOEt, mp. 154–159 °C). IR (ATR): 2924, 1598, 1471 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.39 (3H, s), 2.45 (3H, s), 4.18 (2H, s), 4.28 (2H, s), 6.58 (1H, s), 6.99–7.06 (1H, m), 7.12–7.17 (3H, m), 7.3 (3H, m), 7.28–7.41 (8H, m), 7.71 (2H, d, J = 8.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.6 (CH3), 21.8 (CH3), 50.0 (CH2), 51.9 (CH2), 112.6 (Cq, dd, JC-F = 4.0, 17.8 Hz), 119.4 (Cq, dd, JC-F = 4.0, 11.9 Hz), 124.6 (Cq, m), 127.3 (CH), 127.8 (CH), 127.9 (CH), 127.9 (CH), 128.8 (CH), 129.6 (CH), 129.7 (CH), 130.0 (CH), 130.5 (CH), 135.2 (Cq), 136.0 (Cq), 136.4 (Cq), 137.6 (Cq), 139.7 (Cq), 140.2 (Cq, d, JC-F = 252.6 Hz), 144.0 (Cq), 144.5 (Cq), 151.4 (Cq, d, JC-F = 254.6 Hz); 19F-NMR (376 MHz, CDCl3) δ −135.5–135.6 (m, 1F), −137.0–137.1 (m, 1F), −163.1–163.2 (m, 1F); HRMS (EI) m/z calcd for C30H25F3N2O4S2 [M]+ 598.1208, found 598.1212.
3.15. (Z)-1,4-Ditosyl-2-(4-(trifluoromethyl)benzylidene)-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine[(Z)-3e]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (Z)-3e was prepared from 1a and 2e, with a yield of 61% (26.1 mg, 42.7 µmol), as colorless quadrilaterals (AcOEt, mp. 171–180 °C). IR (ATR): 3015, 1596, 1487 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.35–2.36 (6H, m), 3.67 (1H, d, J = 15.5 Hz), 4.17 (1H, d, J = 15.5 Hz), 4.47 (2H, d, J = 15.5 Hz), 4.53 (2H, d, J = 15.5 Hz), 6.50 (1H, s), 7.03 (2H, d, J = 5.0 Hz), 7.04 (2H, d, J = 5.0 Hz), 7.09 (1H, d, J = 7.0 Hz), 7.14–7.18 (3H, m), 7.22–7.30 (4H, m), 7.38 (2H, d, J = 8.0 Hz), 7.52 (2H, d, J = 8.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.5 (CH3), 21.6 (CH3), 52.0 (CH2), 55.7 (CH2), 123.0 (Cq), 125.1 (CH, d, JC-F = 4.0 Hz), 127.7 (CH, d, JC-F = 9.9 Hz), 128.4 (CH), 128.6 (CH), 129.2 (CH), 129.3 (CH), 129.5 (CH), 129.6 (CH), 130.3 (Cq, d, JC-F = 32.5 Hz), 130.9 (CH), 133.3 (Cq), 134.6 (CH), 136.2 (Cq), 136.3 (Cq), 136.5 (Cq), 137.2 (Cq), 138.0 (Cq), 138.2 (Cq), 143.6 (Cq), 144.4 (Cq); 19F-NMR (376 MHz, CDCl3) δ −65.8 (s, 3F); HRMS (ESI) m/z calcd for C31H27F3N2O4S2 [M]+ 612.1364, found 612.1370.
3.16. (E)-1,4-Ditosyl-2-(4-(trifluoromethyl)benzylidene)-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(E)-3e]
By following the same procedure described with respect to 3a, 1,4-benzodiazepine (E)-3e was prepared from 1a and 2e, with a yield of 38% (16.3 mg, 26.6 µmol), as colorless quadrilaterals (AcOEt, mp. 186–191 °C). IR (ATR): 3037, 1598, 1490 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.38 (3H, s), 2.44 (3H, s), 4.18 (4H, m), 6.75 (1H, s), 7.12 (2H, d, J = 8.5 Hz), 7.25–7.37 (10H, m), 7.62 (2H, d, J = 8.5 Hz), 7.66 (2H, d, J = 8.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.6 (CH3), 21.8 (CH3), 49.7 (CH2), 51.9 (CH2), 125.2 (Cq, q, JC-F = 272.5 Hz), 125.7 (CH, d, JC-F = 3.0 Hz), 127.4 (CH), 127.8 (CH), 128.2 (CH), 128.7 (CH), 129.5 (CH), 129.5 (CH), 129.7 (CH), 129.9 (CH), 130.3 (Cq, q, JC-F = 32.7 Hz), 130.5 (CH), 135.1 (Cq), 135.2 (Cq), 135.4 (Cq), 135.7 (Cq), 137.5 (CH), 139.6 (Cq), 143.7 (Cq), 144.4 (Cq); 19F-NMR (376 MHz, CDCl3) δ −65.9 (s, 3F); HRMS (ESI) m/z calcd for C31H27F3N2O4S2 [M]+ 612.1364, found 612.1364.
3.17. N-(1,3-Diphenylprop-2-yn-1-yl)-4-methyl-N-(2-(((4-methylphenyl)sulfonamido)methyl)phenyl)benzenesulfonamide(6)
To a stirred solution of propargylic carbonate
2g (25.3 mg, 95.2 µmol) in dioxane (0.5 mL) were added
N-tosyl-disubstituted 2-aminobenzylamine
1a (31.5 mg, 73.2 µmol) and Pd(PPh
3)
4 (8.4 mg, 7.3 µmol) at 50 °C. The reaction mixture was stirred for 1 h at the same temperature. After filtration through a small amount of silica gel and concentration under reduced pressure, the crude residue was purified by column chromatography on silica gel (hexane/AcOEt = 4:1
v/
v) to afford
6. Yield: 97% (44.1 mg, 71.0 µmol); colorless quadrilaterals (AcOEt, mp. 173–176 °C); IR (ATR): 3292, 3027, 1598, 1491 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 2.41 (3H, s), 2.53 (3H, s), 3.23 (1H, dd,
J = 13.0, 9.0 Hz), 3.32 (1H, dd,
J = 13.0, 4.5 Hz), 4.70 (1H, dd,
J = 9.0, 4.5 Hz), 6.51 (1H, s), 6.94 (2H, d,
J = 7.5 Hz), 7.03–7.15 (6H, m), 7.22–7.37 (8H, m), 7.42 (2H, d,
J = 8.0 Hz), 7.68 (2H, d,
J = 8.5 Hz), 7.73 (2H, d,
J = 8.5 Hz);
13C-NMR (125 MHz, CDCl
3) δ 21.7 (CH
3), 21.8 (CH
3), 42.5 (CH
2), 56.3 (CH), 85.2 (Cq), 88.5 (Cq), 122.0 (CH), 127.5 (CH), 127.8 (CH), 128.5 (CH), 128.5 (CH), 128.6 (CH), 128.9 (CH), 129.0 (CH), 129.5 (CH), 129.7 (CH), 129.8 (CH), 130.5 (CH), 131.4 (CH), 131.6 (CH), 133.8 (Cq), 135.6 (Cq), 135.7 (Cq), 136.9 (Cq), 139.6 (Cq), 143.3 (Cq), 144.2 (Cq); HRMS (ESI)
m/z calcd for C
36H
32N
2O
4S
2 [M]
+ 620.1803, found 556.2766 [M−SO
2]
+. For single-crystal data, see
Supplementary Materials.
3.18. (Z)-3-Benzylidene-2-phenyl-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(Z)-7g]
To a stirred solution of propargylic carbonate
2g (25.5 mg, 95.6 µmol) in dioxane (0.5 mL) were added
N-tosyl-disubstituted 2-aminobenzylamine
1a (31.6 mg, 73.4 µmol), Pd
2(dba)
3·CHCl
3 (3.8 mg, 3.7 µmol), and DPPP (6.1 mg, 14.7 µmol) at 50 °C. The reaction mixture was stirred for 1 h at the same temperature. After filtration through a small amount of silica gel and concentration under reduced pressure, the crude residue was purified by column chromatography on silica gel (hexane/AcOEt = 4:1
v/
v) to afford
(Z)-7g. Yield: 99% (44.9 mg, 72.3 µmol); colorless quadrilaterals (AcOEt, mp. 183–187 °C); IR (ATR): 3023, 1598, 1492 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 2.37 (6H, m), 4.23 (1H, d,
J = 11.5 Hz), 4.39 (1H, d,
J = 11.5 Hz), 5.79 (1H,s), 6.02 (1H, s), 6.90 (2H, d,
J = 7.5 Hz), 7.01–7.09 (5H, m), 7.20 (4H, d,
J = 8.5 Hz), 7.28 (1H, m), 7.34–7.40 (7H, m), 7.46 (1H, d,
J = 7.5 Hz), 7.59 (2H, d,
J = 8.0 Hz);
13C-NMR (125 MHz, CDCl
3) δ 21.7 (CH
3), 21.7 (CH
3), 50.5 (CH
2), 68.0 (CH), 127.6 (CH), 127.9 (CH), 128.0 (CH), 128.1 (CH), 128.1 (CH), 128.7 (CH), 128.8 (CH), 129.0 (CH), 129.4 (CH), 129.7 (CH), 129.9 (CH), 130.4 (CH), 131.2 (CH), 132.5 (Cq), 133.9 (Cq), 134.3 (Cq), 135.6 (Cq), 137.3 (Cq), 138.8 (Cq), 139.5 (CH), 140.5 (Cq), 143.7 (Cq), 144.0 (Cq); HRMS (ESI)
m/z calcd for C
36H
32N
2O
4S
2 [M]
+ 620.1803, found 556.2747 [M−SO
2]
+. For single-crystal data, see
Supplementary Materials.
3.19. (Z)-2-(4-Methoxyphenyl)-1,4-ditosyl-3-(2,4,6-trifluorobenzylidene)-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(Z)-7h]
By following the same procedure described with respect to (Z)-7g, 1,4-benzodiazepine (Z)-7h was prepared from 1a and 2h, with a yield of 71% (29.3 mg, 41.6 µmol), as colorless quadrilaterals (AcOEt, mp. 196–201 °C). IR (ATR): 2925, 1596, 1495 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.36 (3H, s), 2.47 (3H, s), 3.84 (3H, s), 4.26 (1H, d, J = 11.5 Hz), 4.43 (1H, d, J = 11.5 Hz), 5.69 (1H, s), 5.92 (1H, d, J = 2.0 Hz), 6.42 (2H, t, J = 8.0 Hz), 6.71 (2H, s), 6.89 (2H, d, J = 8.5 Hz), 7.05 (2H, d, J = 8.5 Hz), 7.14–7.17 (4H, m), 7.32–7.36 (4H, m), 7.69 (2H, d, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.7 (CH3), 21.8 (CH3), 50.5 (CH2), 55.5 (CH3), 67.3 (CH), 100.1 (CH, dt, JC-F = 3.0, 25.8 Hz), 109.6 (Cq, dt, JC-F = 4.0, 19.8 Hz), 114.3 (CH), 116.3 (CH), 126.5 (CH), 127.6 (CH), 127.9 (CH), 128.7 (CH), 129.4 (CH), 129.4 (CH), 130.0 (CH), 130.5 (CH), 131.6 (CH), 132.1 (Cq), 134.1 (Cq), 136.0 (Cq), 137.4 (Cq), 138.1 (Cq), 138.5 (Cq), 143.6 (Cq), 144.2 (Cq), 149.6 (Cq), 159.6 (Cq), 160.3 (Cq, dt, JC-F = 14.9, 244.7 Hz); 19F-NMR (376 MHz, CDCl3) δ −108.5 (s, 2F) −111.6–111.7 (m, 1F); HRMS (ESI) m/z calcd for C37H31F3N2O5S2 [M]+ 704.1626, found 640.2001 [M−SO2]+.
3.20. (Z)-2-Phenyl-1,4-Ditosyl-3-(2,4,6-trifluorobenzylidene)-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine [(Z)-7i]
By following the same procedure described with respect to
(Z)-7g, 1,4-benzodiazepine
(Z)-7h was prepared from
1a and
2i, with a yield of 85% (45.2 mg, 66.9 µmol), as colorless quadrilaterals (AcOEt, mp. 182–186 °C). IR (ATR): cm
−1;
1H-NMR (500 MHz, CDCl
3)
1H-NMR (500 MHz, CDCl
3) δ 2.36 (3H, s), 2.47 (3H, s), 4.25 (1H, d
J = 11.5 Hz), 4.45 (1H, d,
J = 11.5 Hz), 6.41 (2H, t,
J = 8.0 Hz), 7.05 (2H, d,
J = 8.0 Hz), 7.27 (1H, s), 7.31–7.37 (9H, m), 7.70 (2H, d,
J = 8.0 Hz);
13C-NMR (125 MHz, CDCl
3) δ 21.7 (CH
3), 21.8 (CH
3), 50.4 (CH
2), 67.7 (CH), 100.1 (CH, d,
JC-F = 25.8 Hz), 109.5 (Cq, dt,
JC-F = 5.0, 18.8 Hz), 126.8 (CH), 127.6 (CH), 127.9 (CH), 128.2 (Cq), 128.4 (CH), 128.7 (CH), 128.9 (CH), 129.4 (CH), 129.5 (CH), 130.0 (CH), 130.5 (CH), 131.7 (CH), 134.1 (Cq), 136.0 (Cq), 137.3 (Cq), 138.2 (Cq), 138.3 (Cq), 140.0 (Cq), 143.7 (Cq), 144.3 (Cq), 160.3 (Cq, ddd,
JC-F = 10.0, 14.9, 254.6 Hz), 162.3 (Cq, dt,
JC-F = 15.9, 249.7 Hz);
19F-NMR (376 MHz, CDCl
3) δ −108.4 (s, 2F) −111.5–111.6 (m, 1F); HRMS (ESI)
m/z calcd for C
36H
29F
3N
2O
4S
2 [M]
+ 674.1521, found 674.1534. For single-crystal data, see
Supplementary Materials.
3.21. 2-Methylene-5-phenyl-1,4-ditosyl-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine (3j)
To a stirred solution of propargylic carbonate 2j (24.6 mg, 129 µmol) in dioxane (0.5 mL) were added N-tosyl-disubstituted 2-aminobenzylamine 1b (50.4 mg, 99.5 µmol) and Pd(PPh3)4 (11.6 mg, 10.0 µmol) at 80 °C. The reaction mixture was stirred for 1 h at the same temperature. After filtration through a small amount of silica gel and concentration under reduced pressure, the crude residue was purified by column chromatography on silica gel (hexane/AcOEt = 5:1 v/v) to produce the 1,4-benzodiazepines 3j. Yield: 62% (33.6 mg, 61.7 µmol); colorless needles (CH2CI2, mp. 186–190 °C); IR (ATR): 3062, 1645, 1595, 1491 cm−1; 1H-NMR (500 MHz, CDCl3) δ 2.30 (3H, s), 2.34 (3H, s), 3.25 (1H, d, J = 15.0 Hz), 4.13 (1H, d, J = 15.0 Hz), 4.92 (1H, s), 5.31 (1H, s), 6.38 (1H, s), 6.98–7.02 (4H, m), 7.05–7.07 (2H, m), 7.10–7.12 (2H, m), 7.20–7.25 (2H, m), 7.36–7.39 (6H, m), 7.63 (1H, d, J = 8.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 21.6 (CH3), 21.7 (CH3), 47.9 (CH2), 64.2 (CH), 119.5 (CH), 127.0 (CH), 127.2 (CH), 127.5 (CH), 127.9 (CH), 129.0 (CH), 129.1 (CH), 129.3 (CH), 129.3 (CH), 129.4 (CH), 132.2 (Cq), 133.8 (Cq), 135.0 (Cq), 138.0 (Cq), 138.9 (Cq), 139.6 (Cq), 143.4 (Cq), 144.2 (Cq); HRMS (ESI) m/z calcd for C30H28N2O4S2 [M]+ 544.1490, found 544.1490.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}