
Ultrasound and Microwave-Assisted Synthesis and Antidiabetic and Hematopoietic Activity of Diphenhydramine Derivatives

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis of Diphenhydramine Derivatives
2.2. Antidiabetic Activity
2.2.1. Inhibitory Activity Against the Enzyme α-Glucosidase
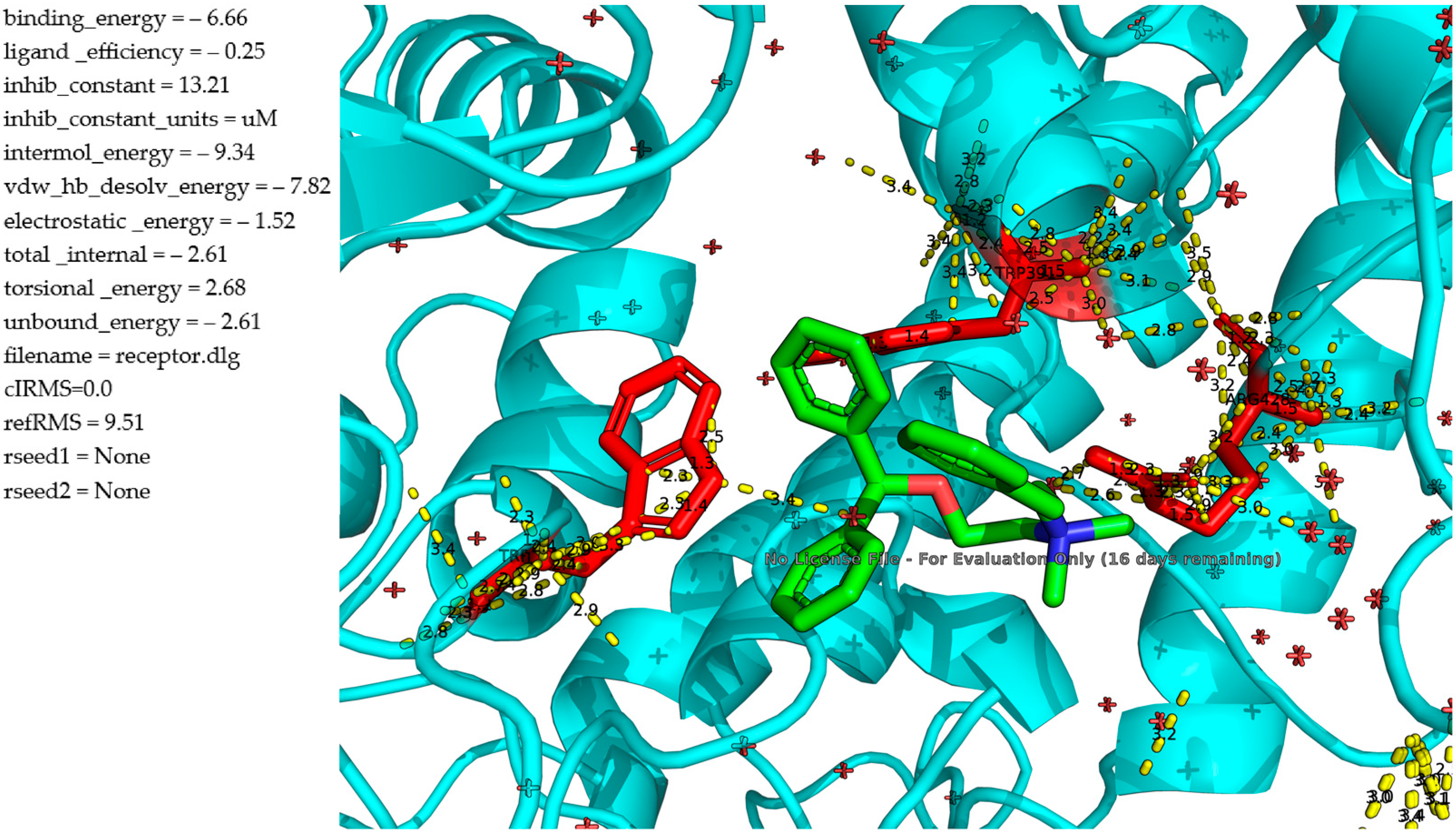
Molecular Docking Study Between α-Glucosidase and Diphenhydramine Derivatives
2.2.2. Inhibitory Activity Against the Enzyme α-Amylase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Code | The Degree of Inhibition of the Activity of α-Amylase, % |
|---|---|---|
| 1 | 1·HCl | 34.7 ± 1.8 |
| 2 | 1a | 31.8 ± 1.1 |
| 3 | 1b | 8.7 ± 1.3 |
| 4 | 1c | No inhibition |
| 5 | 1d | No inhibition |
| 6 | 1e | No inhibition |
| 7 | 1f | No inhibition |
| 8 | 1g | No inhibition |
| 9 | 1h | No inhibition |
| 10 | 1i | No inhibition |
| 11 | 1j | No inhibition |
| 12 | 1k | No inhibition |
| 13 | 1l | No inhibition |
| 14 | 1m | No inhibition |
| 15 | Acarbose | 62.0 ± 0.7 |
2.3. Hematopoietic Activity
3. Materials and Methods
3.1. Chemical Research Methods
3.2. General Procedure of Synthesis
3.3. Structural Confirmation via UV Analysis
3.4. Structural Confirmation via NMR Analysis
- 2-(benzhydryloxy)-N,N,N-trimethylethanaminium iodide (1a)
- 2-(benzhydryloxy)-N-ethyl-N,N-dimethylethanaminium iodide (1b)
- N-(2-(benzhydryloxy)ethyl)-N,N-dimethylpropan-1-aminium iodide (1c)
- N-(2-(benzhydryloxy)ethyl)-N,N-dimethylbutan-1-aminium iodide (1d)
- 2-(benzhydryloxy)-N-(cyanomethyl)-N,N-dimethylethanaminium iodide (1e)
- 2-(benzhydryloxy)-N-(2-hydroxyethyl)-N,N-dimethylethanaminium iodide (1f)
- N-(2-(benzhydryloxy)ethyl)-3-hydroxy-N,N-dimethylpropan-1-aminium iodide (1g)
- N-(2-(benzhydryloxy)ethyl)-4-hydroxy-N,N-dimethylbutan-1-aminium iodide (1h)
- N-(2-(benzhydryloxy)ethyl)-2-ethoxy-N,N-dimethyl-2-oxoethanaminium iodide (1i)
- 2-(benzhydryloxy)-N-(2-carboxyethyl)-N,N-dimethylethanaminium iodide (1j)
- 2-(benzhydryloxy)-N-benzyl-N,N-dimethylethanaminium chloride (1k)
- 2-(benzhydryloxy)-N,N-dimethyl-N-phenethylethanaminium iodide (1l)
- N-(2-(benzhydryloxy)ethyl)-N,N-dimethyl-3-phenylpropan-1-aminium iodide (1m)
3.5. Structural Confirmation via MASS Spectrometry
3.6. Biological Research Methods
3.6.1. Experiment on the Inhibitory Activity Against the Enzyme α-Glucosidase
3.6.2. Experiment on the Inhibitory Activity Against the Enzyme α-Amylase
3.6.3. Animal Studies: Hematopoietic Activity
3.7. Molecular Docking Study
3.8. Statistical Data Processing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| US | Ultrasonic |
| MW | Microwave |
| DM | Diabetes mellitus |
| WBC | White blood cell count |
| NEU | Neutrophil–lymphocyte ratio |
| LYM | Absolute lymphocyte count |
| MON | Monocyte count |
| EO | Eosinophil count |
| BAS | Basophils |
| RBC | Red blood cell count |
| HGB | Hemoglobin |
| HCT | Hematocrit |
| MCV | Mean corpuscular volume |
| MCH | Mean corpuscular hemoglobin |
| MCHC | Mean corpuscular hemoglobin concentration |
| RDW | Red blood cell distribution width |
| PLT | Total platelets volume |
| MPV | Mean platelet volume |
References
- Santos, H.M.; Lodeiro, C.; Capelo-Martinez, J. Ultrasounds in Chemistry: Analytical Applications; Capelo-Martinez, J.-L., Ed.; Wiley: Weinheim, Germany, 2009; p. 171. ISBN 978-3-527-31934-3. [Google Scholar]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed]
- Cravotto, G.; Boffa, L.; Mantegna, S.; Perego, P.; Avogadro, M.; Cintas, P. Improved extraction of vegetable oils under high-intensity ultrasound and/or microwaves. Ultrason. Sonochem. 2008, 15, 898–902. [Google Scholar] [CrossRef]
- Shah, J.J.; Mohanraj, K. Comparison of Conventional and Microwave-assisted Synthesis of Benzotriazole Derivatives. Indian J. Pharm. Sci. 2014, 76, 46–53. [Google Scholar]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Suslick, K.S. Sonochemistry. Science 1990, 247, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- IDF Diabetes Atlas, 6th ed. 2013. Available online: https://diabetesatlas.org/idfawp/resource-files/2010/07/IDF_diabetes_atlas_sixth_edition_en.pdf (accessed on 11 March 2025).
- Ahren, B. New strategy in type 2 diabetes tested in clinical trials: Glucagon-like peptide 1 (GLP-1) affects basic caused of the disease. Lakartidningen 2005, 102, 545–549. [Google Scholar]
- Ahren, B.; Pacini, G.; Foley, J.; Schweizer, A. Improved meal-related (beta)-cell function and insulin sensitivity by the dipeptidyl peptidase-iv inhibitor vildagliptin in metformin-treated patients with type 2 diabetes over 1 year. Diabetes Care 2005, 28, 1936–1940. [Google Scholar] [CrossRef] [PubMed]
- Amori, R.E.; Lau, J.; Pittas, A.G. Efficacy and safety of incretin therapy in type 2 diabetes: Systemic review and meta-analysis. JAMA 2007, 298, 194–206. [Google Scholar] [CrossRef]
- Srinivasan, K.; Ramarao, P. Animal models in type 2 diabetes research: An overview. Indian J. Med. Res. 2007, 125, 451–472. [Google Scholar]
- Risbud, M.V.; Bhonde, R.R. Models of pancreatic regeneration in diabetes. Diabetes Res. Clin. Pr. 2002, 58, 155–165. [Google Scholar] [CrossRef]
- Leiter, E.H.; Prochazka, M.; Shultz, L.D. Effect of immunodeficiency on diabetogenesis in getenocally diabetic (db/db) mice. J. Immunol. 1987, 138, 3224–3229. [Google Scholar] [CrossRef] [PubMed]
- Pliszka, M.; Szablewski, L. Associations between Diabetes Mellitus and Selected Cancers. Int. J. Mol. Sci. 2024, 25, 7476. [Google Scholar] [CrossRef] [PubMed]
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes Mellitus, Fasting Glucose, and Risk of Cause-Specific Death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Barone, B.B.; Yeh, H.C.; Snyder, C.F.; Peairs, K.S.; Stein, K.B.; Derr, R.L.; Wolff, A.C.; Brancati, F.L. Long-Term All-Cause Mortality in Cancer Patients With Preexisting Diabetes Mellitus: A Systematic Review and Meta-Analysis. JAMA 2008, 300, 2754–2764. [Google Scholar] [CrossRef]
- Pavlidakey, P.G.; Brodell, E.E.; Helms, S.E. Diphenhydramine as an alternative local anesthetic agent. J. Clin. Aesthet. Dermatol. 2009, 2, 37–40. [Google Scholar] [PubMed] [PubMed Central]
- Brambilla, G.; Mattioli, F.; Robbiano, L.; Martelli, A. Genotoxicity and carcinogenicity studies of antihistamines. Arch. Toxicol. 2011, 85, 1173–1187. [Google Scholar] [CrossRef]
- Giner, B.; Mergenbayeva, S.; Lomba, L.; Rafikova, K.; Dauletbakov, A.; Belyankova, Y.; Seilkhanov, T.; Zazybin, A. Synthesis and Ecotoxicological Studies of ionic compounds based on Tolperisone, Diphenhydramine and Trimecaine. ChemistrySelect 2020, 5, 12823–12828. [Google Scholar] [CrossRef]
- Chayng, P.J.; Ain, N.; Ambia, K.; Noah, R. Anti-diabetic activity of diphenhydramine in diabetic rats. Int. J. Res. Pharm. Sci. 2020, 11, 5067–5070. [Google Scholar] [CrossRef]
- Telagari, M.; Hullatti, K. In-vitro α-amylase and α-glucosidase inhibitory activity of Adiantum caudatum Linn. and Celosia argentea Linn. extracts and fractions. Indian J. Pharmacol. 2015, 47, 425–429. [Google Scholar] [CrossRef]
- Haiwei, R.; Nana, D.; Xiaoqian, N.; Yonggang, W.; Wenguang, F.N. Inhibitory effects of l-3-phenyllacitc acid on the activity of mushnroom pholyphenol oxidase. Food Sci. Technol. 2021, 41, 343–351. [Google Scholar] [CrossRef]
- Bernfeld, P.; Colowick, S.P.; Kaplan, N.O. Amylase, α and β. In Methods in Enzymology; Academic Press: New York, NY, USA, 1955; pp. 149–158. [Google Scholar] [CrossRef]
- PASS Online. Way2Drug Predictive Service. Available online: https://www.way2drug.com/passonline/ (accessed on 8 April 2025).
- Dauletbakov, A.; Belyankova, Y.; Tursynbek, S.; Anapiayev, B.; Zolotareva, D.; Ten, A.; Zazybin, A. Synthesis and growth-stimulating activity of trimecaine ethyl iodoethanoate. Chem. J. Kaz. 2022, 3, 92–100. [Google Scholar] [CrossRef]
- Giemsa, G. Eine Vereinfachung und Vervollkommnung meiner Methylenazur-Methylenblau-Eosin-Färbemethode zur Erzielung der Romanowsky-Nochtschen Chromatinfärbung. Cent. Bakt. 1904, 37, 308–311. [Google Scholar]
- Order of the Minister of Healthcare of the Republic of Kazakhstan No. KR DSM-181/2020 of 4 November 2020. On approval of the Rules for the Assessment of Materials and Compliance of Preclinical (non-clinical) Studies with Good Laboratory Practice (GLP) Requirements of the Republic of Kazakhstan and/or the Eurasian Economic Union within the Framework of Pharmaceutical Inspection. (Registered with the Ministry of Justice of the Republic of Kazakhstan on 5 November 2020 under No. 21596). Available online: https://adilet.zan.kz/eng/docs/V2000021596 (accessed on 11 March 2025).
- RCSB Protein Data Bank (RCSB PDB). Available online: https://www.rcsb.org/ (accessed on 13 May 2025).
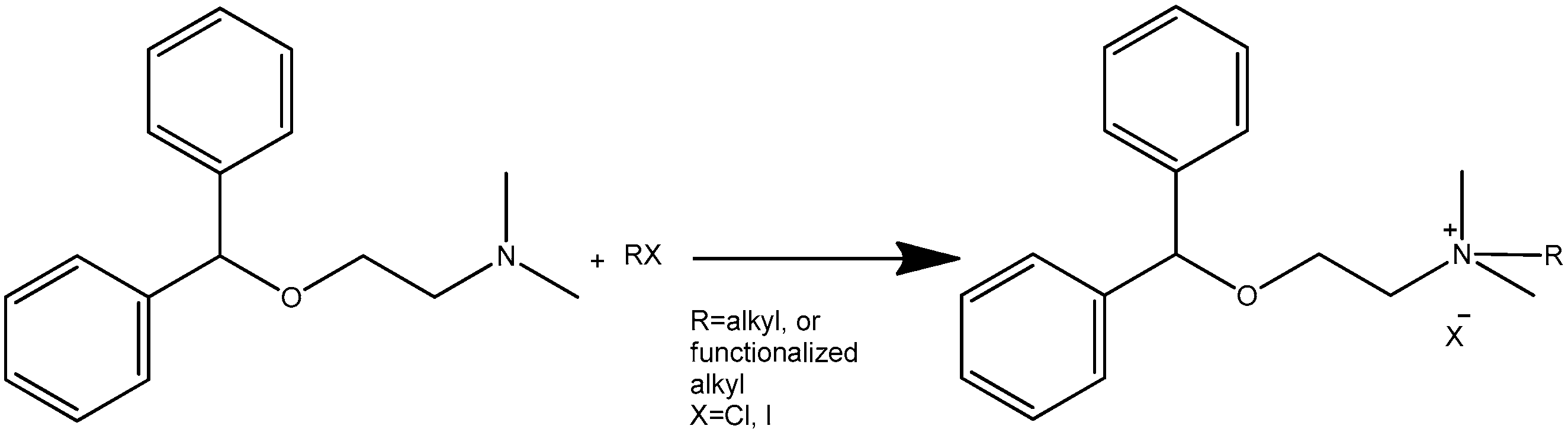


| Products | Reaction Conditions * | Time, Min | Yield, % | Melting Point |
|---|---|---|---|---|
| 1a | Classical | 20 | 85 | 205–207 °C |
| -CH3 | US | 10 | 81 | |
| MW | 1 | 89 | ||
| 1b | Classical | 60 | 78 | 161–163 °C |
| -C2H5 | US | 30 | 62 | |
| MW | 3 | 79 | ||
| 1c | Classical | 120 | 81 | 145–147 °C |
| -n-C3H7 | US | 40 | 65 | |
| MW | 4 | 83 | ||
| 1d | Classical | 180 | 81 | 141–143 °C |
| -n-C4H9 | US | 60 | 70 | |
| MW | 7 | 96 | ||
| 1e | Classical | 90 | 85 | 128–130 °C |
| -CH2CN | US | 30 | 45 | |
| MW | 3 | 96 | ||
| 1f | Classical | 180 | 91 | 106–108 °C |
| -CH2CH2OH | US | 60 | 80 | |
| MW | 6 | 93 | ||
| 1g | Classical | 180 | 87 | 105–107 °C |
| -(CH2)3OH | US | 60 | 79 | |
| MW | 6 | 89 | ||
| 1h | Classical | 210 | 81 | 104–106 °C |
| -(CH2)4OH | US | 120 | 71 | |
| MW | 15 | 85 | ||
| 1i | Classical | 180 | 93 | 147–149 °C |
| -CH2COOEt | US | 60 | 81 | |
| MW | 10 | 94 | ||
| 1j | Classical | 300 | 81 | 108–110 °C |
| -(CH2)3COOH | US | 120 | 71 | |
| MW | 30 | 85 | ||
| 1k | Classical | 120 | 86 | 121–123 °C |
| -CH2C6H5 | US | 60 | 74 | |
| MW | 30 | 85 | ||
| 1l | Classical | 350 | 76 | 116–118 °C |
| -CH2CH2C6H5 | US | 150 | 69 | |
| MW | 50 | 85 | ||
| 1m | Classical | 350 | 78 | 104–106 °C |
| -(CH2)3C6H5 | US | 150 | 72 | |
| MW | 50 | 86 |
| No. | Code | The Degree of Inhibition of the Activity of α-Glucosidase, % |
|---|---|---|
| 1 | 1·HCl | No inhibition |
| 2 | 1a | 13.4 ± 1.6 |
| 3 | 1b | 12.0 ± 1.6 |
| 4 | 1c | 28.9 ± 3.1 |
| 5 | 1d | 35.7 ± 4.4 |
| 6 | 1e | 49.0 ± 0.8 |
| 7 | 1f | 49.8 ± 1.1 |
| 8 | 1g | 38.9 ± 1.1 |
| 9 | 1h | 27.8 ± 2.9 |
| 10 | 1i | 50.2 ± 1.2 |
| 11 | 1j | No inhibition |
| 12 | 1k | 64.2 ± 1.3 |
| 13 | 1l | 20.7 ± 2.1 |
| 14 | 1m | 66.9 ± 2.3 |
| 15 | Acarbose | 51.8 ± 2.1 |
| Parameters | 1·HCl | 1f | 1b | 1k | Control | Placebo | Intact |
|---|---|---|---|---|---|---|---|
| WBC·109/L | 7.22 ± 2.21 ***** | 6.59 ± 1.31 | 11.25 ± 1.45 | 6.65 ± 1.12 | 7.28 ± 1.26 | 3.88 ± 0.92 **** | 10.74 ± 1.11 |
| NEU·109/L | 2.15 ± 1.15 | 3.15 ± 0.90 ****** | 3.44 ± 0.68 | 2.51 ± 0.60 | 2.17 ± 0.64 | 1.72 ± 0.18 | 0.99 ± 0.79 |
| LYM·109/L | 4.67 ± 1.07 | 2.92 ± 0.30 ******* | 6.91 ± 0.31 | 3.72 ± 1.1 ******** | 4.57 ± 0.19 | 1.57 ± 0.13 | 8.84 ± 1.51 |
| MON·109/L | 0.3 ± 0 | 0.19 ± 0 | 0.34 ± 0 | 0.27 ± 0 | 0.31 ± 0 | 0.47 ± 0.24 | 0.29 ± 0 |
| EO·109/L | 0.03 ± 0 | 0.17 ± 0 | 0.41 ± 0 | 0.06 ± 0 | 0.2 ± 0 | 0.07 ± 0 | 0.38 ± 0 |
| BAS·109/L | 0.08 ± 0 | 0.17 ± 0 | 0.12 ± 0 | 0.03 ± 0 | 0.03 ± 0 | 0.03 ± 0 | 0.2 ± 0 |
| NEU% | 29.8 ± 0.24 | 47.6 ± 1.20 | 30.6 ± 0.15 | 37.5 ± 1.10 | 29.8 ± 0.65 | 44.4 ± 1.61 | 9.3 ± 1.05 |
| LYM% | 64.7 ± 1.75 | 44.0 ± 0.96 | 61.5 ± 1.5 | 56.4 ± 0.8 | 62.8 ± 1.75 | 40.6 ± 1.44 | 82.4 ± 3.16 |
| MON% | 4.05 ± 0.75 | 3.9 ± 0.94 | 3.1 ± 0.04 | 4.45 ± 0.41 | 4.2 ± 0.72 | 12.2 ± 0.92 | 2.7 ± 0.07 |
| EO% | 0.5 ± 0 | 2.4 ± 0.2 | 3.65 ± 0.15 | 1.05 ± 0 | 2.8 ± 0 | 2.0 ± 0 | 3.5 ± 0.85 |
| BAS% | 1.3 ± 0 | 2.1 ± 0.42 | 1.1 ± 0 | 0.65 ± 0 | 0.4 ± 0 | 0.8 ± 0 | 2.0 ± 0 |
| RBC 1012/L | 8.3 ± 0.74 | 7.66 ± 0.17 | 6.88 ± 0.44 | 6.43 ± 1.31 | 7.42 ± 1.12 | 4.09 ± 1.64 * | 7.09 ± 1.17 |
| HGB. g/L | 162.0 ± 13.2 | 146.4 ± 16.9 | 126.0 ± 11.6 | 125.5 ± 13.1 | 139.6 ± 2.2 | 71.0 ± 6.04 ** | 158.5 ± 16.5 |
| HCT% | 37.8 ± 1.82 | 33.1 ± 0.91 | 28.45 ± 1.15 | 28.1 ± 1.01 | 30.2 ± 2.34 | 11.0 ± 0.31 *** | 36.95 ± 3.21 |
| MCV | 45.5 ± 1.15 | 43.3 ± 2.32 | 41.3 ± 1.54 | 43.7 ± 1.71 | 40.8 ± 1.02 | 26.9 ± 1.62 | 43.5 ± 2.31 |
| MCH | 19.6 ± 0.6 | 19.1 ± 1.51 | 18.25 ± 0.95 | 19.65 ± 1.42 | 18.7 ± 1.03 | 17.4 ± 0.02 | 19.45 ± 1.65 |
| MCHC. g/L | 430.2 ± 16.4 | 440.5 ± 25.6 | 442.5 ± 17.9 | 448.5 ± 13.1 | 459 ± 22.5 | 647 ± 28.8 | 446.5 ± 16.5 |
| RDWsd | 19.1 ± 4.1 | 21.35 ± 1.65 ********* | 15.2 ± 1.02 | 16.6 ± 1.1 | 10.1 ± 0 | 11.5 ± 0.05 | 19.8 ± 4.65 |
| RDWcv | 18.8 ± 0.8 | 21.9 ± 1.11 ********* | 19.75 ± 0.35 | 18.4 ± 1.04 | 17.0 ± 1.05 | 31.2 ± 0.31 | 20.95 ± 2.05 |
| PLT·109/L | 412.2 ± 24.4 ********** | 524.5 ± 18.9 | 554.5 ± 19.4 | 290.5 ± 21.4 | 340.2 ± 26 | 381 ± 19.6 ** | 561.2 ± 12.2 |
| MPV | 4.2 ± 0 | 3.8 ± 0.2 | 3.9 ± 0.4 | 3.3 ± 0 | 4.1 ± 0 | 3.5 ± 0 | 3.9 ± 0.3 |
| Compound | C (mol/L × 10+5) | λmax (nm) | Amax | ε (L·mol−1·cm−1 × 10−5) |
|---|---|---|---|---|
| 1a | 0.5106 | 194, 223 | 2.48, 0.95 | 4.9613, 1.8952 |
| 1b | 0.5106 | 192, 223 | 1.88, 0.63 | 3.7602, 1.2547 |
| 1c | 0.5106 | 192, 223 | 0.68, 0.21 | 1.3563, 0.4176 |
| 1d | 0.5106 | 192, 223 | 1.39, 0.46 | 2.7759, 0.9229 |
| 1e | 0.5106 | 194, 223 | 2.52, 0.97 | 5.0322, 1.9395 |
| 1f | 0.5106 | 192, 223 | 0.71, 0.24 | 1.4108, 0.4867 |
| 1g | 0.5106 | 192, 223 | 1.87, 0.65 | 3.7328, 1.2922 |
| 1h | 0.5106 | 192, 223 | 1.17, 0.48 | 2.3389, 0.9636 |
| 1i | 0.5106 | 194, 223 | 2.06, 0.82 | 4.1296, 1.6392 |
| 1j | 0.5106 | 192, 223 | 0.70, 0.21 | 1.3974, 0.4279 |
| 1k | 1.105 | 203, 257 | 2.92, 0.14 | 2.9165, 0.1374 |
| 1l | 1.105 | 192, 223 | 1.99, 0.61 | 1.9926, 0.6054 |
| 1m | 1.105 | 190, 223 | 1.18, 0.25 | 1.1791, 0.2485 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dauletbakov, A.; Belyankova, Y.; Assylbekova, S.; Zolotareva, D.; Bayazit, S.; Baktybayeva, L.; Kemelbekov, U.; Yu, V.; Ibragimova, N.; Zazybin, A. Ultrasound and Microwave-Assisted Synthesis and Antidiabetic and Hematopoietic Activity of Diphenhydramine Derivatives. Molecules 2025, 30, 2967. https://doi.org/10.3390/molecules30142967
Dauletbakov A, Belyankova Y, Assylbekova S, Zolotareva D, Bayazit S, Baktybayeva L, Kemelbekov U, Yu V, Ibragimova N, Zazybin A. Ultrasound and Microwave-Assisted Synthesis and Antidiabetic and Hematopoietic Activity of Diphenhydramine Derivatives. Molecules. 2025; 30(14):2967. https://doi.org/10.3390/molecules30142967
Chicago/Turabian StyleDauletbakov, Anuar, Yelizaveta Belyankova, Saniya Assylbekova, Darya Zolotareva, Sarah Bayazit, Layilya Baktybayeva, Ulan Kemelbekov, Valentina Yu, Nailya Ibragimova, and Alexey Zazybin. 2025. "Ultrasound and Microwave-Assisted Synthesis and Antidiabetic and Hematopoietic Activity of Diphenhydramine Derivatives" Molecules 30, no. 14: 2967. https://doi.org/10.3390/molecules30142967
APA StyleDauletbakov, A., Belyankova, Y., Assylbekova, S., Zolotareva, D., Bayazit, S., Baktybayeva, L., Kemelbekov, U., Yu, V., Ibragimova, N., & Zazybin, A. (2025). Ultrasound and Microwave-Assisted Synthesis and Antidiabetic and Hematopoietic Activity of Diphenhydramine Derivatives. Molecules, 30(14), 2967. https://doi.org/10.3390/molecules30142967