
Insights into Binding Mechanisms of Potential Inhibitors Targeting PCSK9 Protein via Molecular Dynamics Simulation and Free Energy Calculation
 ,
,  ,
,
Abstract

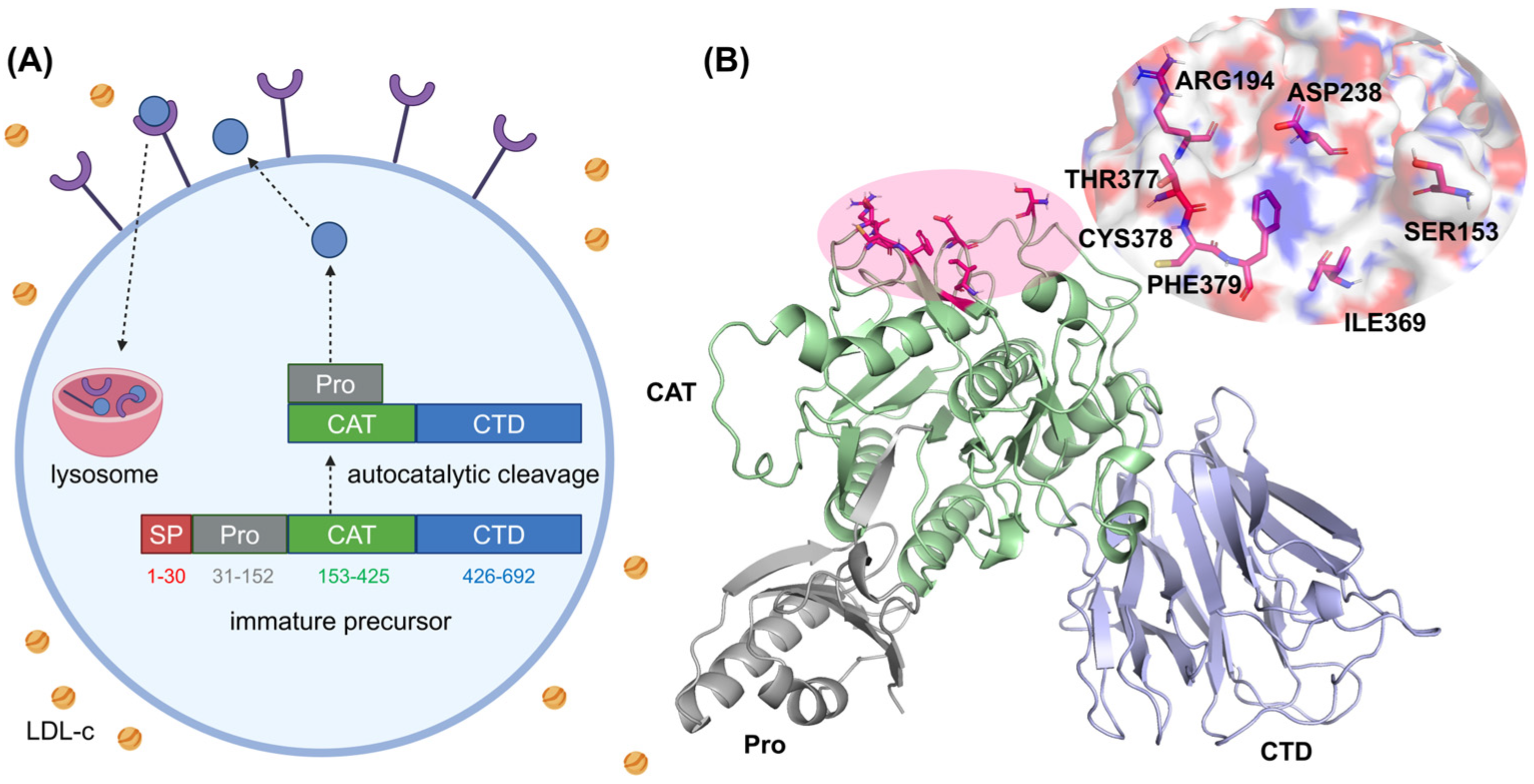
1. Introduction
2. Results and Discussion
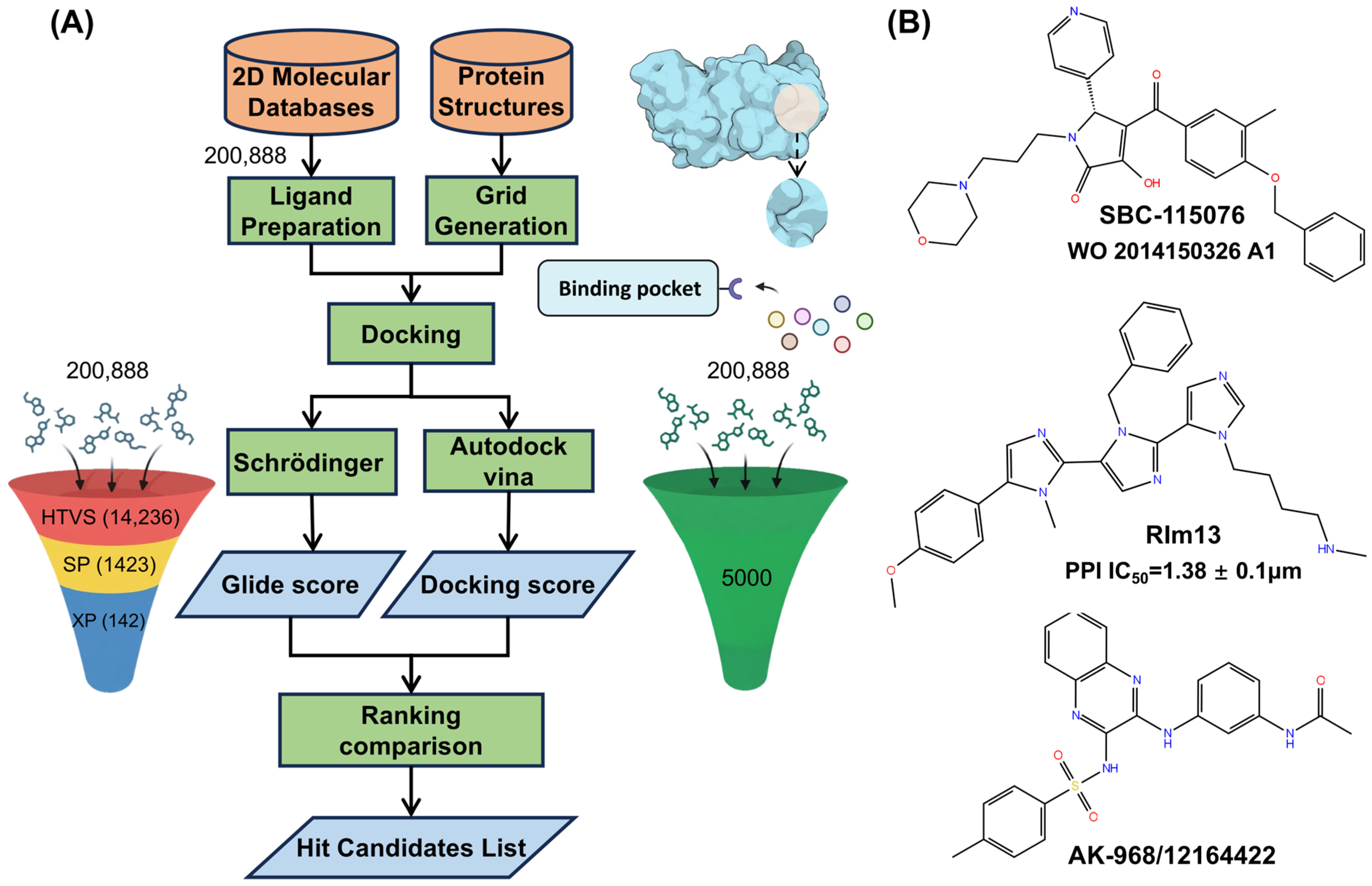
2.1. Virtual Screening
2.2. ADMET Prediction
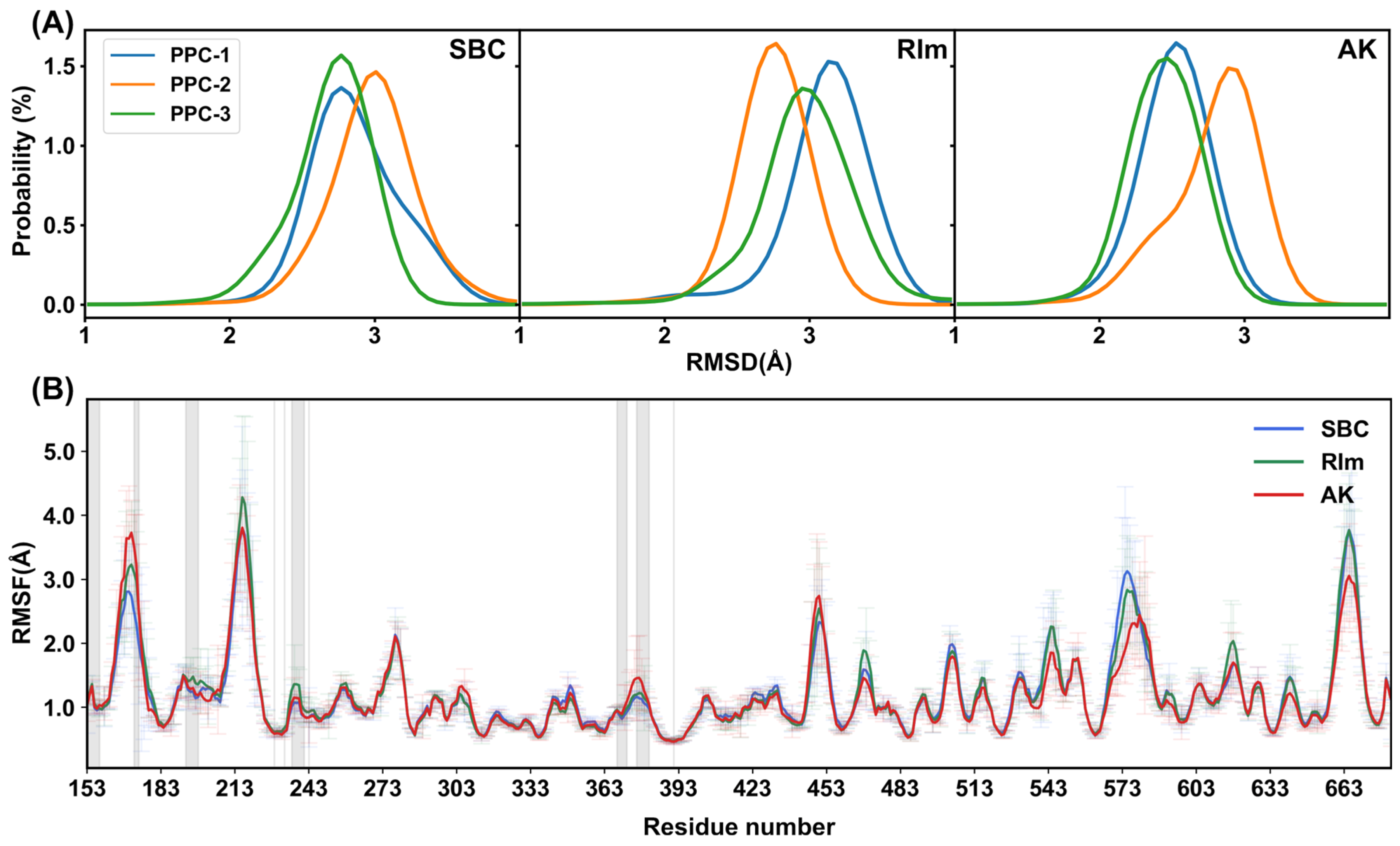
2.3. Structural Stability
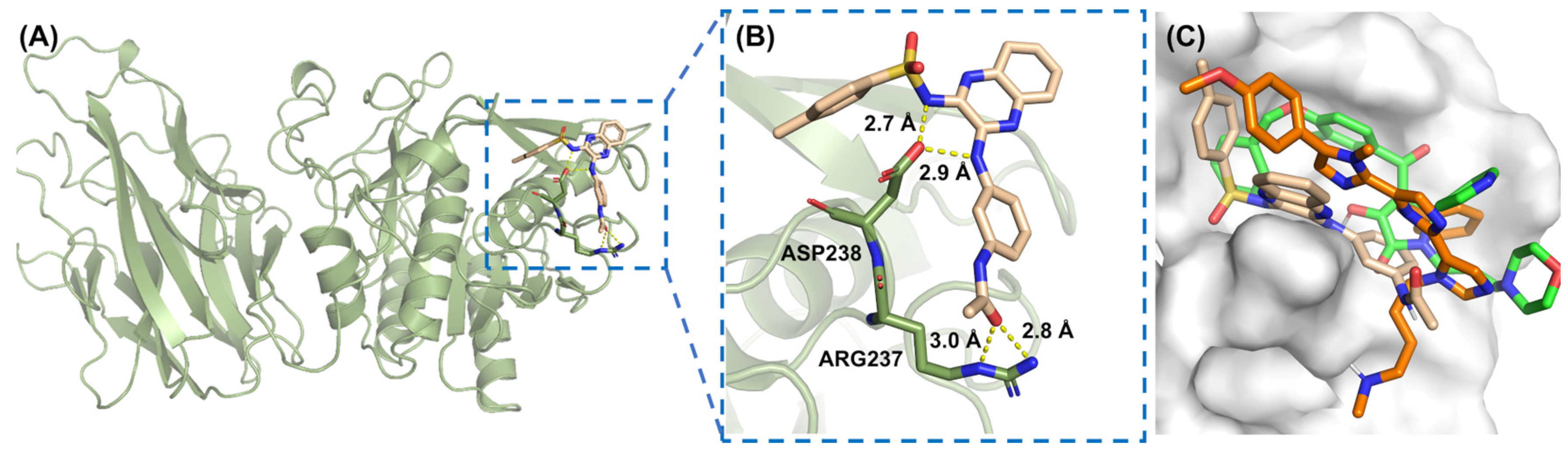
2.4. Interaction Patterns
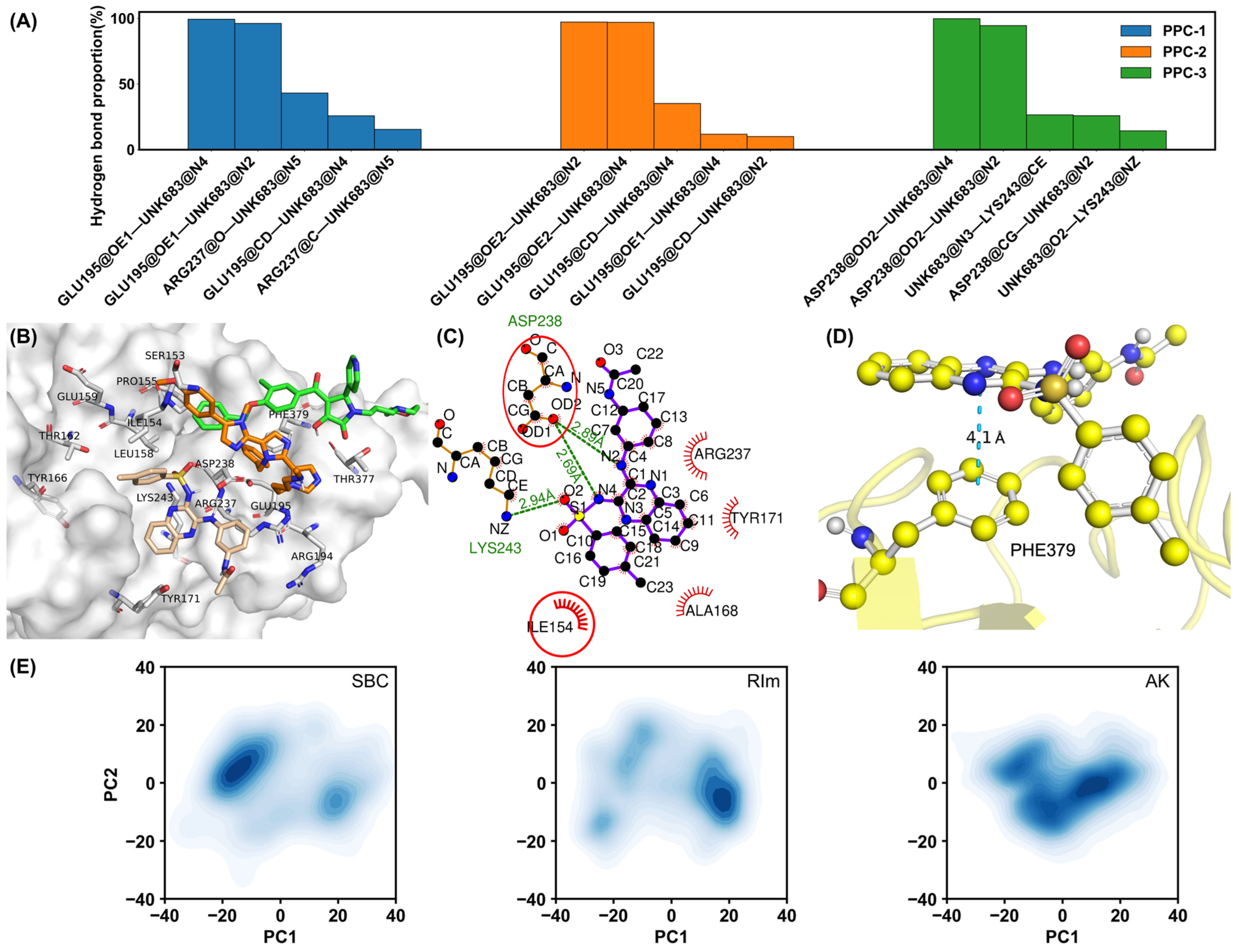
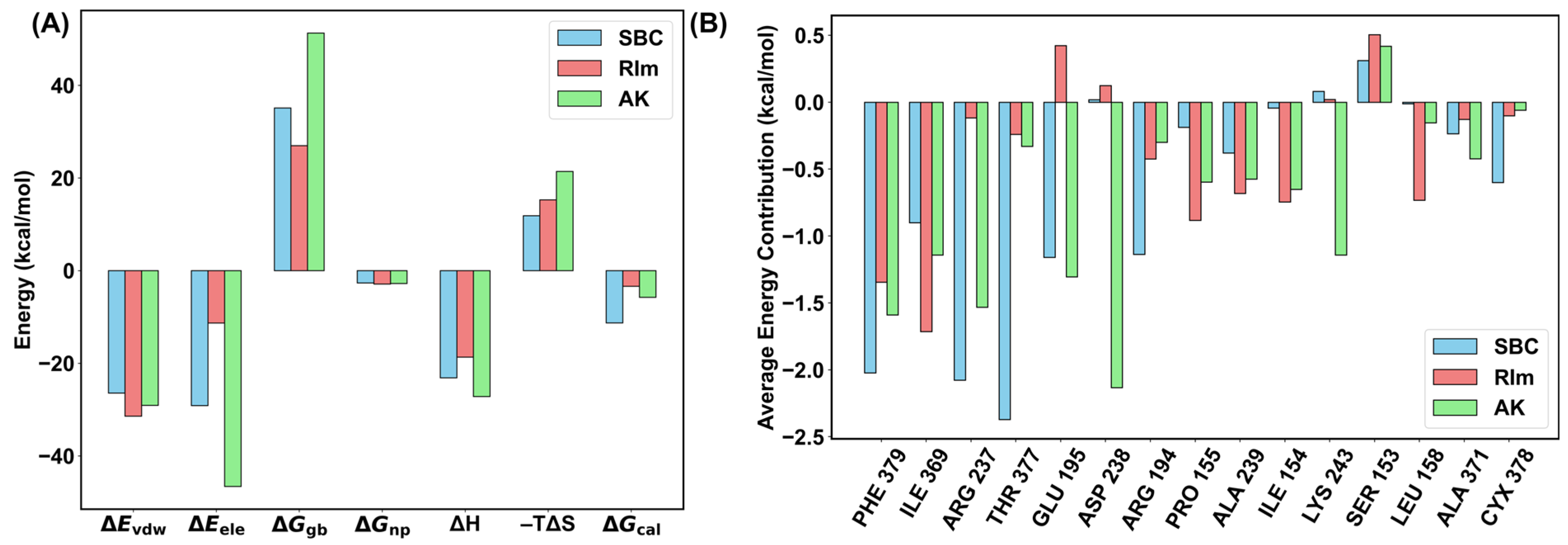
2.5. Binding Free Energy Calculations
3. Methods
3.1. Structures Preparation and Virtual Screening
3.2. Molecular Dynamics Simulation
3.3. Polarized Protein-Specific Charge (PPC)
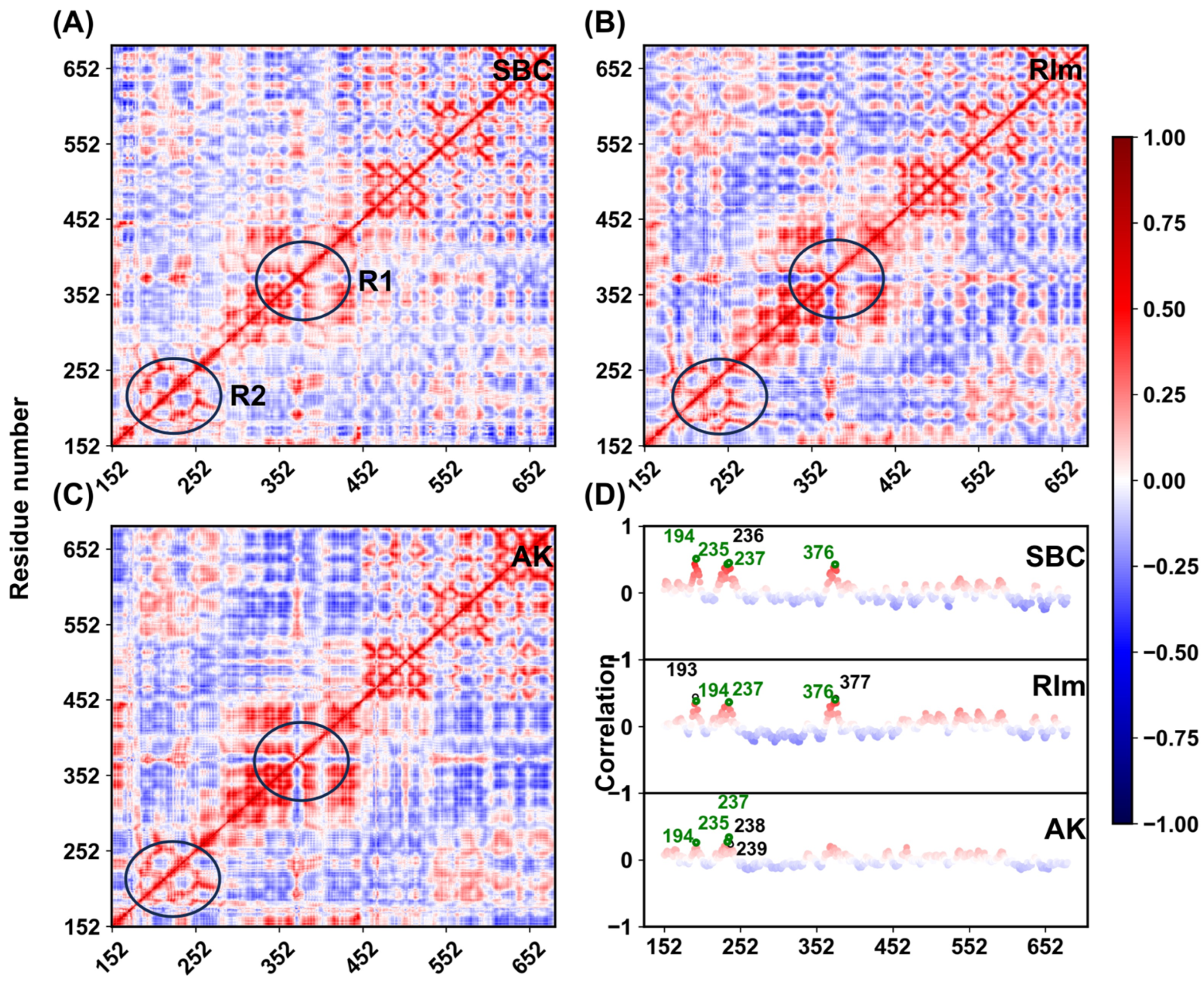
3.4. Dynamic Cross-Correlation Matrices (DCCM)
3.5. The Binding Free Energy Calculation
3.6. M/GBSA
3.7. The Interaction Entropy (IE) Method
3.8. The Alanine Scanning Interaction Entropy (ASIE) Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Schweizer, J.; Nakas, C.T.; Schütz, V.; Westphal, L.P.; Inauen, C.; Pokorny, T.; Luft, A.; Leichtle, A.; Arnold, M.; et al. Lipoprotein(a) Is Associated with Large Artery Atherosclerosis Stroke Aetiology and Stroke Recurrence among Patients below the Age of 60 Years: Results from the BIOSIGNAL Study. Eur. Heart J. 2021, 42, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.J.M.; Demkes, E.J.; Fiolet, A.T.L.; Dekker, M.; Bosch, L.; Van Hout, G.P.J.; Timmers, L.; De Kleijn, D.P.V. Immunomodulation of the NLRP3 Inflammasome in Atherosclerosis, Coronary Artery Disease, and Acute Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the Vulnerable Plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease. 1. Evidence from Genetic, Epidemiologic, and Clinical Studies. A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef] [PubMed]
- Dullaart, R.P.F. PCSK9 Inhibition to Reduce Cardiovascular Events. N. Engl. J. Med. 2017, 376, 1790–1791. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 Cause Autosomal Dominant Hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Piper, D.E.; Jackson, S.; Liu, Q.; Romanow, W.G.; Shetterly, S.; Thibault, S.T.; Shan, B.; Walker, N.P.C. The Crystal Structure of PCSK9: A Regulator of Plasma LDL-Cholesterol. Structure 2007, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Garvie, C.W.; Fraley, C.V.; Elowe, N.H.; Culyba, E.K.; Lemke, C.T.; Hubbard, B.K.; Kaushik, V.K.; Daniels, D.S. Point Mutations at the Catalytic Site of PCSK9 Inhibit Folding, Autoprocessing, and Interaction with the LDL Receptor. Protein Sci. 2016, 25, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Danley, D.E.; Geoghegan, K.F.; Griffor, M.C.; Hawkins, J.L.; Subashi, T.A.; Varghese, A.H.; Ammirati, M.J.; Culp, J.S.; Hoth, L.R.; et al. Structural and Biophysical Studies of PCSK9 and Its Mutants Linked to Familial Hypercholesterolemia. Nat. Struct. Mol. Biol. 2007, 14, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-W.; Garuti, R.; Tang, W.-J.; Cohen, J.C.; Hobbs, H.H. Structural Requirements for PCSK9-Mediated Degradation of the Low-Density Lipoprotein Receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 13045–13050. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Honarpour, N.; Blom, D.J.; Hovingh, G.K.; Xu, F.; Scott, R.; Wasserman, S.M.; Stein, E.A. Inhibition of PCSK9 with Evolocumab in Homozygous Familial Hypercholesterolaemia (TESLA Part B): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2015, 385, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to Watch in 2023. Mabs 2023, 15, 2153410. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Recent Developments in Structure-Based Drug Design. J. Mol. Med. 2000, 78, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Vyas, B.; Choudhary, S.; Singh, P.K.; Singh, A.; Singh, M.; Verma, H.; Singh, H.; Bahadur, R.; Singh, B.; Silakari, O. Molecular Dynamics/Quantum Mechanics Guided Designing of Natural Products Based Prodrugs of Epalrestat. J. Mol. Struct. 2018, 1171, 556–563. [Google Scholar] [CrossRef]
- Chen, J.; Duan, L.; Ji, C.; Zhang, J.Z.H. Computational Study of PCSK9-EGFA Complex with Effective Polarizable Bond Force Field. Front. Mol. Biosci. 2018, 4, 101. [Google Scholar] [CrossRef] [PubMed]
- Taechalertpaisarn, J.; Zhao, B.; Liang, X.; Burgess, K. Small Molecule Inhibitors of the PCSK9·LDLR Interaction. J. Am. Chem. Soc. 2018, 140, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, J.; Liu, S.; Zhou, X.; Dai, L.; Chen, C.; Xu, Q.; Wen, X.; Cheng, K.; Sun, H.; et al. Discovery of Novel Small Molecule Inhibitors Disrupting the PCSK9-LDLR Interaction. J. Chem. Inf. Model. 2021, 61, 5269–5279. [Google Scholar] [CrossRef] [PubMed]
- Lammi, C.; Sgrignani, J.; Arnoldi, A.; Lesma, G.; Spatti, C.; Silvani, A.; Grazioso, G. Computationally Driven Structure Optimization, Synthesis, and Biological Evaluation of Imidazole-Based Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) Inhibitors. J. Med. Chem. 2019, 62, 6163–6174. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Luo, S.; Zhu, Z.; Xu, J. Small Molecules as Inhibitors of PCSK9: Current Status and Future Challenges. Eur. J. Med. Chem. 2019, 162, 212–233. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.-H.; Christoffer, C.W.; Kihara, D. In Silico Structure-Based Approaches to Discover Protein-Protein Interaction-Targeting Drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of Folded Proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Bioinf. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Lagace, T.A.; McNutt, M.C.; Horton, J.D.; Deisenhofer, J. Molecular Basis for LDL Receptor Recognition by PCSK9. Proc. Natl. Acad. Sci. USA 2008, 105, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Mei, Y. Some Practical Approaches to Treating Electrostatic Polarization of Proteins. Acc. Chem. Res. 2014, 47, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Gamian, A.; Sroka, Z. A Statistically Supported Antioxidant Activity DFT Benchmark—The Effects of Hartree-Fock Exchange and Basis Set Selection on Accuracy and Resources Uptake. Molecules 2021, 26, 5058. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin Stabilization of Molecular Dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An Analysis of the Accuracy of Langevin and Molecular Dynamics Algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Ji, C.; Mei, Y.; Zhang, J.Z.H. Developing Polarized Protein-Specific Charges for Protein Dynamics: MD Free Energy Calculation of pKa Shifts for Asp26/Asp20 in Thioredoxin. Biophys. J. 2008, 95, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Zhang, J.Z.H. Molecular Fractionation with Conjugate Caps for Full Quantum Mechanical Calculation of Protein-Molecule Interaction Energy. J. Chem. Phys. 2003, 119, 3599–3605. [Google Scholar] [CrossRef]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Honig, B.; Ringnalda, M.; Goddard, W.A.I. Accurate First Principles Calculation of Molecular Charge Distributions and Solvation Energies from Ab Initio Quantum Mechanics and Continuum Dielectric Theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Raghavan, S.S.; Iqbal, S.; Ayyadurai, N.; Gunasekaran, K. Insights in the Structural Understanding of Amyloidogenicity and Mutation-Led Conformational Dynamics of Amyloid Beta (Aβ) through Molecular Dynamics Simulations and Principal Component Analysis. J. Biomol. Struct. Dyn. 2021, 40, 5577–5587. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 4. Accuracies of MM/PBSA and MM/GBSA Methodologies Evaluated by Various Simulation Protocols Using PDBbind Data Set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Liu, X.; Zhang, J.Z.H. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F.; Olson, A.J.; Spehner, J.-C. Reduced Surface: An Efficient Way to Compute Molecular Surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Xu, X.; Luo, S.; Zhao, X.; Tang, B.; Zhang, E.; Liu, J.; Duan, L. Computational Analysis of PD-L1 Dimerization Mechanism Induced by Small Molecules and Potential Dynamical Properties. Int. J. Biol. Macromol. 2024, 265, 130921. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Dong, S.; Huang, K.; Cong, Y.; Luo, S.; Zhang, J.Z.H. Computational Analysis of Binding Free Energies, Hotspots and the Binding Mechanism of Bcl-xL/Bcl-2 Binding to Bad/Bax. Phys. Chem. Chem. Phys. 2021, 23, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yang, M.; Ji, C.G.; Zhang, J.Z.H. Interaction Entropy for Computational Alanine Scanning. J. Chem. Inf. Model. 2017, 57, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lu, X.; Duan, L.L.; Zhang, J.Z.H.; Mei, Y. Polarization of Intraprotein Hydrogen Bond Is Critical to Thermal Stability of Short Helix. J. Phys. Chem. B 2012, 116, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.L.; Gao, Y.; Mei, Y.; Zhang, Q.G.; Tang, B.; Zhang, J.Z.H. Folding of a Helix Is Critically Stabilized by Polarization of Backbone Hydrogen Bonds: Study in Explicit Water. J. Phys. Chem. B 2012, 116, 3430–3435. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Guo, M.; Mei, Y.; Zhang, J.Z.H. Protein–Water Hydrogen Bonds Are Stabilized by Electrostatic Polarization. Mol. Phys. 2012, 110, 595–604. [Google Scholar] [CrossRef]
- Xia, X.; Peng, Z.; Gu, H.; Wang, M.; Wang, G.; Zhang, D. Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8, 764038. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | MW (g/mol) | AMES Toxicity | Carcinogenicity | Acute Oral Toxicity | Rat Acute Toxicity (LD50, mol/kg) | Rotatable Bonds | H-Bond Acceptors | H-Bond Donors | ESOL Log S | TPSA | W Log P | Log Kp (cm/s) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SBC | 527.61 | None | None | III | 2.70 | 10 | 8 | 1 | −5.17 | 92.2 | 3.03 | −6.84 |
| RIm | 495.62 | None | None | III | 2.76 | 11 | 5 | 1 | −4.64 | 74.72 | 4.87 | −7.16 |
| AK | 447.52 | None | None | III | 2.23 | 7 | 8 | 3 | −5.29 | 121.46 | 3.31 | −6.56 |
| System | Force Field Replica | ΔEvdw | ΔEele | ΔGgb | ΔGnp | ΔH | −TΔS | ΔGbind | |
|---|---|---|---|---|---|---|---|---|---|
| SBC | AMBER | No. 1 | −23.21 ± 0.16 | −20.72 ± 0.25 | 26.85 ± 0.16 | −2.36 ± 0.01 | −19.43 ± 0.11 | 15.97 ± 0.55 | −3.46 ± 0.33 |
| No. 2 | −23.24 ± 0.16 | −19.95 ± 0.24 | 26.09 ± 0.13 | −2.29 ± 0.01 | −19.38 ± 0.11 | 16.09 ± 0.38 | −3.29 ± 0.24 | ||
| No. 3 | −28.08 ± 0.15 | −20.77 ± 0.59 | 30.66 ± 0.42 | −2.70 ± 0.01 | −20.89 ± 0.17 | 25.95 ± 0.56 | 5.06 ± 0.37 | ||
| Average | −24.84 ± 0.16 | −20.48 ± 0.36 | 27.87 ± 0.24 | −2.45 ± 0.01 | −19.90 ± 0.13 | 19.34 ± 0.50 | −0.56 ± 0.31 | ||
| PPC | No. 1 | −36.22 ± 0.20 | −27.99 ± 0.17 | 40.06 ± 0.14 | −3.29 ± 0.02 | −27.44 ± 0.14 | 8.63 ± 0.29 | −18.81 ± 0.21 | |
| No. 2 | −20.51 ± 0.14 | −23.45 ± 0.16 | 27.09 ± 0.12 | −2.25 ± 0.01 | −19.11 ± 0.10 | 14.57 ± 0.59 | −4.54 ± 0.34 | ||
| No. 3 | −22.60 ± 0.15 | −35.96 ± 0.20 | 38.14 ± 0.14 | −2.48 ± 0.01 | −22.89 ± 0.12 | 12.39 ± 0.57 | −10.50 ± 0.34 | ||
| Average | −26.44 ± 0.16 | −29.13 ± 0.18 | 35.10 ± 0.13 | −2.67 ± 0.01 | −23.15 ± 0.12 | 11.86 ± 0.48 | −11.28 ± 0.30 | ||
| RIm | AMBER | No. 1 | −28.83 ± 0.17 | −26.81 ± 0.49 | 59.51 ± 0.56 | −2.37 ± 0.01 | 1.50 ± 0.15 | 42.55 ± 0.59 | 44.05 ± 0.37 |
| No. 2 | −26.08 ± 0.18 | −15.52 ± 0.59 | 43.28 ± 0.67 | −2.08 ± 0.01 | −0.40 ± 0.16 | 50.48 ± 0.54 | 50.08 ± 0.35 | ||
| No. 3 | −29.28 ± 0.23 | −16.61 ± 0.56 | 50.96 ± 0.53 | −2.46 ± 0.02 | 2.62 ± 0.26 | 48.65 ± 0.48 | 51.27 ± 0.37 | ||
| Average | −28.06 ± 0.19 | −19.64 ± 0.54 | 51.25 ± 0.59 | −2.30 ± 0.01 | 1.24 ± 0.19 | 47.23 ± 0.54 | 48.47 ± 0.36 | ||
| PPC | No. 1 | −26.64 ± 0.15 | −11.02 ± 0.14 | 24.11 ± 0.16 | −2.59 ± 0.02 | −16.13 ± 0.11 | 10.51 ± 0.57 | −5.62 ± 0.34 | |
| No. 2 | −34.39 ± 0.17 | −9.08 ± 0.21 | 29.81 ± 0.22 | −3.16 ± 0.01 | −16.83 ± 0.12 | 13.48 ± 0.51 | −3.35 ± 0.32 | ||
| No. 3 | −33.26 ± 0.25 | −13.86 ± 0.19 | 27.01 ± 0.20 | −2.95 ± 0.02 | −23.07 ± 0.23 | 21.85 ± 0.50 | −1.22 ± 0.36 | ||
| Average | −31.43 ± 0.19 | −11.32 ± 0.18 | 26.98 ± 0.19 | −2.90 ± 0.02 | −18.67 ± 0.15 | 15.28 ± 0.53 | −3.39 ± 0.34 | ||
| AK | AMBER | No. 1 | −26.00 ± 0.12 | −28.67 ± 0.38 | 42.42 ± 0.34 | −2.39 ± 0.01 | −14.65 ± 0.11 | 24.79 ± 0.48 | 10.14 ± 0.30 |
| No. 2 | −35.80 ± 0.15 | −33.01 ± 0.43 | 48.15 ± 0.40 | −3.31 ± 0.01 | −23.96 ± 0.17 | 28.41 ± 0.59 | 4.45 ± 0.38 | ||
| No. 3 | −24.58 ± 0.17 | −55.48 ± 0.56 | 52.01 ± 0.48 | −2.43 ± 0.01 | −30.48 ± 0.21 | 37.82 ± 0.59 | 7.34 ± 0.40 | ||
| Average | −28.80 ± 0.15 | −39.05 ± 0.46 | 47.53 ± 0.41 | −2.71 ± 0.01 | −23.03 ± 0.16 | 30.34 ± 0.55 | 7.31 ± 0.36 | ||
| PPC | No. 1 | −26.72 ± 0.15 | −42.13 ± 0.48 | 45.72 ± 0.43 | −2.29 ± 0.01 | −25.43 ± 0.15 | 23.43 ± 0.42 | −1.99 ± 0.29 | |
| No. 2 | −26.69 ± 0.17 | −56.91 ± 0.37 | 58.96 ± 0.31 | −2.79 ± 0.01 | −27.43 ± 0.17 | 23.71 ± 0.35 | −3.72 ± 0.26 | ||
| No. 3 | −33.89 ± 0.16 | −40.74 ± 0.35 | 49.17 ± 0.31 | −3.23 ± 0.01 | −28.69 ± 0.15 | 17.08 ± 0.42 | −11.61 ± 0.29 | ||
| Average | −29.10 ± 0.16 | −46.59 ± 0.40 | 51.28 ± 0.35 | −2.77 ± 0.01 | −27.18 ± 0.16 | 21.41 ± 0.40 | −5.77 ± 0.28 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Zhu, X.; Fang, M.; Qi, F.; Yin, Z.; Zhang, J.Z.H.; Luo, S.; Zhu, T.; Gao, Y. Insights into Binding Mechanisms of Potential Inhibitors Targeting PCSK9 Protein via Molecular Dynamics Simulation and Free Energy Calculation. Molecules 2025, 30, 2962. https://doi.org/10.3390/molecules30142962
Wu X, Zhu X, Fang M, Qi F, Yin Z, Zhang JZH, Luo S, Zhu T, Gao Y. Insights into Binding Mechanisms of Potential Inhibitors Targeting PCSK9 Protein via Molecular Dynamics Simulation and Free Energy Calculation. Molecules. 2025; 30(14):2962. https://doi.org/10.3390/molecules30142962
Chicago/Turabian StyleWu, Xingyu, Xi Zhu, Min Fang, Fenghua Qi, Zhixiang Yin, John Z.H. Zhang, Shihua Luo, Tong Zhu, and Ya Gao. 2025. "Insights into Binding Mechanisms of Potential Inhibitors Targeting PCSK9 Protein via Molecular Dynamics Simulation and Free Energy Calculation" Molecules 30, no. 14: 2962. https://doi.org/10.3390/molecules30142962
APA StyleWu, X., Zhu, X., Fang, M., Qi, F., Yin, Z., Zhang, J. Z. H., Luo, S., Zhu, T., & Gao, Y. (2025). Insights into Binding Mechanisms of Potential Inhibitors Targeting PCSK9 Protein via Molecular Dynamics Simulation and Free Energy Calculation. Molecules, 30(14), 2962. https://doi.org/10.3390/molecules30142962