

Optimization of Kerosene-like Fuels Produced via Catalytic Pyrolysis of Packaging Plastic Waste via Central Composite Design and Response Surface Methodology: Performance of Iron-Doped Dolomite and Activated Carbon


Abstract
1. Introduction
2. Results and Discussions
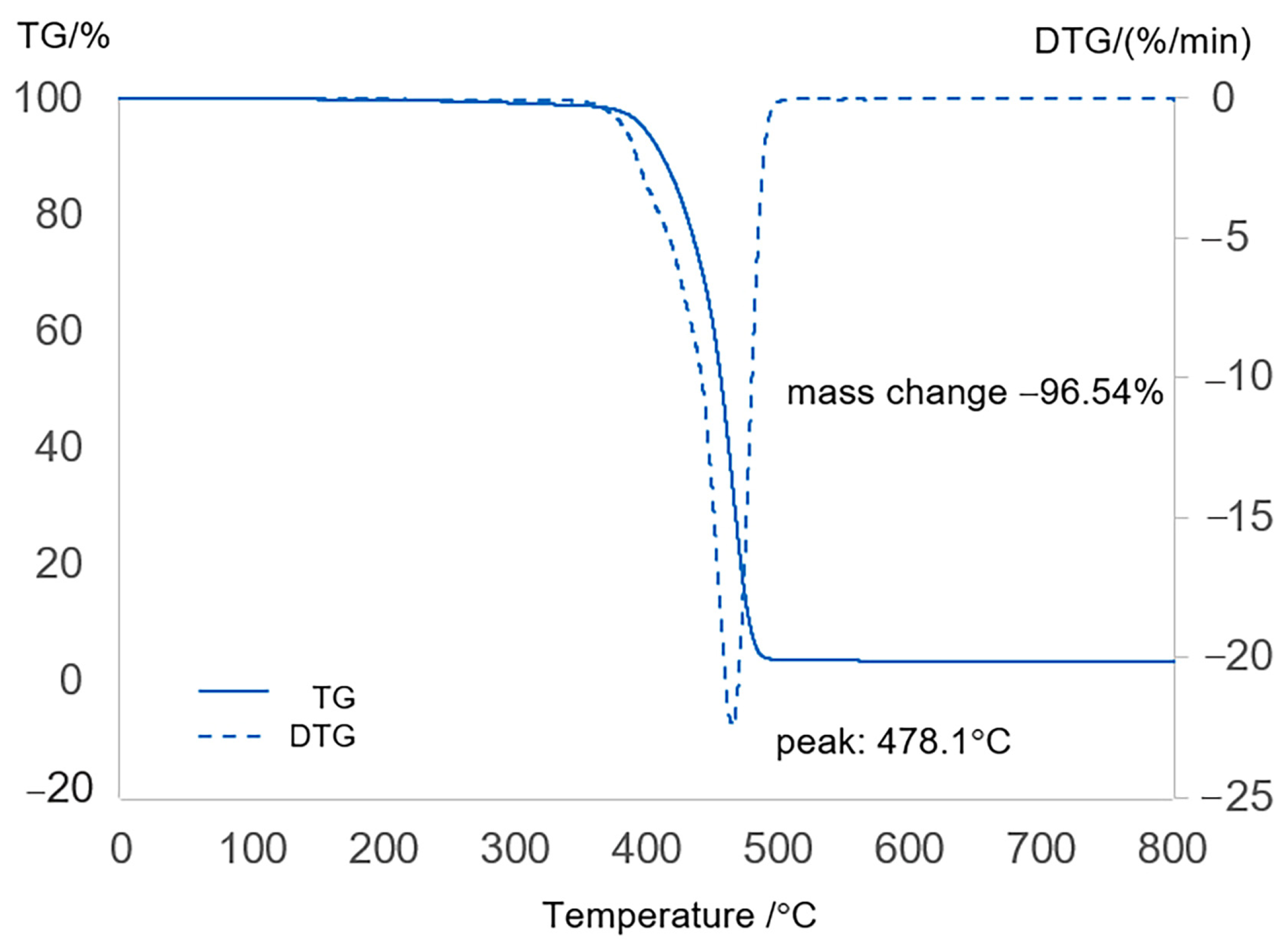
2.1. Characterization of Feedstock
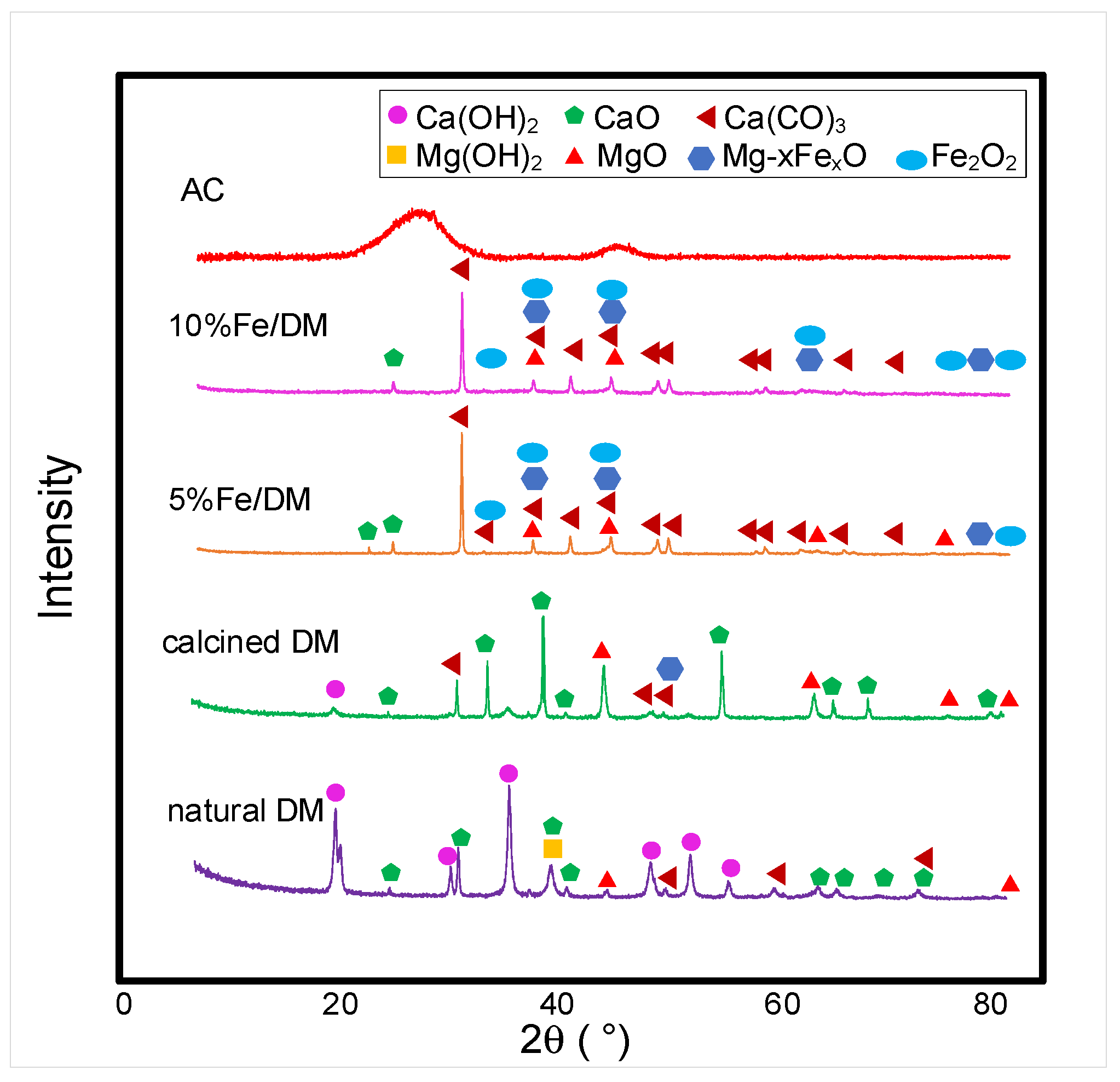
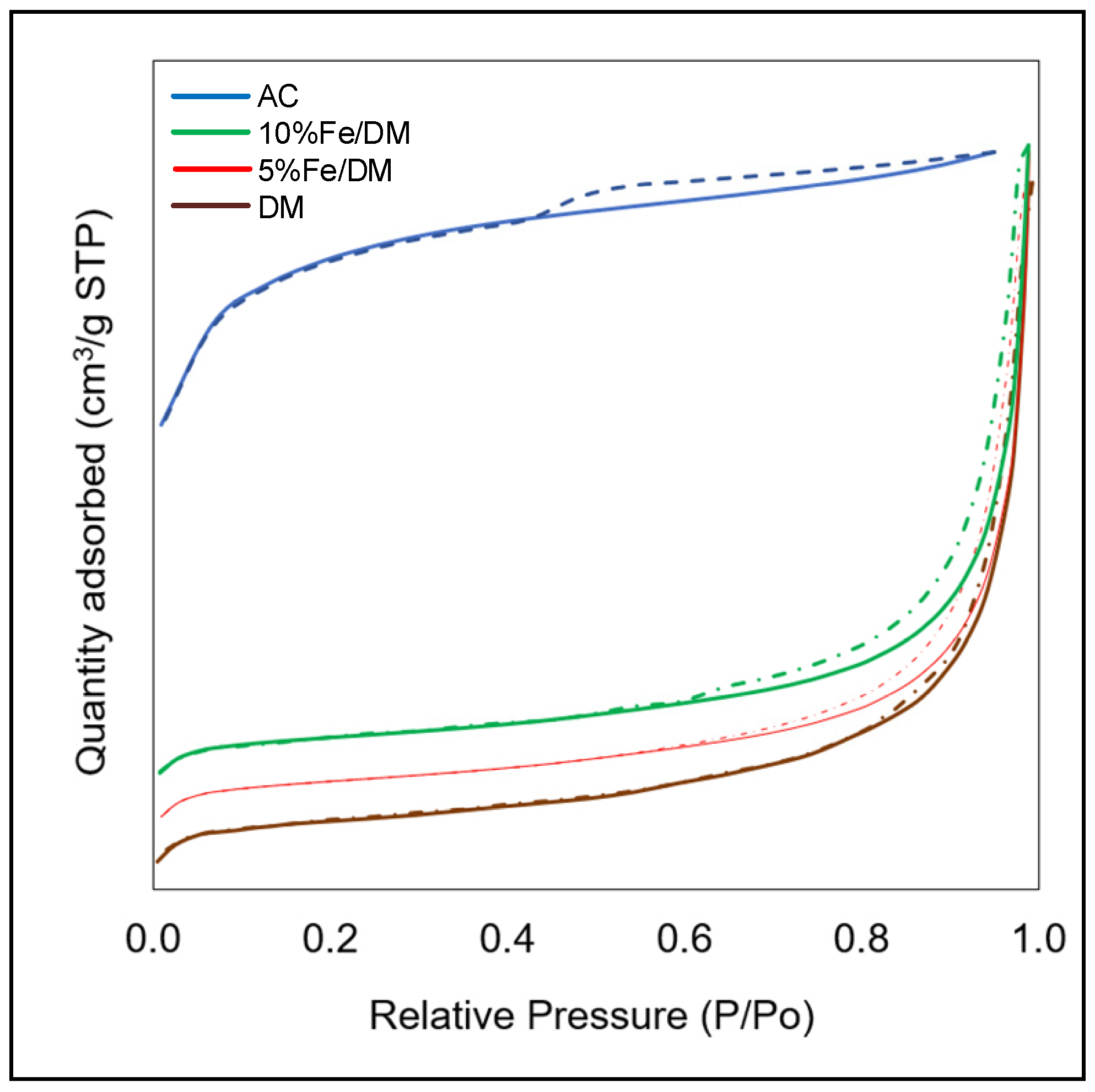
2.2. Characterization of the Catalysts
2.3. Univariate Experiment
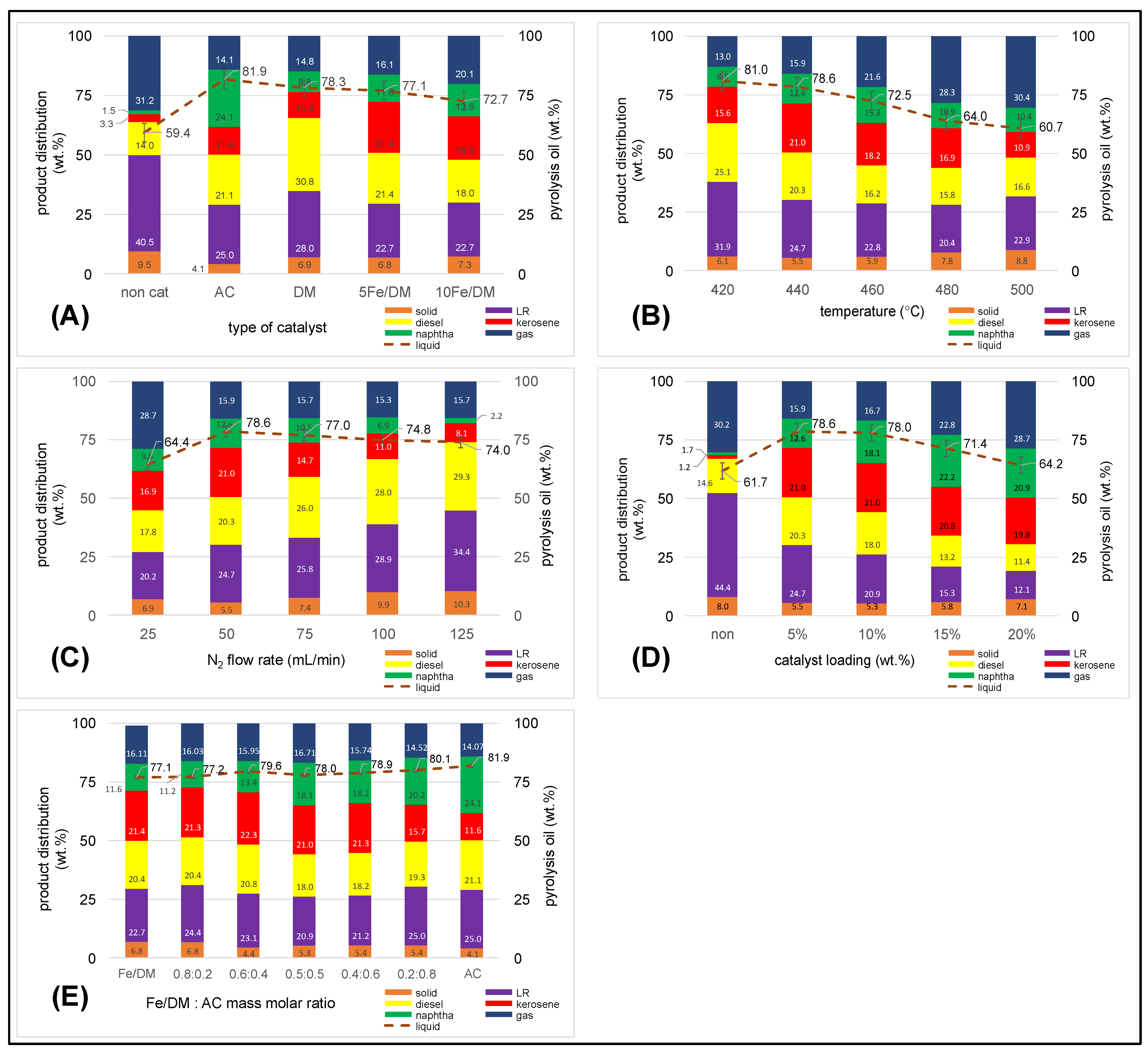
2.3.1. Influence of the Catalyst Active Site
2.3.2. Influence of Temperature
2.3.3. Influence of the Inert N2 Flow Rate
2.3.4. Influence of Catalyst Loading
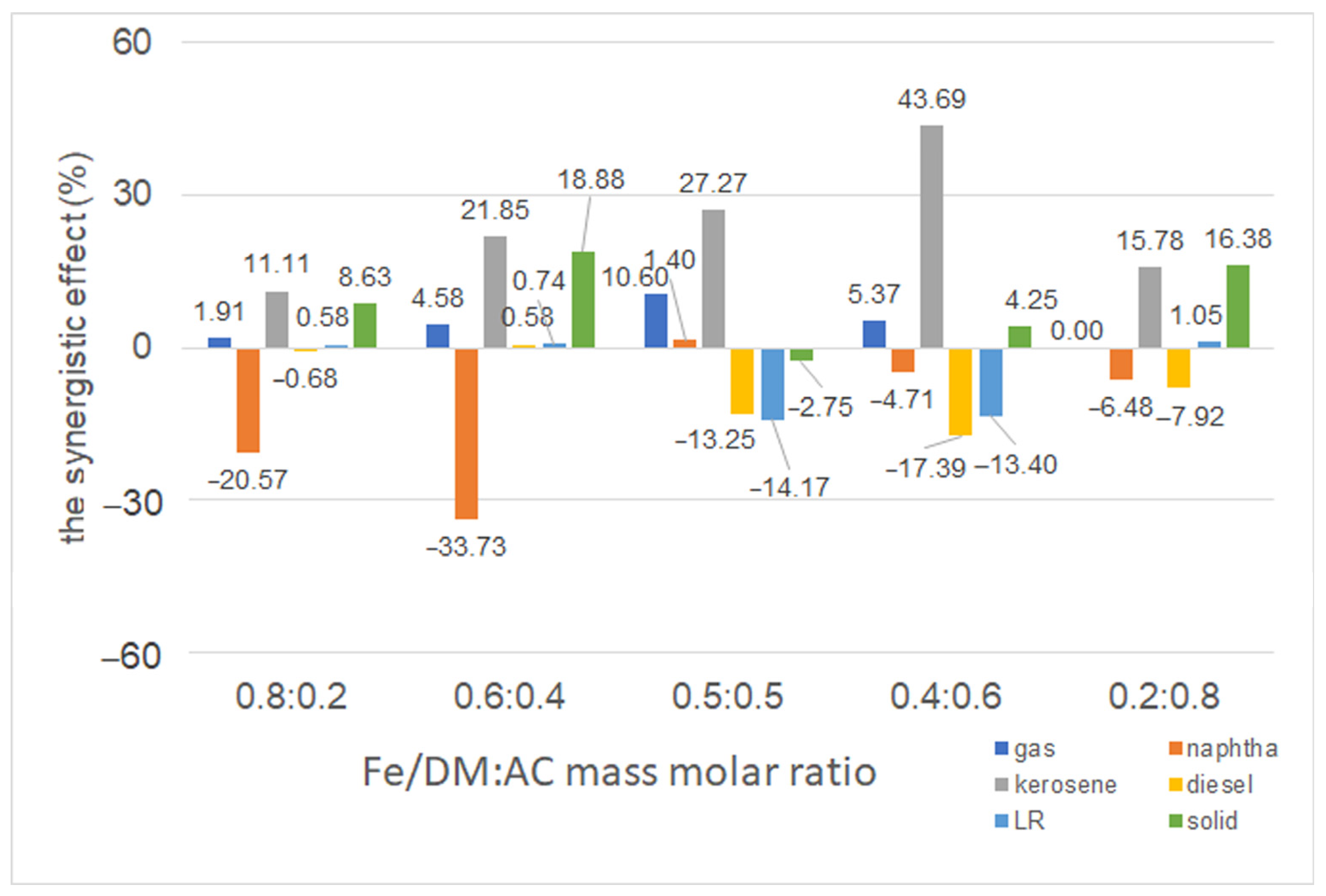
2.3.5. Influence of a Dual Catalyst at Different Mass Ratios of Fe/DM and AC Catalysts
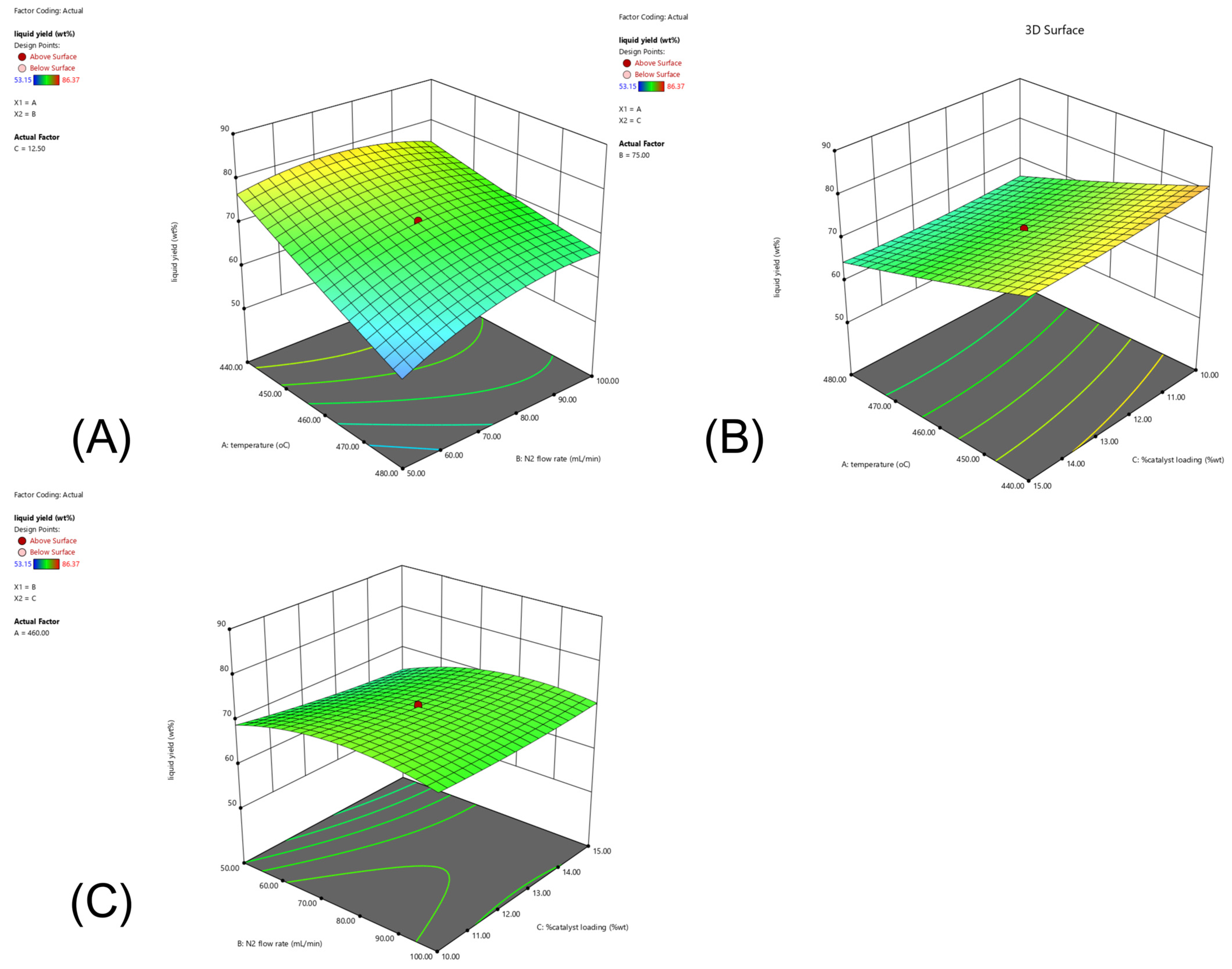
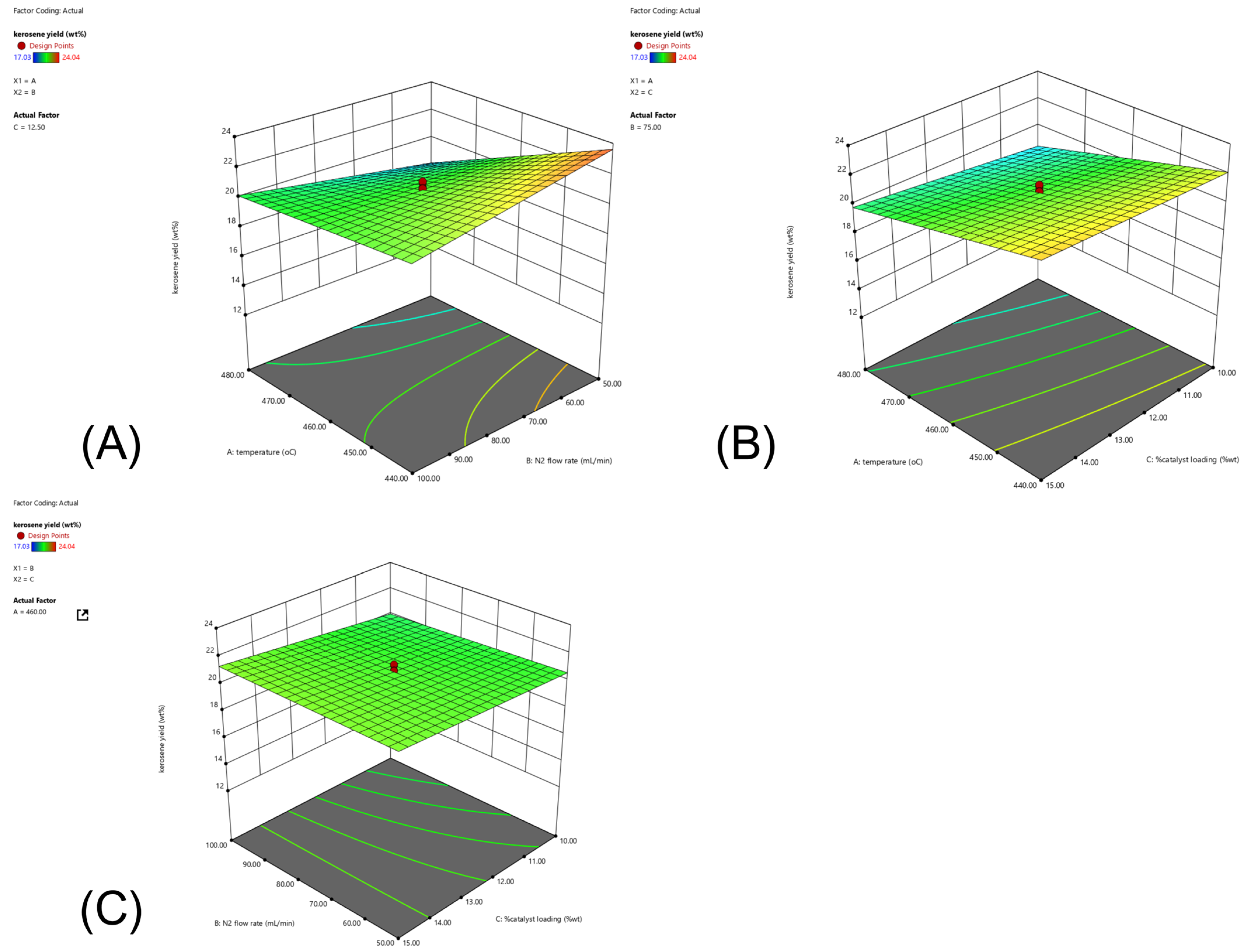
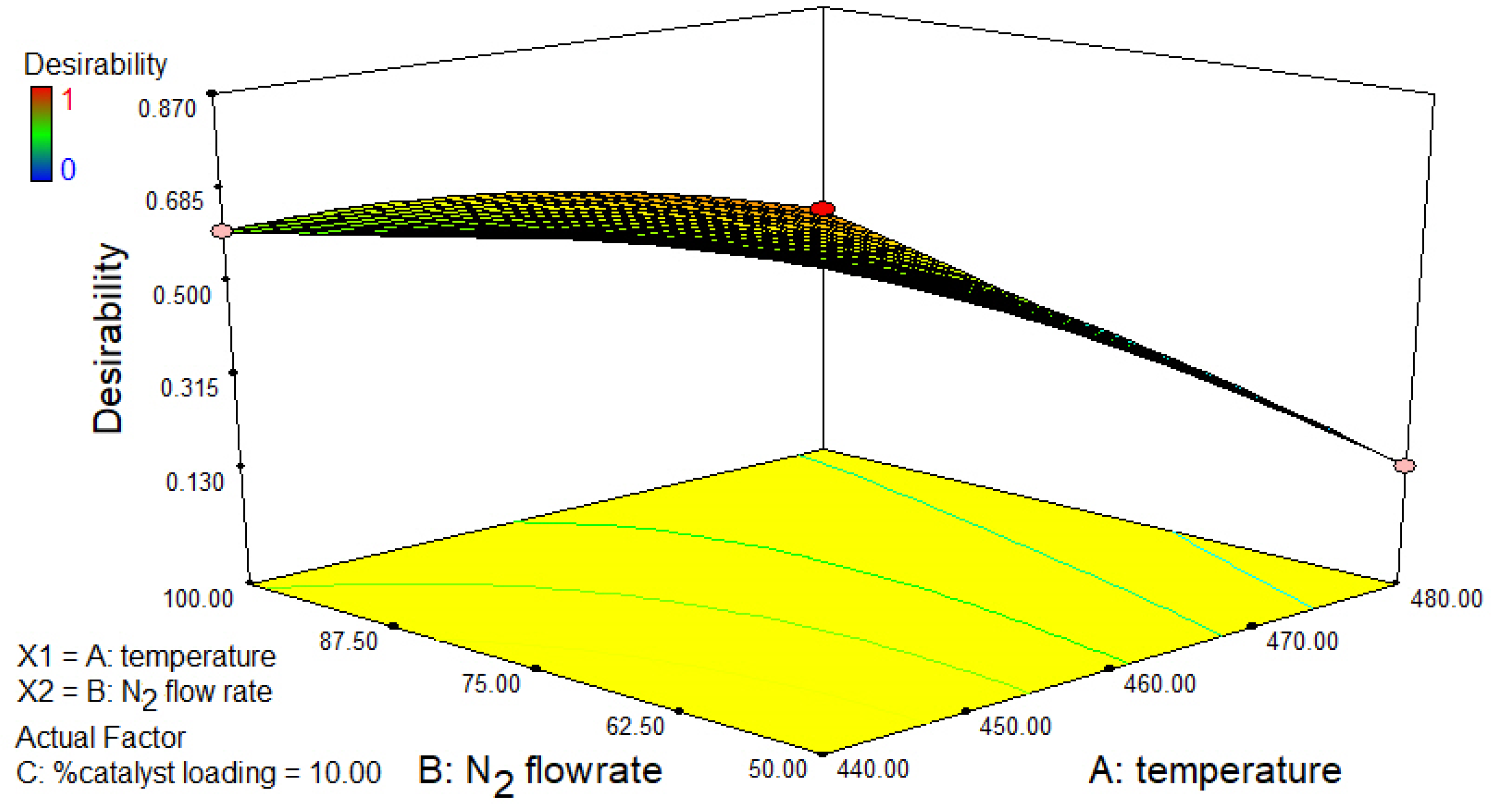
2.4. CCD and RSM Optimization of the Experimental Results
0.451AC + 1.061BC + 0.318A2 − 3.255B2 + 0.602C2
8.275(% catalyst loading) + 0.006(temperature ×
N2 flow rate) + 0.009(temperature × % catalyst loading)
+ 0.017(N2 flow rate × % catalyst loading) +
0.001(temperature)2 − 0.005(N2 flow rate)2 +
0.096(% catalyst loading)2
0.1925AC + 0.1675BC
8.275(% catalyst loading) +
0.006(temperature × N2 flow rate) +
0.009(temperature × % catalyst loading) +
0.017(N2 flow rate × % catalyst loading)
2.5. Characterization of Pyrolysis Oil
2.5.1. Chemical Compounds via GC/MS
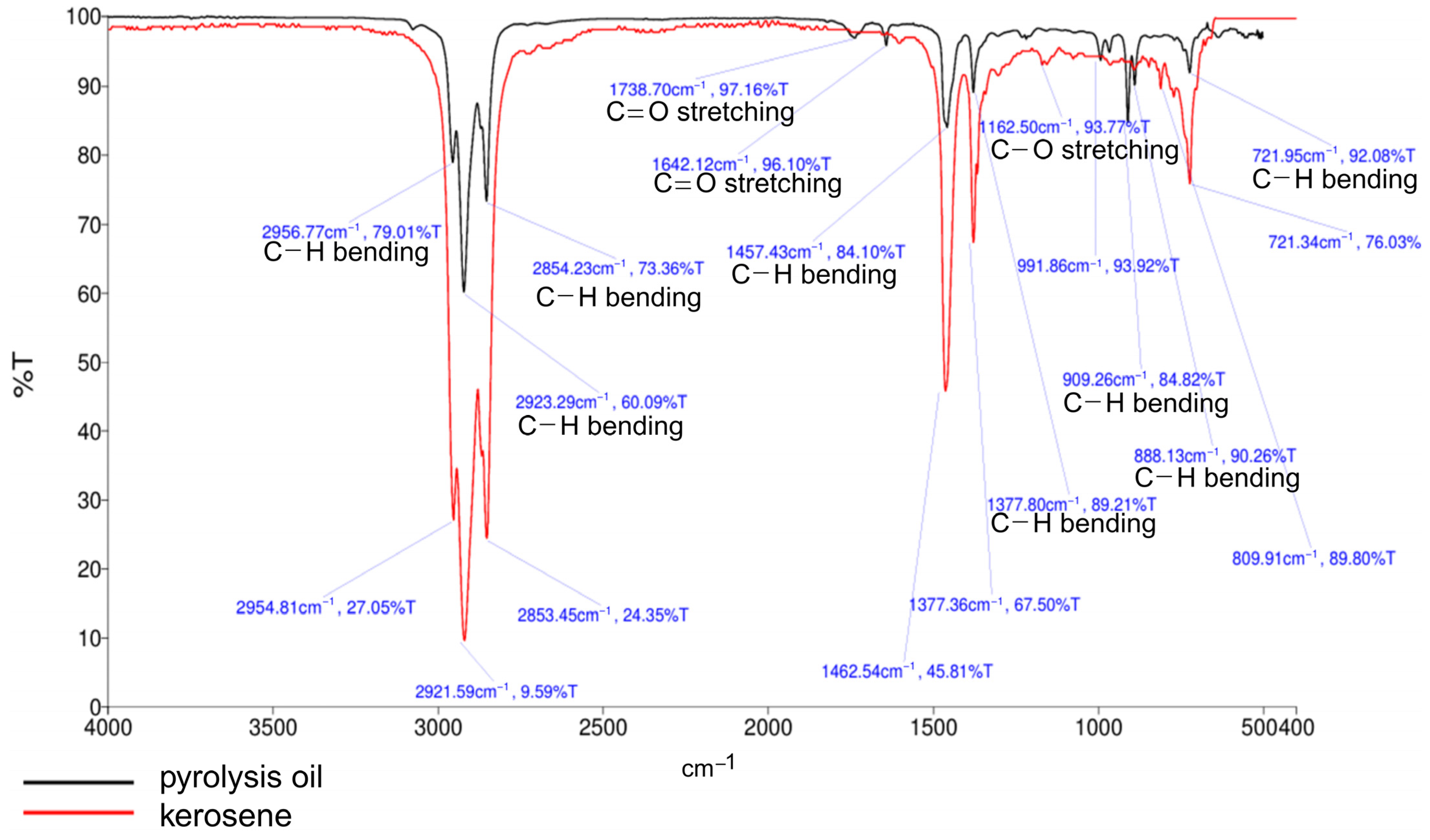
2.5.2. FT-IR Analyses
2.5.3. Physicochemical Analyses
3. Materials and Methods
3.1. Feedstock
3.2. Catalyst Preparation and Characterization
3.3. Experimental Design
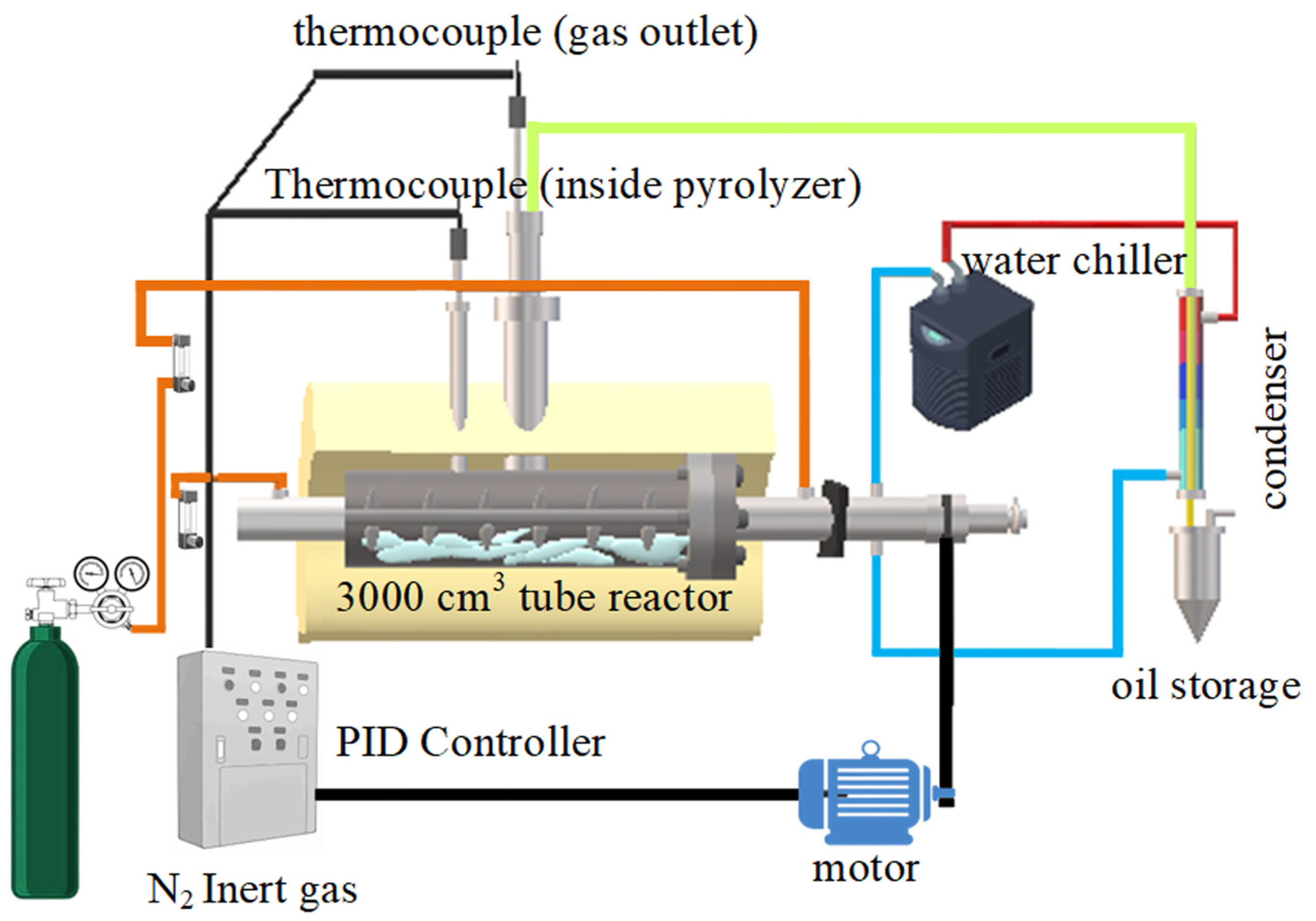
3.4. Experimental Procedure
3.5. Product Yield and Composition Analysis
3.6. Synergy of Dual Catalytic Pyrolysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pal, S.K.; Prabhudesai, V.S.; Vinu, R. Catalytic Upcycling of Post-Consumer Multilayered Plastic Packaging Wastes for the Selective Production of Monoaromatic Hydrocarbons. J. Environ. Manag. 2024, 351, 119630. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Z.; Shi, L.; Liu, Q. Pyrolysis of COVID-19 Disposable Masks and Catalytic Cracking of the Volatiles. J. Anal. Appl. Pyrolysis 2022, 162, 105481. [Google Scholar] [CrossRef] [PubMed]
- Seyed Mousavi, S.A.H.; Sadrameli, S.M.; Saeedi Dehaghani, A.H. Catalytic Pyrolysis of Municipal Plastic Waste over Nano MIL-53 (Cu) Derived @Zeolite Y for Gasoline, Jet Fuel, and Diesel Range Fuel Production. Process Saf. Environ. Prot. 2022, 164, 449–467. [Google Scholar] [CrossRef]
- Kim, D.; Lee, S.; Woo, S.-Y.; Park, K.Y. Enhanced Hydrogen Production Through Temperature-Optimized Pyrolysis of Mixed Plastic Waste for Sustainable Energy Recovery. Process Saf. Environ. Prot. 2025, 196, 106934. [Google Scholar] [CrossRef]
- Till, Z.; Varga, T.; Sója, J.; Miskolczi, N.; Chován, T. Kinetic Identification of Plastic Waste Pyrolysis on Zeolite-Based Catalysts. Energy Convers. Manag. 2018, 173, 320–330. [Google Scholar] [CrossRef]
- Mahapatra, P.M.; Aech, S.; Mandal, P.K.; Panda, A.K. Pyrolysis of Multilayered Plastic Packets to Hydrocarbon Fuel Feedstock: Thermal Degradation Behaviour, Kinetics and Thermodynamic Analysis, and Semi-Batch Studies. J. Energy Inst. 2024, 115, 101690. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Zhang, J.-L.; Zhang, X.; Luan, X.; Chen, H.-Z.; Hu, B.; Zhao, L.; Wu, Y.-L.; Lu, Q. Catalytic Depolymerization of Lignin via Transfer Hydrogenation Strategy over Skeletal CuZnAl Catalyst. Fuel Process. Technol. 2022, 237, 107448. [Google Scholar] [CrossRef]
- Dai, L.; Lata, S.; Cobb, K.; Zou, R.; Lei, H.; Chen, P.; Ruan, R. Recent Advances in Polyolefinic Plastic Pyrolysis to Produce Fuels and Chemicals. J. Anal. Appl. Pyrolysis 2024, 180, 106551. [Google Scholar] [CrossRef]
- Wang, L.; Han, F.; Li, G.; Zheng, M.; Wang, A.; Wang, X.; Zhang, T.; Cong, Y.; Li, N. Direct Synthesis of a High-Density Aviation Fuel Using a Polycarbonate. Green Chem. 2021, 23, 912–919. [Google Scholar] [CrossRef]
- Moonsin, P.; Roschat, W.; Phewphong, S.; Watthanalao, S.; Chaona, P.; Maneerat, B.; Arthan, S.; Thammayod, A.; Leelatam, T.; Duanguppama, K.; et al. The Physicochemical Characterization of Diesel-Like Fuels Derived from Plastic Waste Pyrolysis. J. Taiwan Inst. Chem. Eng. 2025, 171, 106040. [Google Scholar] [CrossRef]
- Hasan, M.M.; Haque, R.; Jahirul, M.I.; Rasul, M.G. Pyrolysis of Plastic Waste for Sustainable Energy Recovery: Technological Advancements and Environmental Impacts. Energy Convers. Manag. 2025, 326, 119511. [Google Scholar] [CrossRef]
- Vellaiyan, S.; Chandran, D.; Venkatachalam, R.; Ramalingam, K.; Rao, R.; Raviadaran, R. Maximizing Waste Plastic Oil Yield and Enhancing Energy and Environmental Metrics Through Pyrolysis Process Optimization and Fuel Modification. Results Eng. 2024, 22, 102066. [Google Scholar] [CrossRef]
- Hegedüs, B.; Hegedüs, B.; Palotás, Á.B.; Muránszky, G.; Dobó, Z. Investigation of Gasoline-Like Transportation Fuel Obtained by Plastic Waste Pyrolysis and Distillation. J. Clean. Prod. 2024, 447, 141500. [Google Scholar] [CrossRef]
- Karaba, A.; Le, T.A.; Patera, J.; Suková, M.; Suchopa, R.; Herink, T.; Zámostný, P. Waste Plastic Pyrolysis Oils Are Promising Feedstock for Sustainable Monomers Production via Steam Cracking Process. J. Anal. Appl. Pyrolysis 2025, 186, 106950. [Google Scholar] [CrossRef]
- Charusiri, W.; Vitidsant, T. Upgrading Bio-Oil Produced from the Catalytic Pyrolysis of Sugarcane (Saccharum officinarum L.) Straw Using Calcined Dolomite. Sustain. Chem. Pharm. 2017, 6, 114–123. [Google Scholar] [CrossRef]
- Li, B.; Mbeugang, C.F.M.; Xie, X.; Wei, J.; Zhang, S.; Zhang, L.; Samahy, A.A.E.; Xu, D.; Wang, Q.; Zhang, S.; et al. Catalysis/CO2 Sorption Enhanced Pyrolysis-Gasification of Biomass for H2-Rich Gas Production: Effects of Activated Carbon, NiO Active Component and Calcined Dolomite. Fuel 2023, 334, 126842. [Google Scholar] [CrossRef]
- Wu, T.; Dang, Q.; Wu, Y.; Lei, T.; Yu, J. Catalytic Hydropyrolysis of Biomass over NiMo Bimetallic Carbon-Based Catalysts. J. Environ. Chem. Eng. 2023, 11, 110024. [Google Scholar] [CrossRef]
- Bae, M.-G.; Lee, Y.-L.; Kim, H.-M.; Son, J.-W.; Cheon, B.-S.; Jeong, D.-W. Optimization of Ni Loading Amount in Ni-CeZrO2 Catalyst for Dodecane Steam Reforming for the Upcycling of Plastic Pyrolysis Oil. Fuel Process. Technol. 2025, 272, 108218. [Google Scholar] [CrossRef]
- Valizadeh, B.; Valizadeh, S.; Kim, H.; Choi, Y.J.; Seo, M.W.; Yoo, K.S.; Lin, K.-Y.A.; Hussain, M.; Park, Y.-K. Production of Light Olefins and Monocyclic Aromatic Hydrocarbons from the Pyrolysis of Waste Plastic Straws over High-Silica Zeolite-Based Catalysts. Environ. Res. 2024, 245, 118076. [Google Scholar] [CrossRef]
- Ji, X.; Bowen, W.; Zhen, H.; Haoquan, H.; Lijun, J. Catalytic Conversion of Biomass Pyrolysis Volatiles over Composite Catalysts of Activated Carbon and HY Zeolite. J. Fuel Chem. Technol. 2024, 52, 1318–1326. [Google Scholar] [CrossRef]
- Nazarloo, N.H.; Shirvanimoghaddam, K.; Zabihi, O.; Naebe, M. Catalytic Pyrolysis of Plastic Waste Using Activated Carbon from Cotton Waste. J. Anal. Appl. Pyrolysis 2024, 182, 106692. [Google Scholar] [CrossRef]
- Xing, G.; Liu, S.; Guan, Q.; Li, W. Investigation on Hydroisomerization and Hydrocracking of C15–C18 n-Alkanes Utilizing a Hollow Tubular Ni-Mo/SAPO-11 Catalyst with High Selectivity of Jet Fuel. Catal. Today 2019, 330, 109–116. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhang, D.; Chen, W.; Zhang, T.; Huang, Y.; Gu, L.; Deng, H.; Tian, D. Box–Behnken Design Approach Towards Optimization of Activated Carbon Synthesized by Co-Pyrolysis of Waste Polyester Textiles and MgCl2. Appl. Surf. Sci. 2018, 427, 340–348. [Google Scholar] [CrossRef]
- Andalia, W.; Pratiwi, I.; Afrah, B.D.; Akbar, I.; Ammarullah, M.I. Optimized Pyrolysis of Household Plastic Waste in Indonesia for Oil Production: Hydrocarbon Profiling, Fuel Alternative, and Potential Biomedical Application. Results Chem. 2025, 15, 102247. [Google Scholar] [CrossRef]
- Amrullah, A.; Farobie, O.; Irawansyah, H.; Lutfi, M.; Haty, L.N. Synergistic Enhancement of Bio-Oil Production, Quality, and Optimization from Co-Pyrolysis Purun Tikus (Eleocharis dulcis) and Plastic Waste Using Response Surface Methodology. Process Saf. Environ. Prot. 2024, 187, 471–482. [Google Scholar] [CrossRef]
- Palizdar, A.; Sadrameli, S.M. Catalytic upgrading of beech wood pyrolysis oil over iron- and zinc-promoted hierarchical MFI zeolites. Fuel 2020, 264, 116813. [Google Scholar] [CrossRef]
- Fu, H.; Li, X.; Shao, S.; Cai, Y. Jet fuel range hydrocarbon generation from catalytic pyrolysis of lignin and polypropylene with iron-modified activated carbon. J. Anal. Appl. Pyrolysis 2024, 177, 106360. [Google Scholar] [CrossRef]
- Fu, W.; Cheng, Y.W.; Wang, Y.; Zhang, Y.; Wang, C.-H. Counteractive catalytic effects of FeNi- versus Fe- and Ni- in plastic pyrolysis for advanced-quality jet fuel production. Chem. Eng. J. 2024, 494, 153078. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, D.; Guo, Y.; Lin, X.; Cai, H. From wood plastic composite waste to high-value aromatics via catalytic pyrolysis over an activated carbon. J. Anal. Appl. Pyrolysis 2022, 176, 105688. [Google Scholar] [CrossRef]
- Kollman, M.S.; Jiang, X.; Sun, R.; Zhang, X.; Li, W.; Chang, H.; Jameel, H. Towards jet fuel from technical lignins: Feedstock-catalyst-product interactions revealed during catalytic hydrogenolysis. Chem. Eng. J. 2023, 451, 138464. [Google Scholar] [CrossRef]
- Chen, J.; Qu, Q.; Lu, J.; Wang, H.; Gu, F.; Zhu, M. The structural characteristics and pyrolysis products properties based on cotton stalks and husks. Ind. Crops Prod. 2025, 227, 120858. [Google Scholar] [CrossRef]
- Munir, D.; Abdullah, F.; Piepenbreier, F.; Usman, M.R. Hydrocracking of a plastic mixture over various micro-mesoporous composite zeolites. Powder Technol. 2017, 316, 542–550. [Google Scholar] [CrossRef]
- Nie, G.; Dai, Y.; Xie, J.; Zhang, X.; Pan, L.; Zou, J.-J. Improving low-temperature properties of lignin-derived jet-fuel-ranged hydrocarbons via hydroisomerization. Catal. Today 2021, 365, 235–240. [Google Scholar] [CrossRef]
- Ahmad, N.; Ahmad, N.; Maafa, I.M.; Ahmed, U.; Akhter, P.; Shehzad, N.; Amjad, U.; Hussain, M. Thermal conversion of polystyrene plastic waste to liquid fuel via ethanolysis. Fuel 2020, 279, 118498. [Google Scholar] [CrossRef]
- Sipra, A.T.; Gao, N.; Zhang, L.; Chu, H.; Quan, C. The application of spent catalysts from catalytic pyrolysis of plastic waste as solid functional materials. Carbon Capture Sci. Technol. 2024, 13, 100285. [Google Scholar] [CrossRef]
- Sorino, D.; de Vizia, P.; Baldelli, M.; Bartolucci, L.; Cordiner, S.; Falsetti, A.; Lombardi, F.; Mulone, V. Pyrolysis of mixed contaminated plastic wastes: Assessing the influence of polymers composition, temperature and residence time. Waste Manag. 2025, 201, 114793. [Google Scholar] [CrossRef]
- Çelebi, S.; Kocakulak, T.; Demir, U.; Ergen, G.; Yilmaz, E. Optimizing the effect of a mixture of light naphtha, diesel and gasoline fuels on engine performance and emission values on an HCCI engine. Appl. Energy 2023, 330, 120349. [Google Scholar] [CrossRef]
- Ndiaye, N.K.; Derkyi, N.S.A.; Amankwah, E. Pyrolysis of plastic waste into diesel engine-grade oil. Sci. Afr. 2023, 21, e01836. [Google Scholar] [CrossRef]
- Wang, K.; Ban, Y.; Wu, Y.; Jin, L.; Hu, H. Synergistic effect and chlorine migration behavior in co-pyrolysis of Pingshuo coal and polyvinyl chloride and directional chlorine enrichment using calcium oxide. Fuel 2023, 349, 128749. [Google Scholar] [CrossRef]
- Charusiri, W.; Phowan, N.; Vitidsant, T. Catalytic copyrolysis of sugarcane leaves and low-density polyethylene waste to produce bio-oil and chemicals using copper-doped HZSM-35. Energy Fuel 2022, 36, 2639–2655. [Google Scholar] [CrossRef]
- Hu, Y.; Li, M.; Zhou, N.; Yuan, H.; Guo, Q.; Jiao, L.; Ma, Z. Catalytic stepwise pyrolysis for dechlorination and chemical recycling of PVC-containing mixed plastic wastes: Influence of temperature, heating rate, and catalyst. Sci. Total Environ. 2024, 908, 168344. [Google Scholar] [CrossRef] [PubMed]
- Vellaiyan, S. Optimization of pyrolysis process parameters for a higher yield of plastic oil with enhanced physicochemical properties derived from medical plastic wastes. Sustain. Chem. Pharm. 2023, 36, 101310. [Google Scholar] [CrossRef]
- Chen, W.-H.; Biswas, P.P.; Kwon, E.E.; Park, Y.-K.; Rajendran, S.; Gnanasekaran, L.; Chang, J.-S. Optimization of the process parameters of catalytic plastic pyrolysis for oil production using design of experiment approaches: A review. J. Chem. Eng. 2023, 471, 144695. [Google Scholar] [CrossRef]
- Kasetsupsin, P.; Vitidsant, T.; Permpoonwiwat, A.; Phowan, N.; Charusiri, W. Combined activated carbon with spent fluid catalytic cracking catalyst and MgO for the catalytic conversion of waste polyethylene wax into diesel-like hydrocarbon fuels. ACS Omega 2022, 7, 20306–20320. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | PPW | PE | PP | Standard Method |
|---|---|---|---|---|
| proximate analysis (wt.%) | ASTM D7582 | |||
| volatile | 90.28 | 96.13 | 97.24 | |
| fixed carbon | 2.89 | 3.87 | 2.76 | |
| ash | 6.83 | n.d. | n.d. | |
| ultimate analysis (wt.%) | ASTM D7583 | |||
| C | 83.35 | 83.24 | 84.11 | |
| H | 14.73 | 16.76 | 15.89 | |
| O a | 1.92 | n.d. | n.d. | |
| N | n.d. b | n.d. | n.d. | |
| S | n.d. | n.d. | n.d. | |
| H/C (mol/mol) | 2.12 | 2.42 | 2.27 | calculation |
| O/C (mol/mol) | 0.02 | n.a. c | n.a. | calculation |
| HHV | 39.08 | 42.85 | 41.82 | ASTM D5865 |
| Element | Compound | Concentration (wt.%) | |||
|---|---|---|---|---|---|
| AC | DM | 5% Fe/DM | 10% Fe/DM | ||
| Al | Al2O3 | n.d. | 0.19 | 0.22 | 0.18 |
| Si | SiO2 | 0.08 | 0.39 | 0.67 | 0.55 |
| Ti | TiO2 | n.d. | 0.06 | 0.06 | n.d. |
| Na | Na2O | 0.42 | 0.33 | 0.41 | 0.83 |
| Mg | MgO | 0.16 | 33.01 | 31.82 | 31.72 |
| P | P2O5 | 0.02 | 0.02 | 0.02 | n.d. |
| K | K2O | 0.54 | n.d. a | n.d. | n.d. |
| Ca | CaO | 0.08 | 59.82 | 54.85 | 53.19 |
| S | SO3 | 0.03 | 0.06 | 0.11 | 0.11 |
| Fe | Fe2O3 | 0.01 | 0.23 | 5.82 | 9.14 |
| other | Other | 1.91 | 5.89 | 6.02 | 4.28 |
| Element | Oxide | Concentration of Fe-Doped (wt.%) | ||
|---|---|---|---|---|
| DM | 5%Fe/DM | 10%Fe/DM | ||
| Fe | Fe2O3 | 0.04 | 4.77 | 7.82 |
| Catalyst | BET Surface Area (m2/g) | Total Pore Volume (cm3/g) | Total Pore Size (nm) |
|---|---|---|---|
| AC | 830.77 | 0.05 | 2.37 |
| calcined DM | 19.95 | 0.10 | 18.79 |
| 5 wt.%Fe/DM | 18.77 | 0.10 | 19.46 |
| 10 wt.%Fe/DM | 17.03 | 0.10 | 25.80 |
| Std | Variables | Product Distribution (wt.%) | |||
|---|---|---|---|---|---|
| Temperature (°C) | N2 Flowing Rate (mL/min) | % Catalyst Loading (wt.%) | Liquid Yield | Kerosene Fraction | |
| 1 | 440 | 50 | 10 | 82.47 | 24.04 |
| 2 | 480 | 50 | 10 | 64.63 | 17.03 |
| 3 | 440 | 100 | 10 | 75.37 | 21.25 |
| 4 | 480 | 100 | 10 | 68.76 | 19.02 |
| 5 | 440 | 50 | 15 | 75.11 | 24.03 |
| 6 | 480 | 50 | 15 | 59.18 | 18.15 |
| 7 | 440 | 100 | 15 | 72.36 | 22.27 |
| 8 | 480 | 100 | 15 | 67.45 | 20.45 |
| 9 | 426 | 75 | 12 | 86.37 | 22.52 |
| 10 | 493 | 75 | 12 | 54.63 | 19.57 |
| 11 | 460 | 32 | 12 | 53.15 | 20.72 |
| 12 | 460 | 117 | 12 | 67.64 | 19.93 |
| 13 | 460 | 75 | 8 | 70.68 | 19.54 |
| 14 | 460 | 75 | 16 | 71.93 | 21.25 |
| 15 | 460 | 75 | 12 | 71.31 | 20.92 |
| 16 | 460 | 75 | 12 | 72.04 | 21.27 |
| 17 | 460 | 75 | 12 | 71.63 | 21.34 |
| 18 | 460 | 75 | 12 | 71.68 | 20.87 |
| 19 | 460 | 75 | 12 | 72.02 | 20.91 |
| 20 | 460 | 75 | 12 | 71.56 | 20.88 |
| Source | Sum of Squares | df | Mean Square | F Value | p Value Prob > F | |
|---|---|---|---|---|---|---|
| Model | 1024.711 | 9 | 113.857 | 7.0709 | 0.0026 | significant |
| A-temperature | 712.887 | 1 | 712.887 | 44.2725 | <0.0001 | |
| B-N2 flow rate | 53.061 | 1 | 53.061 | 3.2952 | 0.0995 | |
| C-%catalyst loading | 16.536 | 1 | 16.536 | 1.0270 | 0.3348 | |
| AB | 61.883 | 1 | 61.883 | 3.8431 | 0.0784 | |
| AC | 1.629 | 1 | 1.629 | 0.1012 | 0.7570 | |
| BC | 9.010 | 1 | 9.010 | 0.5596 | 0.4717 | |
| A2 | 1.455 | 1 | 1.455 | 0.0904 | 0.7699 | |
| B2 | 152.678 | 1 | 152.678 | 9.4818 | 0.0117 | |
| C2 | 5.229 | 1 | 5.229 | 0.3248 | 0.5813 | |
| Residual | 161.022 | 10 | 16.102 | |||
| Lack of Fit | 160.628 | 5 | 32.126 | 406.9271 | <0.0001 | significant |
| Pure Error | 0.395 | 5 | 0.079 | |||
| Cor Total | 1185.733 | 19 | ||||
| Std. Dev. | 4.013 | R-Squared | 0.864 | |||
| Mean | 69.999 | Adj R-Squared | 0.742 | |||
| C.V.% | 5.733 | Pred R-Squared | −0.028 | |||
| PRESS | 1218.850 | Adeq Precision | 10.246 | |||
| Source | Sum of Squares | df | Mean Square | F Value | p Value Prob > F | |
|---|---|---|---|---|---|---|
| Model | 48.630 | 6 | 8.105 | 15.574 | <0.0001 | significant |
| A-temperature | 35.123 | 1 | 35.123 | 67.489 | <0.0001 | |
| B-N2 flow rate | 0.185 | 1 | 0.185 | 0.355 | 0.5615 | |
| C-%catalyst loading | 3.033 | 1 | 3.033 | 5.828 | 0.0312 | |
| AB | 9.768 | 1 | 9.768 | 18.770 | 0.0008 | |
| AC | 0.296 | 1 | 0.296 | 0.570 | 0.4639 | |
| BC | 0.224 | 1 | 0.224 | 0.431 | 0.5228 | |
| A2 | 6.766 | 13 | 0.520 | |||
| B2 | 6.537 | 8 | 0.817 | 17.898 | 0.0028 | significant |
| C2 | 0.228 | 5 | 0.046 | |||
| Residual | 55.395 | 19 | ||||
| Lack of Fit | 48.630 | 6 | 8.105 | 15.574 | <0.0001 | significant |
| Pure Error | 35.123 | 1 | 35.123 | 67.489 | <0.0001 | |
| Cor Total | 0.185 | 1 | 0.185 | 0.355 | 0.5615 | |
| Std. Dev. | 0.721 | R-Squared | 0.8779 | |||
| Mean | 20.798 | Adj R-Squared | 0.8215 | |||
| C.V.% | 3.469 | Pred R-Squared | 0.5386 | |||
| PRESS | 25.557 | Adeq Precision | 14.1168 | |||
| R.T (min) | A Relative Percentage Area Peak (%) | Chemical Compounds | Molecular Formular | ||||
|---|---|---|---|---|---|---|---|
| Non Cat | DM | Fe/DM | AC | Fe/DM-AC | |||
| Straight aliphatic hydrocarbon compounds | |||||||
| 1.576 | 1.444 | 1.993 | 1.303 | 2.887 | Pentane | C5H12 | |
| 1.945 | 1.048 | n-Hexane | C6H14 | ||||
| 4.267 | 1.016 | 1.741 | 1.611 | 1.465 | 1.935 | Octane | C8H18 |
| 6.029 | 1.014 | 1.772 | 1.827 | 1.521 | 3.232 | Nonane | C9H20 |
| 7.774 | 1.234 | 2.957 | 1.887 | 7.025 | 4.116 | Decane | C10H22 |
| 9.416 | 2.112 | 2.752 | 1.739 | 4.322 | Undecane | C11H24 | |
| 10.939 | 1.386 | 2.190 | 6.097 | 1.837 | 4.329 | Dodecane | C12H26 |
| 12.372 | 1.697 | 2.439 | 5.958 | 2.077 | 4.226 | Tridecane | C13H28 |
| 13.706 | 1.384 | 3.881 | 4.449 | 2.068 | 6.727 | Tetradecane | C14H30 |
| 14.881 | 1.242 | 2.587 | 4.252 | 1.980 | 10.935 | Pentadecane | C15H32 |
| 16.168 | 1.169 | 2.936 | 2.716 | 1.682 | 8.981 | Hexadecane | C16H34 |
| 17.297 | 1.511 | 1.682 | 3.571 | 2.419 | 3.164 | Heptadecane | C17H36 |
| 18.372 | 1.554 | 2.791 | 2.462 | 2.004 | 1.645 | Octadecane | C18H38 |
| 19.387 | 1.431 | 2.934 | 2.109 | 2.291 | 1.847 | Nonadecane | C19H40 |
| 20.360 | 2.868 | 2.652 | 0.384 | 0.262 | Eicosane | C20H42 | |
| 21.283 | 2.225 | 1.477 | 0.119 | 0.511 | Heneicosane | C21H44 | |
| 22.172 | 2.016 | 1.197 | 0.302 | 1.641 | Docosane | C22H46 | |
| 23.025 | 2.842 | 1.494 | 0.035 | 1.555 | Tricosane | C23H48 | |
| 23.845 | 4.316 | 0.456 | 0.173 | 0.131 | Tetracosane | C24H50 | |
| 24.635 | 4.360 | 0.299 | 0.546 | Pentacosane | C25H52 | ||
| 25.395 | 2.461 | 0.815 | 0.127 | 0.432 | Hexacosane | C26H54 | |
| 26.132 | 1.249 | 0.124 | 0.207 | 0.410 | Heptacosane | C27H56 | |
| 26.842 | 1.031 | 0.557 | 0.721 | 0.401 | Octacosane | C28H58 | |
| 27.529 | 1.379 | Nonacosane | C29H60 | ||||
| Branched aliphatic hydrocarbon compounds | |||||||
| 0.034 | Butane, 2-methyl- | C5H12 | |||||
| 1.792 | 0.556 | 0.564 | 0.507 | 0.558 | Pentane, 2-methyl- | C6H14 | |
| 1.855 | 0.064 | 0.030 | 1.037 | Pentane, 3-methyl- | C6H14 | ||
| 2.535 | 0.055 | 0.059 | 0.068 | 0.061 | Hexane, 3-methyl- | C7H16 | |
| 2.807 | 0.892 | 1.568 | 1.383 | 1.249 | 1.302 | Heptane | C7H16 |
| 3.729 | 1.508 | 1.456 | 1.317 | 0.845 | Heptane, 4-methyl- | C8H18 | |
| 4.632 | 0.303 | 0.498 | 0.397 | 0.584 | Hexane, 2,3,5-trimethyl- | C9H20 | |
| 6.121 | 0.511 | 1-Ethyl-4-methylcyclohexane | C9H18 | ||||
| 7.916 | 0.813 | 1.589 | 0.565 | 0.526 | 1.268 | Nonane, 2,6-dimethyl- | C11H24 |
| 7.986 | 0.891 | 0.589 | 0.738 | 0.666 | Decane, 2-methyl- | C11H24 | |
| 11.446 | 0.204 | Dodecane, 4,6-dimethyl- | C14H30 | ||||
| 16.072 | 1.871 | 2.845 | 2.436 | 1.716 | 2.847 | Cetene | C16H32 |
| 17.200 | 1.832 | 2.776 | 1.881 | 2.521 | 1.227 | 1-Heptadecene | C17H34 |
| 17.204 | 1.543 | 1.729 | 1-Nonadecene | C19H38 | |||
| 18.027 | 2.035 | Heptadecane, 3-methyl- | C18H38 | ||||
| Olefin hydrocarbon compounds | |||||||
| 1.629 | 0.206 | 0.169 | 0.271 | 2-Methyl-1-butene | C5H10 | ||
| 1.689 | 0.057 | 0.178 | 0.055 | 0.106 | 1,4-Pentadiene | C5H8 | |
| 1.897 | 1.593 | 1.952 | 2.317 | 2.692 | 3.244 | 1-Pentene, 2-methyl- | C6H12 |
| 1.998 | 0.698 | 0.894 | 0.820 | 0.769 | 2-Butene, 2,3-dimethyl- | C6H12 | |
| 2.044 | 0.148 | 2-Hexene | C6H12 | ||||
| 2.087 | 0.073 | 0.136 | 0.083 | 0.083 | 2-Pentene, 3-methyl- | C6H12 | |
| 2.203 | 0.276 | 0.244 | 0.151 | 0.168 | 2,4-Hexadiene, (E,E)- | C6H10 | |
| 2.236 | 0.258 | 0.308 | 1.104 | 1.132 | Cyclopentene, 1-methyl- | C6H10 | |
| 2.280 | 0.350 | 0.358 | 0.284 | 0.257 | 1-Pentene, 2,4-dimethyl- | C7H14 | |
| 2.674 | 0.163 | 1-Hexene, 2-methyl- | C7H14 | ||||
| 2.706 | 1.898 | 1.618 | 1.594 | 1.531 | 1-Heptene | C7H14 | |
| 2.854 | 0.774 | 0.762 | 0.597 | 1,4-Hexadiene, 2-methyl- | C7H12 | ||
| 2.857 | 0.819 | 1,4-Hexadiene, 5-methyl- | C7H12 | ||||
| 2.890 | 0.195 | 0.166 | 0.147 | (Z)-2-Heptene | C7H14 | ||
| 2.946 | 0.065 | 0.096 | 0.075 | 0.070 | 1,4-Hexadiene, 5-methyl- | C7H12 | |
| 3.361 | 0.252 | 0.337 | 0.309 | 2,4-Hexadiene, 2-methyl- | C7H12 | ||
| 4.124 | 2.258 | 2.103 | 1.935 | 0.384 | 1-Octene | C8H16 | |
| 4.207 | 0.328 | 0.264 | 0.253 | 1,4-Pentadiene, 2,3,3-trimethyl- | C8H14 | ||
| 4.366 | 0.197 | 0.167 | 0.140 | 2-Octene, (Z)- | C8H16 | ||
| 4.410 | 0.678 | 0.572 | 0.518 | 0.439 | 2,2-Dimethyl-3-heptene trans | C9H18 | |
| 4.874 | 1.615 | 1.654 | 6.759 | 2,3-Dimethyl-2-heptene | C9H18 | ||
| 5.007 | 4.779 | 3.742 | 1.864 | 3.391 | 4.331 | 2,4-Dimethyl-1-heptene | C9H18 |
| 5.139 | 0.278 | 1,3-Hexadiene, 3-ethyl-2-methyl-, (Z)- | C9H16 | ||||
| 5.491 | 0.520 | 1,3-Heptadiene, 2,3-dimethyl- | C9H16 | ||||
| 5.650 | 0.228 | 6,6-Dimethylhepta-2,4-diene | C9H16 | ||||
| 5.873 | 1.540 | 2.562 | 1.371 | 2.178 | 1.689 | 1-Nonene | C9H18 |
| 5.909 | 0.333 | 0.311 | 2,4,6-Trimethyl-3-heptene | C10H20 | |||
| 6.261 | 0.234 | 1,6-Octadiene, 2,5-dimethyl-, (E)- | C10H18 | ||||
| 7.509 | 0.318 | 5-Decene, (E)- | C10H20 | ||||
| 7.628 | 2.084 | 2.328 | 1-Decene | C10H20 | |||
| 7.677 | 0.409 | 2-Decene, 7-methyl-, (Z)- | C11H22 | ||||
| 7.837 | 0.248 | cis-4-Decene | C10H20 | ||||
| 7.850 | 0.350 | 0.313 | 2-Decene, (Z)- | C10H20 | |||
| 7.853 | 0.341 | 4-Decene | C10H20 | ||||
| 8.490 | 0.116 | 1-Decene, 5-methyl- | C11H22 | ||||
| 8.776 | 0.569 | 0.637 | 0.199 | 2-Decene, 2,4-dimethyl- | C12H24 | ||
| 8.832 | 0.528 | 0.680 | 0.537 | 3-Decene, 2,2-dimethyl-, (E)- | C12H24 | ||
| 9.087 | 1.439 | 5-Ethyl-1-nonene | C11H22 | ||||
| 9.157 | 1.433 | 1-Decene, 2,4-dimethyl- | C12H24 | ||||
| 9.283 | 3.140 | 2.087 | 1.462 | 5.189 | 1-Undecene | C11H22 | |
| 9.476 | 0.280 | 0.447 | 0.242 | 2-Undecene, (Z)- | C11H22 | ||
| 9.480 | 0.144 | 5-Undecene | C11H22 | ||||
| 10.816 | 1.795 | 3.223 | 2.736 | 2.371 | 5.865 | 1-Dodecene | C12H24 |
| 10.992 | 0.288 | 0.353 | 0.351 | 0.323 | 2-Dodecene, (Z)- | C12H24 | |
| 12.140 | 0.538 | 6-Tridecene, (Z)- | C13H26 | ||||
| 12.143 | 0.751 | 0.584 | 4-Nonene, 5-butyl- | C13H26 | |||
| 12.253 | 1.765 | 3.255 | 6.491 | 4.577 | 5.443 | 1-Tridecene | C13H26 |
| 13.490 | 0.818 | 5-Tetradecene, (Z)- | C14H28 | ||||
| 13.587 | 1.755 | 4.259 | 3.039 | 2.642 | 2.415 | 1-Tetradecene | C14H28 |
| 13.749 | 0.313 | 7-Tetradecene | C14H28 | ||||
| 13.756 | 1.389 | 0.489 | 2-Tetradecene, (E)- | C14H28 | |||
| 13.875 | 0.183 | 3-Tetradecene, (E)- | C14H28 | ||||
| 13.879 | 1.796 | 2.720 | 0.229 | 2.416 | 2.753 | 1-Pentadecene | C15H30 |
| 18.276 | 1.615 | 1.365 | 1.887 | 1-Octadecene | C18H36 | ||
| 19.298 | 1.456 | 2.015 | 0.958 | 1.891 | 1-Nonadecene | C19H38 | |
| 20.277 | 1.304 | 0.294 | 1-Eicosene | C20H40 | |||
| 23.808 | 4.268 | 0.333 | 1-Tetracosene | C24H48 | |||
| Cycloalkane hydrocarboon compounds | |||||||
| 1.755 | 0.120 | 0.222 | 0.145 | 0.215 | Cyclopentene | C5H8 | |
| 2.144 | 0.248 | 0.466 | 0.316 | 0.355 | Cyclopentane, methyl- | C6H12 | |
| 3.544 | 0.173 | 0.258 | 0.237 | 0.204 | Cyclobutane, (1-methylethylidene)- | C7H12 | |
| 4.499 | 0.159 | 0.136 | Cyclopropane, (2,2-dimethylpropylidene)- | C8H14 | |||
| 4.662 | 0.321 | 0.428 | 0.284 | 0.295 | 1-Methyl-2-methylenecyclohexane | C8H14 | |
| 4.788 | 1.493 | 0.756 | 0.798 | 0.753 | Cyclohexane, 1,2,4-trimethyl- | C9H18 | |
| 4.821 | 0.345 | 0.316 | 0.255 | 0.431 | Cyclohexane, ethyl- | C8H16 | |
| 5.222 | 1.946 | 1.514 | 0.245 | 1.448 | Cyclohexane, 1,3,5-trimethyl- | C9H18 | |
| 5.912 | 0.462 | 0.337 | Cyclopentane, 1,2,3,4,5-pentamethyl | C10H20 | |||
| 6.118 | 0.423 | Cyclohexane, 1,2,3-trimethyl- | C9H18 | ||||
| 6.845 | 0.388 | Cyclohexane, (1-methylethylidene)- | C9H16 | ||||
| 8.968 | 0.151 | 1-Isopropyl-1,4,5-trimethylcyclohexane | C12H24 | ||||
| 9.605 | 0.163 | Cyclopentane, 1,2-dipropyl- | C11H22 | ||||
| 10.242 | 0.326 | Cyclohexane, (2,2-dimethylcyclopentyl)- | C13H24 | ||||
| 10.378 | 0.201 | 1-Isopropyl-1,4,5-trimethylcyclohexane | C12H24 | ||||
| 12.047 | 1.454 | 0.657 | Cyclopentadecane | C15H30 | |||
| Cycloalkene hydrocarbon compound | |||||||
| 2.601 | 0.195 | 0.253 | 0.201 | 0.198 | Cyclohexene | C6H10 | |
| 2.983 | 0.147 | 0.272 | 0.242 | Cyclopentene, 1,5-dimethyl- | C7H12 | ||
| 3.056 | 0.308 | 0.189 | 0.206 | Cyclopentane, 1-methyl-2-methylene- | C7H12 | ||
| 3.096 | 0.493 | 0.861 | 0.683 | 0.660 | Cyclohexane, methyl- | C7H14 | |
| 3.245 | 0.206 | 0.358 | 0.312 | 0.280 | Cyclopentane, ethyl- | C7H14 | |
| 3.321 | 0.270 | 0.296 | 0.252 | 0.253 | Cyclohexene, 3-methyl- | C7H12 | |
| 3.573 | 0.715 | 0.649 | 1-Ethylcyclopentene | C7H12 | |||
| 3.779 | 0.408 | 0.696 | 0.569 | 0.517 | 0.432 | Cyclohexene, 1-methyl- | C7H12 |
| 3.932 | 0.180 | 0.220 | 0.193 | 0.151 | 3-Methylenecyclohexene | C7H10 | |
| 5.070 | 0.150 | 0.134 | 1-Propylcyclopentene | C8H14 | |||
| 5.123 | 0.253 | 0.181 | 0.185 | 1-Methyl-2-methylenecyclohexane | C8H14 | ||
| 6.317 | 0.246 | Cyclopentene, 1,4-dimethyl-5-(1-methylethyl)- | C10H18 | ||||
| 6.841 | 0.264 | 0.392 | 0.285 | 0.422 | Cyclopentene, 1-butyl- | C9H16 | |
| Aromatic hydrocarbon compounds | |||||||
| 5.312 | 0.106 | 0.151 | 0.485 | 0.368 | Ethylbenzene | C8H10 | |
| 7.087 | 0.489 | 0.715 | 0.882 | Benzene, 1-ethyl-4-methyl- | C9H12 | ||
| 7.203 | 0.241 | Benzene, 1-ethyl-3-methyl- | C9H12 | ||||
| 7.400 | 0.122 | 0.761 | Benzene, 1-ethyl-2-methyl- | C9H12 | |||
| 7.402 | 0.217 | 0.293 | Mesitylene | C9H12 | |||
| Elemental Analyses | Non Catalyst | AC | Fe/DM | Fe/DM-AC |
|---|---|---|---|---|
| C | 80.43 | 82.38 | 82.48 | 82.65 |
| H | 19.57 | 17.62 | 17.52 | 17.35 |
| H/C (mol/mol) | 2.92 | 2.57 | 2.55 | 2.52 |
| Kinematic viscosity (mm2/s) | 36.14 | 6.37 | 6.69 | 4.31 |
| HHV (MJ/kg) | 41.27 | 42.84 | 44.73 | 44.92 |
| Parameter | Experiment Factor | Level | ||||
|---|---|---|---|---|---|---|
| −α | −1 | 0 | 1 | +α | ||
| Temperature (°C) | A | 426.36 | 440 | 460 | 480 | 493.64 |
| N2 flow rate (mL/min) | B | 32.96 | 50 | 75 | 100 | 117.04 |
| catalyst loading (%) | C | 8.30 | 10 | 12.50 | 15 | 16.70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arjharnwong, O.; Vitidsant, T.; Permpoonwiwat, A.; Phowan, N.; Charusiri, W. Optimization of Kerosene-like Fuels Produced via Catalytic Pyrolysis of Packaging Plastic Waste via Central Composite Design and Response Surface Methodology: Performance of Iron-Doped Dolomite and Activated Carbon. Molecules 2025, 30, 2884. https://doi.org/10.3390/molecules30132884
Arjharnwong O, Vitidsant T, Permpoonwiwat A, Phowan N, Charusiri W. Optimization of Kerosene-like Fuels Produced via Catalytic Pyrolysis of Packaging Plastic Waste via Central Composite Design and Response Surface Methodology: Performance of Iron-Doped Dolomite and Activated Carbon. Molecules. 2025; 30(13):2884. https://doi.org/10.3390/molecules30132884
Chicago/Turabian StyleArjharnwong, Oratepin, Tharapong Vitidsant, Aminta Permpoonwiwat, Naphat Phowan, and Witchakorn Charusiri. 2025. "Optimization of Kerosene-like Fuels Produced via Catalytic Pyrolysis of Packaging Plastic Waste via Central Composite Design and Response Surface Methodology: Performance of Iron-Doped Dolomite and Activated Carbon" Molecules 30, no. 13: 2884. https://doi.org/10.3390/molecules30132884
APA StyleArjharnwong, O., Vitidsant, T., Permpoonwiwat, A., Phowan, N., & Charusiri, W. (2025). Optimization of Kerosene-like Fuels Produced via Catalytic Pyrolysis of Packaging Plastic Waste via Central Composite Design and Response Surface Methodology: Performance of Iron-Doped Dolomite and Activated Carbon. Molecules, 30(13), 2884. https://doi.org/10.3390/molecules30132884