
Integrating Graph Convolution and Attention Mechanism for Kinase Inhibition Prediction




Abstract
1. Introduction
2. Results and Discussion
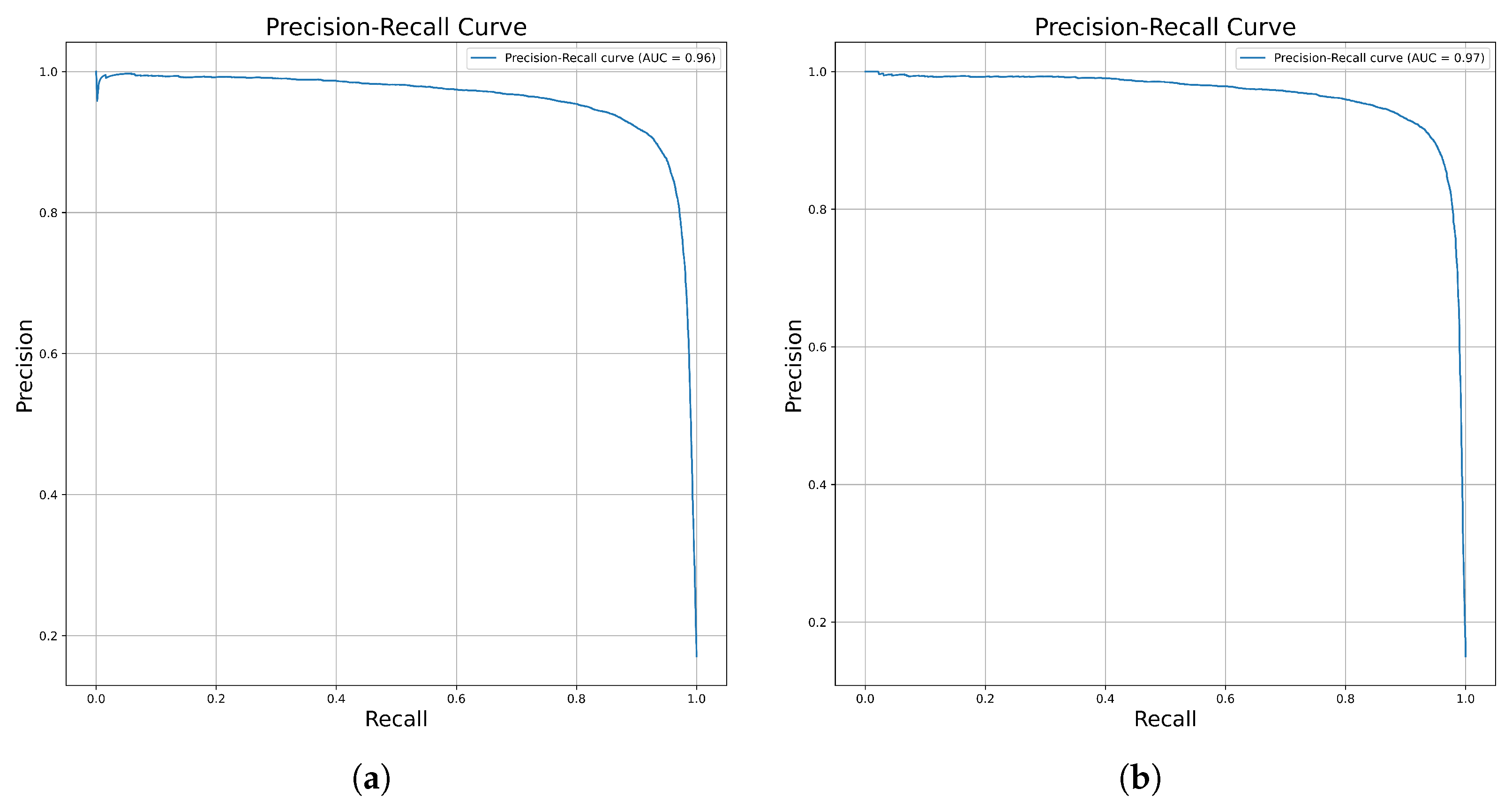
2.1. Cross-Validation Performance of the Proposed Model
2.2. Model Performance on the Independent Dataset
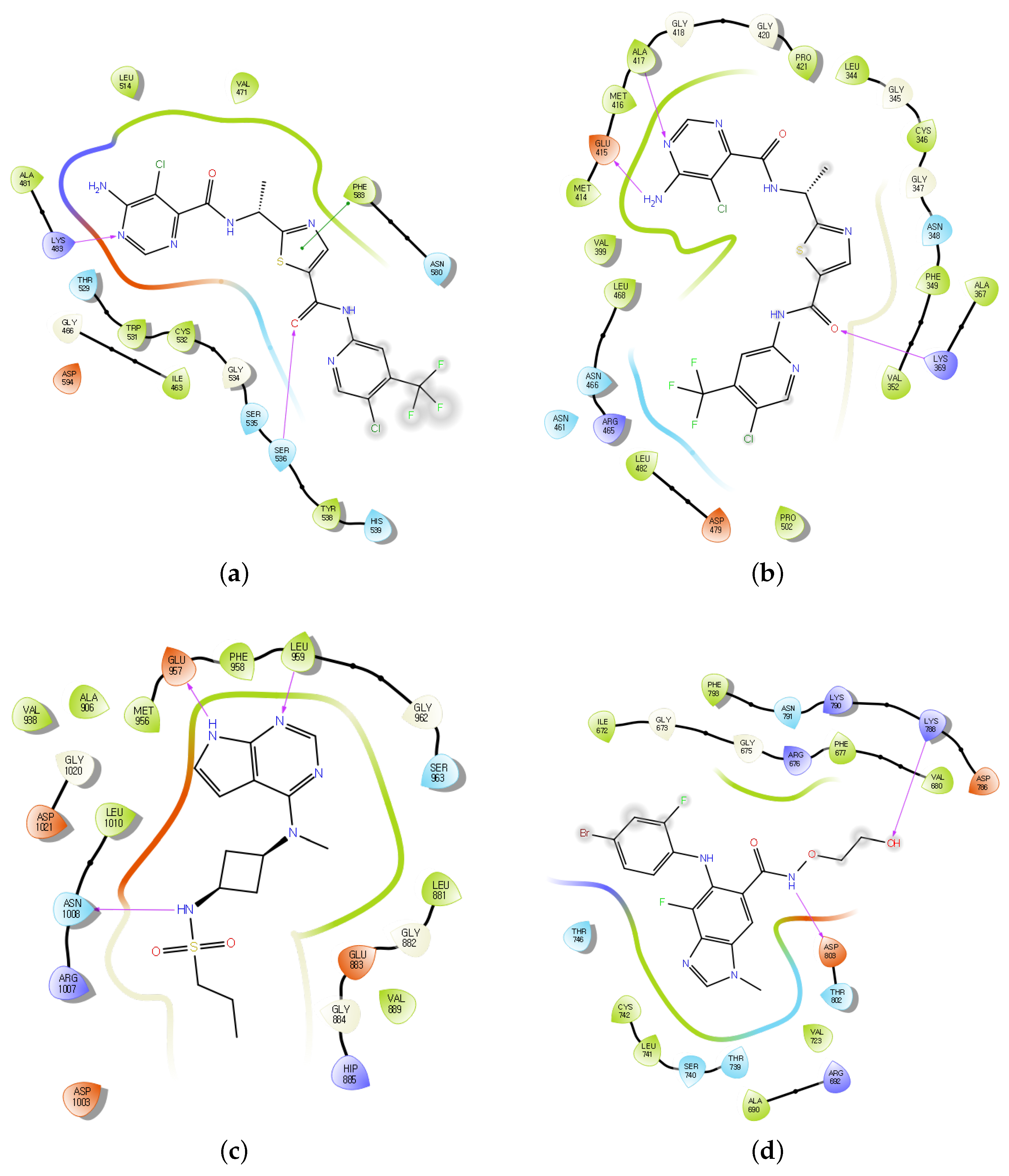
2.3. Experimental Validation of Kinase Inhibitors Using Deep Learning and Molecular Docking
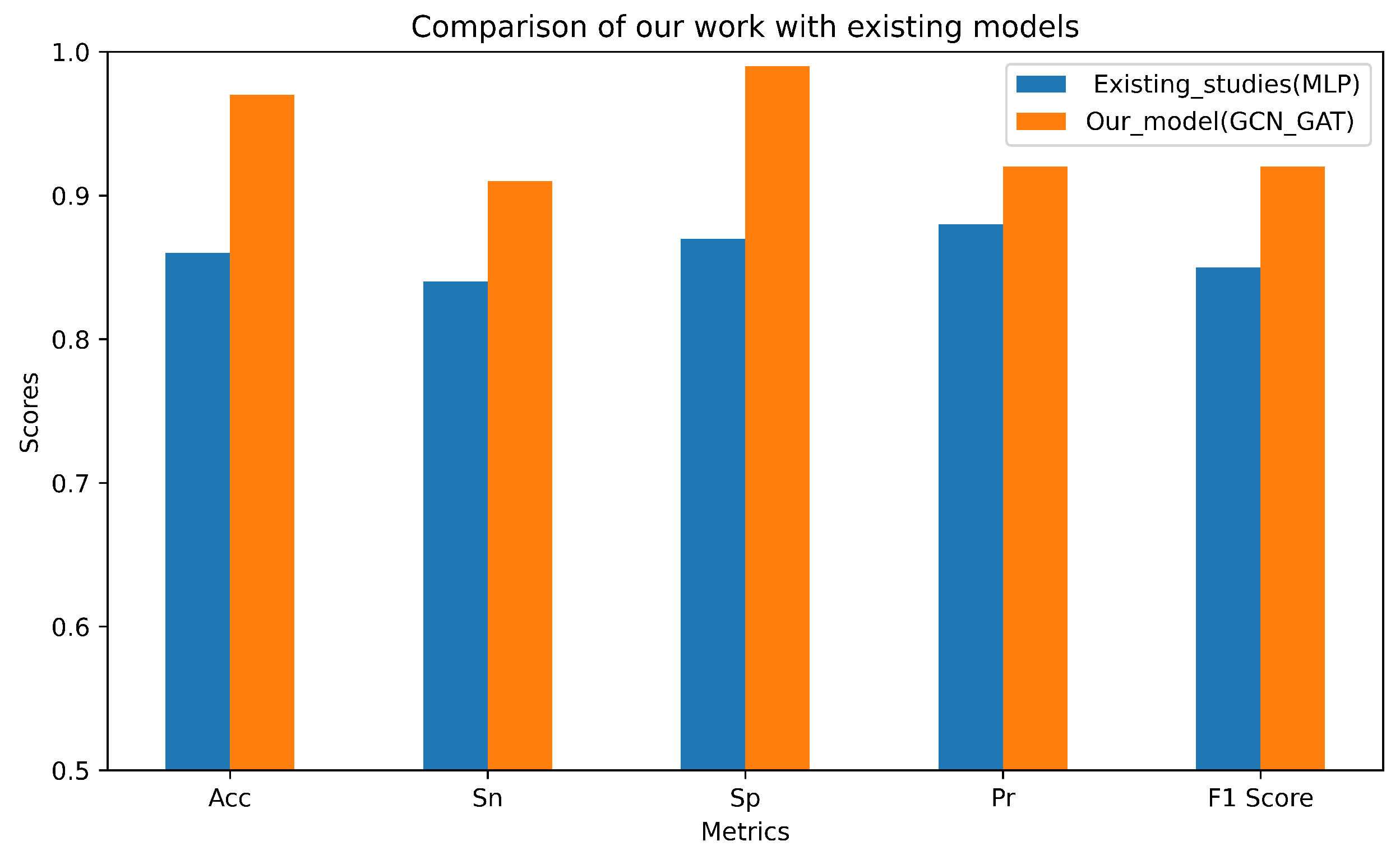
2.4. Comparison with Established Models
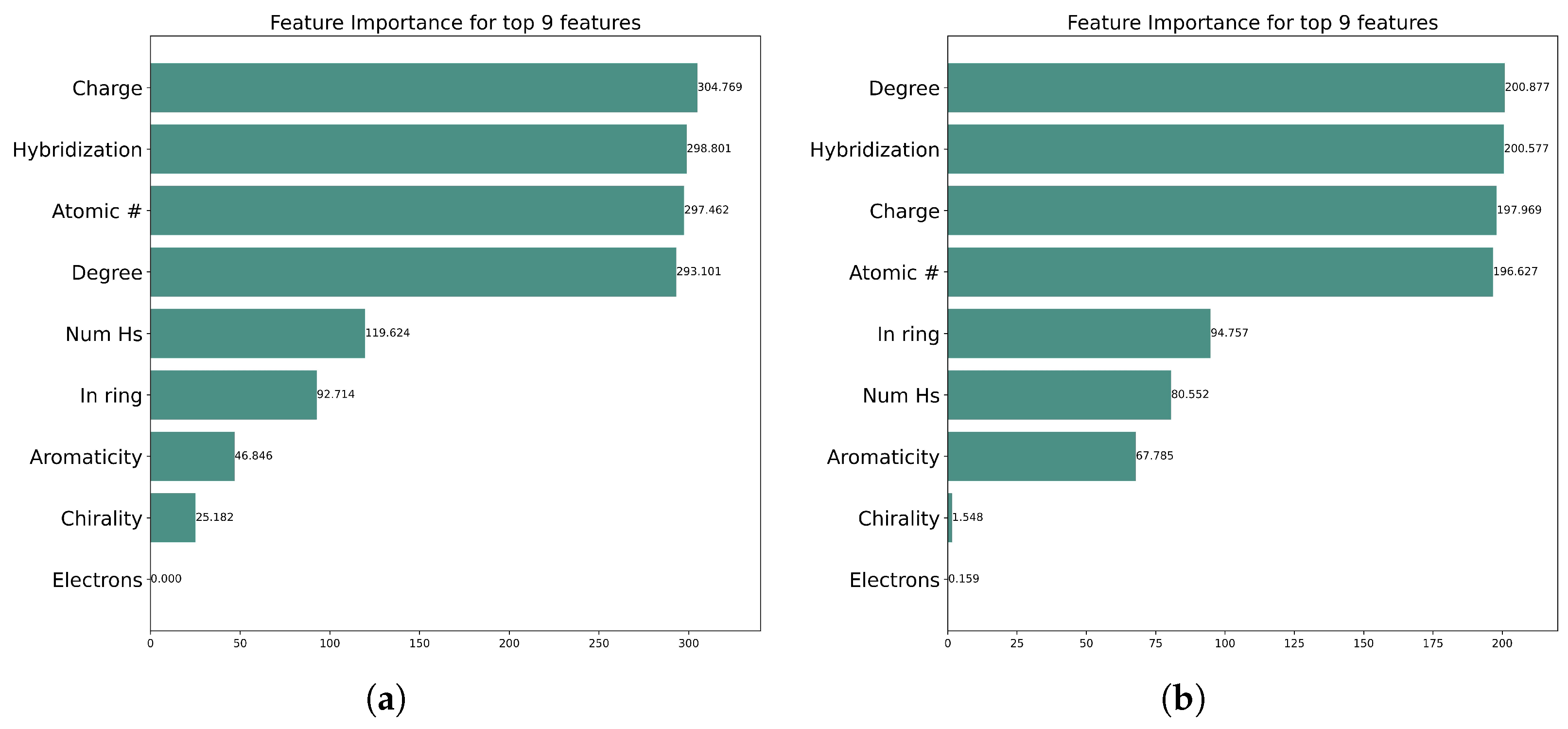
2.5. Graph Explainability and Feature Importance
3. Materials and Methods
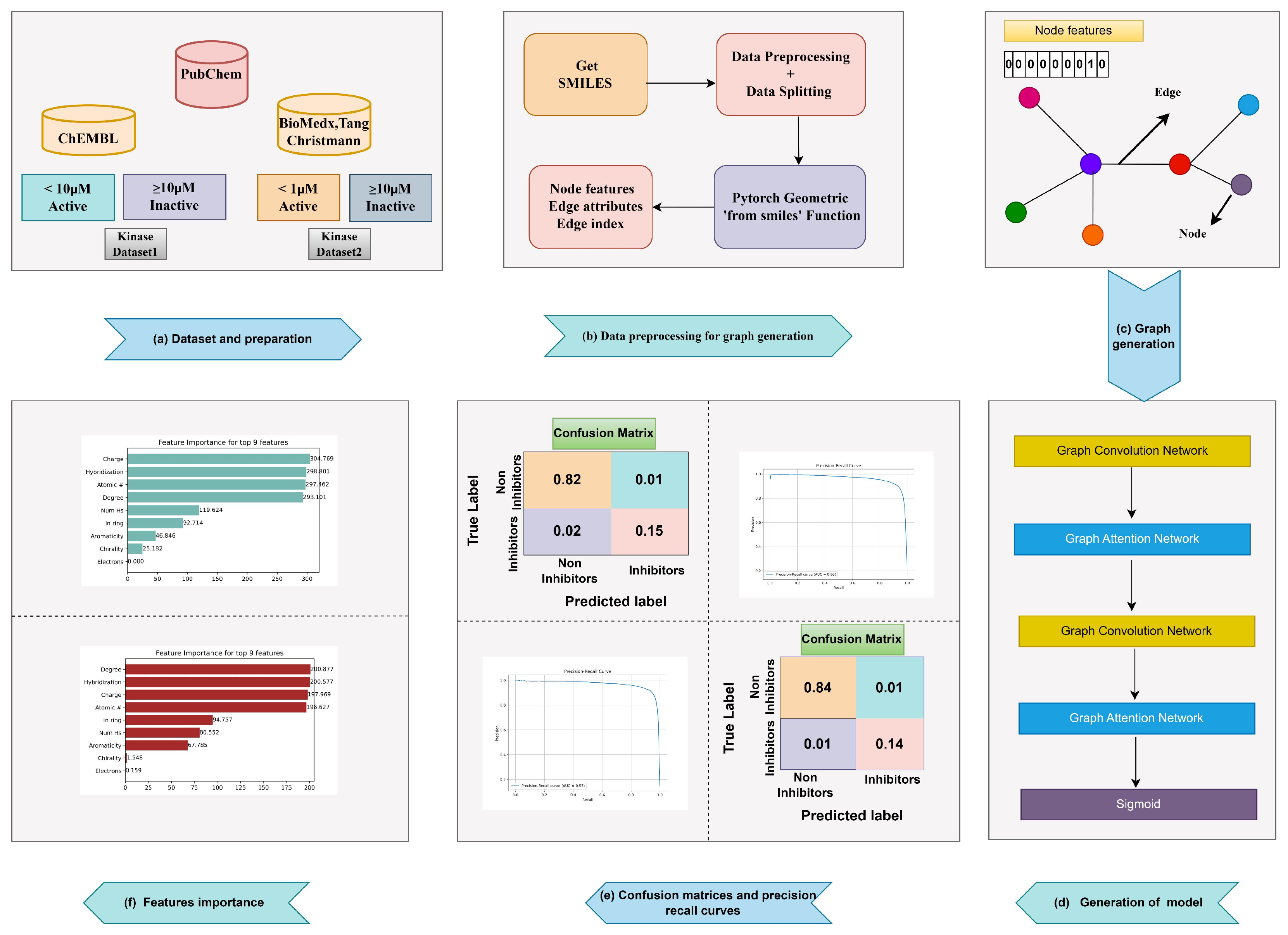
3.1. Data Collection and Preparation
3.2. Model Evaluation Measures
3.3. Graph Generation and Feature Extraction
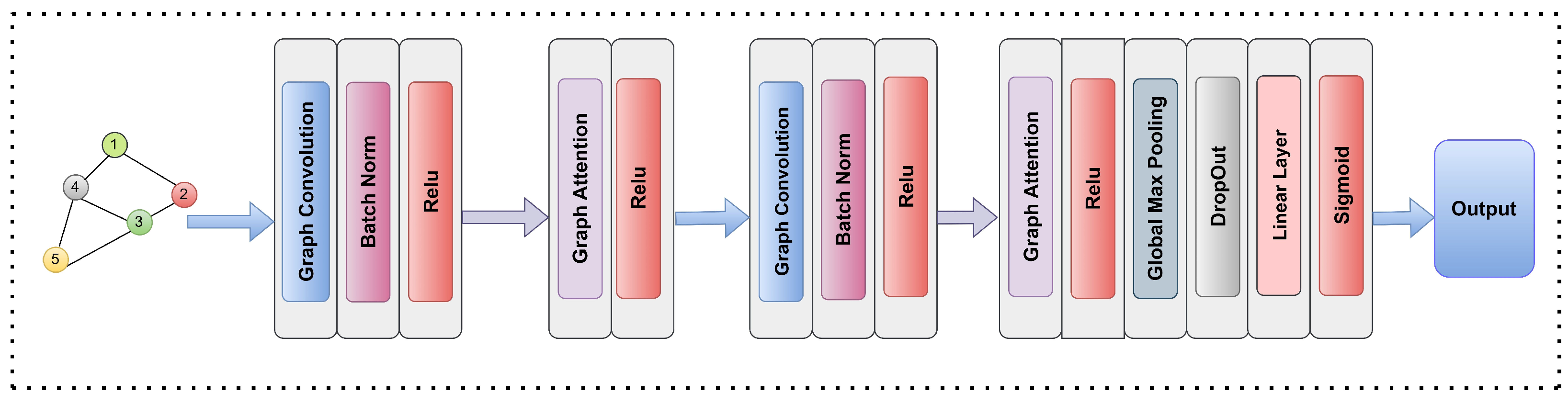
3.4. Graph Based Models Selection
3.4.1. Graph Convolutional Networks
- Every graph convolution layer transforms the node features X by aggregating the information from other neighboring nodes and itself using the adjacency matrix. The expression of the GCN can be written as follows:where X represents the node features matrix, represents the degree of the adjacency matrix with self-loop added, is the trainable weight matrix, is the activation function, represents the adjacency matrix with self-loop added, and I is the identity matrix.
3.4.2. Graph Attention Networks
- Consider a set of nodes each having some input node features. These features are passed as input to the GAT layer. The set of features for all the input nodes can be represented aswhere is the feature vector of node i with features F.
- The GAT layer produces a new set of node features as output, denoted aswhere is the updated feature of node i with node features .
- To transform the input features into higher-level features a learnable linear transformation using a shared matrix is applied to every node, where Next, a self-attention mechanism “a” is used to calculate the attention coefficient which determines the importance of neighboring node j’s features to node i, after which the raw attention scores are normalized using the softmax function as follows:where is the neighboring node of node i.
- For each neighbor j, the features of the nodes are first transformed using the weight matrix . These transformed features are then multiplied by the normalized attention scores . Summing these weighted scores and applying a nonlinearity yields new features for node i, as provided in the following equation:
3.5. Model Interpretation and Hyperparameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Grant, S. Therapeutic protein kinase inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Singha, M.; Pu, L.; Srivastava, G.; Ni, X.; Stanfield, B.A.; Uche, I.K.; Rider, P.J.; Kousoulas, K.G.; Ramanujam, J.; Brylinski, M. Unlocking the potential of kinase targets in cancer: Insights from CancerOmicsNet, an AI-driven approach to drug response prediction in cancer. Cancers 2023, 15, 4050. [Google Scholar] [CrossRef]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Lien, S.T.; Lin, T.E.; Hsieh, J.H.; Sung, T.Y.; Chen, J.H.; Hsu, K.C. Establishment of extensive artificial intelligence models for kinase inhibitor prediction: Identification of novel PDGFRB inhibitors. Comput. Biol. Med. 2023, 156, 106722. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Hessler, G.; Baringhaus, K.H. Artificial intelligence in drug design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Ai, D.; Xie, G.; Qadri, S.F.; Song, H.; Huang, Y.; Wang, Y.; Yang, J. Deep belief network modeling for automatic liver segmentation. IEEE Access 2019, 7, 20585–20595. [Google Scholar] [CrossRef]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Wu, X.; Xiong, Z.; Yang, T.; Fu, Z.; Liu, X.; Tan, X.; Zhong, F.; Wan, X.; et al. Deep learning enhancing kinome-wide polypharmacology profiling: Model construction and experiment validation. J. Med. Chem. 2019, 63, 8723–8737. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, R.; Bajorath, J. Multitask machine learning for classifying highly and weakly potent kinase inhibitors. ACS Omega 2019, 4, 4367–4375. [Google Scholar] [CrossRef]
- Hu, J.; Allen, B.K.; Stathias, V.; Ayad, N.G.; Schürer, S.C. Kinome-Wide Virtual Screening by Multi-Task Deep Learning. Int. J. Mol. Sci. 2024, 25, 2538. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Liu, X.; Fu, Z.; Xiong, Z.; Wu, X.; Tan, X.; Zhao, J.; Zhong, F.; Wan, X.; et al. KinomeX: A web application for predicting kinome-wide polypharmacology effect of small molecules. Bioinformatics 2019, 35, 5354–5356. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Zhang, X.; Zhao, A.; Li, X.; Chen, H.; Li, J. Recent Developments in GNNs for Drug Discovery. arXiv 2025, arXiv:2506.01302. [Google Scholar]
- Zhang, O.; Lin, H.; Zhang, X.; Wang, X.; Wu, Z.; Ye, Q.; Zhao, W.; Wang, J.; Ying, K.; Kang, Y.; et al. Graph Neural Networks in Modern AI-aided Drug Discovery. arXiv 2025, arXiv:2506.06915. [Google Scholar]
- Ng, S.S.; Lu, Y. Evaluating the use of graph neural networks and transfer learning for oral bioavailability prediction. J. Chem. Inf. Model. 2023, 63, 5035–5044. [Google Scholar] [CrossRef]
- Li, W.; Ma, W.; Yang, M.; Tang, X. Drug repurposing based on the DTD-GNN graph neural network: Revealing the relationships among drugs, targets and diseases. BMC Genom. 2024, 25, 584. [Google Scholar] [CrossRef]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Sitaula, C.; Ghimire, N. An analysis of early stopping and dropout regularization in deep learning. Int. J. Concept. Comput. Inf. Technol. 2017, 5, 17–20. [Google Scholar]
- Mahsereci, M.; Balles, L.; Lassner, C.; Hennig, P. Early stopping without a validation set. arXiv 2017, arXiv:1703.09580. [Google Scholar]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2025 update. Pharmacol. Res. 2025, 216, 107723. [Google Scholar] [CrossRef]
- Maryam; Rehman, M.U.; Hussain, I.; Tayara, H.; Chong, K.T. A graph neural network approach for predicting drug susceptibility in the human microbiome. Comput. Biol. Med. 2024, 179, 108729. [Google Scholar] [CrossRef]
- Jeevan, K.; Palistha, S.; Tayara, H.; Chong, K.T. PUResNetV2. 0: A deep learning model leveraging sparse representation for improved ligand binding site prediction. J. Cheminform. 2024, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Bourgeois, D.; You, J.; Zitnik, M.; Leskovec, J. GNNExplainer: Generating explanations for graph neural networks. Adv. Neural Inf. Process. Syst. 2019, 32, 9240–9251. [Google Scholar] [PubMed]
- Amara, K.; Ying, R.; Zhang, Z.; Han, Z.; Shan, Y.; Brandes, U.; Schemm, S.; Zhang, C. Graphframex: Towards systematic evaluation of explainability methods for graph neural networks. arXiv 2022, arXiv:2206.09677. [Google Scholar]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Drewry, D.H.; Willson, T.M.; Zuercher, W.J. Seeding collaborations to advance kinase science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top. Med. Chem. 2014, 14, 340–342. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef]
- Merget, B.; Turk, S.; Eid, S.; Rippmann, F.; Fulle, S. Profiling prediction of kinase inhibitors: Toward the virtual assay. J. Med. Chem. 2017, 60, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Christmann-Franck, S.; van Westen, G.J.; Papadatos, G.; Beltran Escudie, F.; Roberts, A.; Overington, J.P.; Domine, D. Unprecedently large-scale kinase inhibitor set enabling the accurate prediction of compound–kinase activities: A way toward selective promiscuity by design? J. Chem. Inf. Model. 2016, 56, 1654–1675. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum 2013, 8, 5281. [Google Scholar]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Ahmad, W.; Tayara, H.; Shim, H.; Chong, K.T. SolPredictor: Predicting Solubility with Residual Gated Graph Neural Network. Int. J. Mol. Sci. 2024, 25, 715. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Zahid, H.; Tayara, H.; Chong, K.T. Harnessing machine learning to predict cytochrome P450 inhibition through molecular properties. Arch. Toxicol. 2024, 98, 2647–2658. [Google Scholar] [CrossRef]
- Hassan, M.T.; Tayara, H.; Chong, K.T. Possum: Identification and interpretation of potassium ion inhibitors using probabilistic feature vectors. Arch. Toxicol. 2025, 99, 225–235. [Google Scholar] [CrossRef]
- Chicco, D.; Tötsch, N.; Jurman, G. The Matthews correlation coefficient (MCC) is more reliable than balanced accuracy, bookmaker informedness, and markedness in two-class confusion matrix evaluation. BioData Min. 2021, 14, 13. [Google Scholar] [CrossRef]
- Okoe, M.; Jianu, R.; Kobourov, S. Node-link or adjacency matrices: Old question, new insights. IEEE Trans. Vis. Comput. Graph. 2018, 25, 2940–2952. [Google Scholar] [CrossRef]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. [Google Scholar] [CrossRef]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Nguyen, T.; Le, H.; Quinn, T.P.; Nguyen, T.; Le, T.D.; Venkatesh, S. GraphDTA: Predicting drug–target binding affinity with graph neural networks. Bioinformatics 2021, 37, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Grattarola, D.; Zambon, D.; Bianchi, F.M.; Alippi, C. Understanding pooling in graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2022, 35, 2708–2718. [Google Scholar] [CrossRef]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Shyam Sunder, S.; Sharma, U.C.; Pokharel, S. Adverse effects of tyrosine kinase inhibitors in cancer therapy: Pathophysiology, mechanisms and clinical management. Signal Transduct. Target. Ther. 2023, 8, 262. [Google Scholar] [CrossRef]
- Purushotham, S.; Tripathy, B. Evaluation of classifier models using stratified tenfold cross validation techniques. In Global Trends in Information Systems and Software Applications, Proceedings of the 4th International Conference on Computing and Communication Systems, Vellore, TN, India, 9–11 December 2011; Springer: Berlin/Heidelberg, Germany, 2011; pp. 680–690. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Performance on the 10 Folds | Performance on Holdout Data | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc | Ba | MCC | Sn | Sp | Pr | F1 | Acc | Ba | MCC | Sn | Sp | Pr | F1 | |
| Dataset 1 (GCN_GAT) | 0.97 | 0.94 | 0.89 | 0.90 | 0.98 | 0.92 | 0.91 | 0.96 | 0.94 | 0.89 | 0.90 | 0.98 | 0.91 | 0.90 |
| Dataset 2 (GCN_GAT) | 0.97 | 0.95 | 0.90 | 0.91 | 0.98 | 0.92 | 0.91 | 0.97 | 0.95 | 0.90 | 0.91 | 0.99 | 0.92 | 0.92 |
| Dataset 1 (GCN) | 0.92 | 0.84 | 0.73 | 0.80 | 0.97 | 0.83 | 0.77 | 0.92 | 0.84 | 0.72 | 0.71 | 0.97 | 0.83 | 0.76 |
| Dataset 2 (GCN) | 0.94 | 0.85 | 0.74 | 0.72 | 0.97 | 0.83 | 0.77 | 0.93 | 0.84 | 0.74 | 0.72 | 0.97 | 0.83 | 0.77 |
| Node Features | Range and Description |
|---|---|
| Hybridization | S, SP, SP3, etc. |
| Degree | 00–11 |
| Formal charge | −05–07 |
| No. of Hs | 00–09 |
| No. of radical electrons | 00–05 |
| Atomic number | 01–119 |
| Is aromatic | Boolean |
| Is in ring | Boolean |
| Chirality | Atom chirality |
| Hyperparameters | Values |
|---|---|
| Learning rate | 0.0001 |
| Batch size | 32 |
| GCN layers | 2 |
| GAT layers | 2 |
| Dropout rate | 0.4 |
| Pooling layer | global max |
| Activation function | Relu |
| Optimizer | Adam |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahid, H.; Chong, K.T.; Tayara, H. Integrating Graph Convolution and Attention Mechanism for Kinase Inhibition Prediction. Molecules 2025, 30, 2871. https://doi.org/10.3390/molecules30132871
Zahid H, Chong KT, Tayara H. Integrating Graph Convolution and Attention Mechanism for Kinase Inhibition Prediction. Molecules. 2025; 30(13):2871. https://doi.org/10.3390/molecules30132871
Chicago/Turabian StyleZahid, Hamza, Kil To Chong, and Hilal Tayara. 2025. "Integrating Graph Convolution and Attention Mechanism for Kinase Inhibition Prediction" Molecules 30, no. 13: 2871. https://doi.org/10.3390/molecules30132871
APA StyleZahid, H., Chong, K. T., & Tayara, H. (2025). Integrating Graph Convolution and Attention Mechanism for Kinase Inhibition Prediction. Molecules, 30(13), 2871. https://doi.org/10.3390/molecules30132871